

Abstract
Purpose
Upregulation of sphingosine-1-phosphate receptor 1 (S1PR1) expression in multiple sclerosis (MS) lesions is associated with neuroinflammatory response. This study investigated the correlation between neuroinflammation and S1PR1 expression in the spinal cord of an experimental autoimmune encephalomyelitis (EAE) rat model of MS, using the S1PR1 positron emission tomography (PET) radiotracer [11C]TZ3321.
Procedures
MicroPET imaging studies of [11C]TZ3321 were performed to measure uptake of [11C]TZ3321 in the spinal cord of EAE rats. Immunohistochemical staining was performed to confirm the overexpression of S1PR1 and other inflammatory biomarkers.
Results
MicroPET imaging demonstrated a 20–30 % increase in [11C]TZ3321 uptake in the lumbar spinal cord of EAE rats versus sham controls at 35–60 min post injection. The increased uptake of [11C]TZ3321 was correlated with the overexpression of S1PR1 in the lumbar spinal cord of EAE rats that was confirmed by immunohistochemical staining. Upregulated S1PR1 expression was associated with glial cell activation and immune cell infiltration.
Conclusions
MicroPET imaging modality with a specific radioligand [11C]TZ3321 is able to assess the expression of S1PR1 in EAE rat lumbar spinal cord. This may provide a new approach to the assessment of neuroinflammatory response in MS and other inflammatory diseases.
Keywords: Sphingosine-1-phosphate receptor 1, Multiple sclerosis, Radiotracer, PET imaging, Neuroinflammation, Experimental autoimmune encephalomyelitis
Introduction
Multiple sclerosis (MS) is a neuroinflammatory demyelinating disease of the central nervous system (CNS) [1, 2] which affects 2.5 million people worldwide [3]. The mechanisms of MS pathogenesis and progression are incompletely understood. Sphingolipids, which play an important role in the nervous system, were linked to MS several decades ago; sphingosine-1-phosphate (S1P) and its receptors were recently identified as a critical target for research on pathophysiology of MS and for the development of therapeutics. S1P is a membrane-derived lysophospholipid that binds with five subtypes of G protein-coupled receptors, S1PR1-5. Activated S1PRs participate in key biological signaling processes including activities in the CNS and immune response [4]. The immunomodulator fingolimod (FTY-720, Gilenya) was the first oral disease-modifying drug approved by the US Food and Drug Administration (FDA) for treating relapsing-remitting MS in 2010 [5]. Following in vivo phosphorylation, fingolimod sequesters lymphocytes within the lymph nodes, thus preventing their trafficking to the CNS by the binding of the phosphorylated drug to S1PR1, S1PR3, S1PR4, and S1PR5 but not S1PR2 [6]. The success of treating relapsing-remitting MS using the non-selective modulator fingolimod has inspired significant research efforts devoted to investigating the role of individual S1PR subtypes, especially S1PR1 [7–9], in the pathogenesis and progression of MS.
A strong increase in S1PR1 expression is detected in MS lesions in postmortem brain tissues [10]. The major cell sources of S1PR1 in MS lesions are glial cells and infiltrating T cells; both types of cells produce pro-inflammatory cytokines [11, 12]. In the most commonly used rodent model of MS, experimental autoimmune encephalomyelitis (EAE), studies using knockout mice have demonstrated that pro-inflammatory cytokines such as interleukin (IL)-6 and chemokine (C-C motif) ligand 2 (CCL2) play an important role in progression of EAE [13, 14]. S1P-S1PR1 signaling has been reported to directly activate the differentiation of interleukin 17 (IL-17)-producing T cells, which is a key determinant of disease severity in MS, and promoted IL-17 production [11]. Fingolimod has a marked S1PR1-dependent therapeutic effect on EAE by reducing infiltration of IL-17-producing T cells into the spinal cord of mice [15–17]. These observations collectively indicate that the expression and activation of S1PR1 reflects the progression of neuroinflammation in MS and suggest that S1PR1 is a potential biomarker for monitoring progression of neuroinflammation in patients with MS.
Positron emission tomography (PET) with suitable radiotracers provides a highly sensitive non-invasive imaging methodology, and it can directly quantify the expression of target biomolecules in living subjects. Several groups have reported their efforts on identifying promising S1P receptor tracers, including BZM055, W146, and other FTY-720 analogs [18–20]. However, these ligands either suffer from low rate of phosphorylation which is required for binding to S1P [18, 20] or display fast in vivo defluorination [19]. Currently, the implementation of PET imaging modality for S1PR1 target is hampered due to lack of a suitable specific S1PR1 radiotracer. Our group previously reported the radiosynthesis of [11C]TZ3321 and in vivo evaluation in a mouse model of neointimal hyperplasia [21]. TZ3321 has a highly binding potency for S1PR1 (IC50 = 2.13 ± 1.63 nM) and does not bind to S1PR2 (IC50 > 1000 nM) or S1PR3 (IC50 > 1000 nM); the uptake of [11C]TZ3321 correlated well with inflammatory response at the site of neointimal hyperplasia [21]. In the current study, we explored the implementation of [11C]TZ3321 for assessment of S1PR1 expression in inflammatory lesions in an EAE rat model of MS.
Methods
Radiosynthesis
The radiotracer [11C]TZ3321 was produced as reported recently by our group [21]. Briefly, the radiosynthesis of [11C]TZ3321 was accomplished by alkylating the precursor with [11C]methyl triflate in acetonitrile at 60 °C for 5 min, followed by removal of the t-butyl group using trifluoroacetic acid, and was purified using reverse phase high-performance liquid chromatography (HPLC). The radiosynthesis of [11C]TZ3321 was accomplished in 50–55 min; the radiochemical yield was 50–70 % (decay corrected, n > 10), radiochemical purity > 98 %, and specific activity (SA) > 2 Ci/μmol (at the end of synthesis (EOS)).
Induction of EAE in Female Lewis Rats
All animal experiments were conducted in compliance with the Guidelines for the Care and Use of Research Animals under protocols approved by Washington University’s Animal Studies Committee. Female Lewis rats (Charles River Laboratories, Inc., Wilmington, MA), weighing 100–125 g on arrival, were acclimated in the animal facility for 1 week before use.
To test the feasibility of in vivo imaging S1PR1 using PET, an EAE rat model of MS characterized by acute neuroinflammation induced by injection of myelin basic protein (MBP) was generated according to literature reports [22]. Before each immunization, an emulsion of MBP fragment (MBP68-86, AnaSpec Inc., San Jose, CA) was freshly prepared by mixing equal volumes of MBP68-86 (0.5 mg/ml in phosphate-buffered saline (PBS)) and complete Freund’s adjuvant (CFA) containing 1.0 mg/ml heat-inactivated Mycobacterium tuberculosis H37Ra (Difco Laboratories, Detroit, MI). The emulsion for sham control rats was freshly prepared by similarly emulsifying equal volumes of PBS and CFA containing 1 mg/ml heat-inactivated M. tuberculosis. Rats were anesthetized with 2–3 % isoflurane in O2, and 200 μl of emulsion was injected into the foot pad, divided equally between the both hind feet.
Following immunization, animal weight and neurological deficits were monitored daily. To grade neurological impairment in EAE rats, the following scoring system was used according to the literature [22]. Total six grades are used to describe the severity of EAE, 0 representing the baseline and 5 represents the most severe condition of EAE: 0, no symptoms; 1, flaccid tail; 2, hindlimb weakness; 3, paraparesis; 3.5, unilateral hindlimb paralysis; 4, bilateral hindlimb paralysis; and 5, bilateral hindlimb paralysis and incontinence. Approximately 2 weeks, post-immunization rats weighed 150–190 g. For rats used in the current study, sham controls were asymptomatic while EAE-induced rats exhibited scores of 3.5–4.0.
MicroPET Scan and Data Acquisition
EAE rats with a score of 3.5–4.0 (n = 4) and age-matched sham control rats (n = 4) were used for microPET study. Small animal imaging was performed using an Inveon PET/CT system or a F220 scanner (Siemens Inc., Knoxville, TN). Rats were anesthetized in an induction chamber with 2–3 % isoflurane in oxygen, and a catheter was placed in the lateral tail vein. Each rat was secured to a custom-designed acrylic bed equipped with a nosecone for gas anesthesia. PET/CT scans were conducted under 1.5–2 % isoflurane anesthesia with the rat placed in transaxial position with the lumbar spines centered in the field of view. Following a CT scan in the Inveon for anatomical coregistration, the rat was moved to the F220 if needed for a transmission scan or remained on the Inveon for the PET scan. This allowed imaging of two rats from each production of the 11C-labeled radiopharmaceutical. A list mode protocol was used with 60-min dynamic data acquisition. Animals received a bolus injection of [11C]TZ3321 (0.35–0.5 mCi) via the tail vein catheter; body temperature was maintained with heat lamps during the imaging session. For histological and immunohistochemical analysis, the rats were euthanized immediately following the PET scan; the spinal cords of rats were harvested for staining studies.
PET image data was processed using filter back projection algorithm with attenuation and scatter corrections. The data were reconstructed per time frame employing an iterative reconstruction algorithm (three-dimensional ordered subset expectation maximization (3D-OSEM), maximum a posteriori (MAP)) and corrected for decay, random coincidences, scatter, and attenuation. The list mode data of the emission scans were reframed into a dynamic sequence of 1 × 3, 6 × 2, 9 × 5, 6 × 10, 4 × 30, 2 × 60 s, 2 × 2, 10 × 5 min. Image data was analyzed using Inveon Research Workstation software IRW 4.2 (Siemens Inc., Knoxville, TN). Regions of interest (ROIs), including the lumbar spinal cord and abdominal aorta, were drawn over the coregistered PET/CT images (details shown in Fig. S1). Time-activity curves (TACs) were obtained and were expressed as dimensionless standardized uptake values (SUVs). SUV is defined as [tissue activity concentration (mCi/g) × body weight (g)/injected dose (mCi)]. Tracer uptake in spinal cord was calculated by Logan Reference (LoganREF) model [23–25] with the abdominal aorta (blood) as the reference region. The distribution volume ratio (DVR) was estimated for each ROI using IRW 4.2 program. The SUV ratio (SUVR) of lumbar spinal cord to abdominal aorta was also calculated as a modeling-independent parameter for evaluating tracer uptake. One-tailed t test was used for the comparison of the tracer uptake in EAE rats versus sham control animals. A P value less than 0.05 was considered statistically significant.
Histological Analyses
To further explore the S1PR1 expression and neuroinflammatory status in EAE rat spinal cord, both immunohistochemical and immunofluorescence analyses were performed. Immediately following microPET imaging, rat spinal cord samples (n = 2 for EAE and sham control) were fixed in 10 % formalin, then embedded in paraffin and cut into 5-μm sections. Hematoxylin and eosin (H&E) staining and Luxol fast blue staining were performed to visually check lesion morphology and demyelination surrounding spinal cord lesions. For immunohistochemical analysis of S1PR1 expression, glial activation, and inflammatory cell infiltration, sections were deparaffinized in xylene and rehydrated through a graded alcohol series to water. Endogenous peroxidase activity was quenched with a solution of 3 % H2O2 in PBS for 5 min. Slides were incubated in blocking buffer for 30 min before incubating with the primary antibody overnight at 4 °C. The primary antibodies used in this study included (1) a rabbit anti-rat S1PR1 antibody (1:50, Santa Cruz Biotechnology, Santa Cruz, CA), (2) a mouse anti-rat glial fibrillary acidic protein (GFAP) antibody (1:400, Millipore, Billerica, MA), (3) a mouse anti-rat ionized calcium-binding adapter molecule 1 (Iba-1) antibody (1:300, Millipore, Billerica, MA), and (4) a rabbit anti-rat IL-17 antibody (1:100, Abcam, Cambridge, MA). Primary antibody binding was detected using an anti-rabbit horseradish peroxidase/3,3-diaminobenzidine (HRP-DAB) staining kit (R&D Systems, Minneapolis, MN) following the manufacturer’s instructions. To further identify cell types expressing S1PR1, immunofluorescence was performed on deparaffinized and hydrated slides. Endogenous peroxidases were quenched using 0.3 % hydrogen peroxide in PBS. Sections were incubated in blocking buffer for 60 min, following deparaffinization, rehydration, and endogenous peroxidase deactivation. The primary antibodies were added as a mixture of the rabbit anti-rat S1PR1 antibody (1:50) and the mouse anti-rat GFAP (1:400)/Iba-1 (1:300) antibodies and incubated with the slides at 4 °C overnight. The secondary fluorescently labeled antibodies (fluorescein-conjugated goat anti-mouse IgG antibody (1:100, Jackson ImmunoResearch Lab, West Grove, PA) and rhodamine-conjugated donkey anti-rabbit IgG antibody (1:100, Jackson ImmunoResearch Lab, West Grove, PA)) were added and incubated in the dark for 60 min. A Nikon E600 microscope with a charge-coupled device camera was used to obtain all photomicrographs.
Results
The microPET scans were performed in EAE rats with a score of 3.5–4.0 and asymptomatic age-matched sham controls. The summed images of PET scans (0–60 min) in EAE and sham rats are shown in Fig. 1. The PET/CT images clearly visualize high accumulation of the S1PR1-specific radiotracer [11C]TZ3321 in L1–L6 of the EAE-induced rat lumbar spinal cord. The time–activity curves of SUVs for the lumbar spinal cord and abdominal aorta regions are shown in Fig. 2 and Fig. S2. Tracer uptake in the lumbar spinal cord reached steady state within 20 min after tracer injection and was higher than that in abdominal aorta (blood), in both EAE and sham rats. There was no significant difference of the abdominal aorta SUV between EAE and sham (Table 1); thus, the abdominal aorta was used as a reference region for comparison with tracer uptake in lumbar spinal cords, as well as for the kinetic modeling. Normalized SUVR curves showing the SUV of lumbar spinal cord divided by SUV of abdominal aorta (the reference region) revealed higher tracer accumulation in EAE rats than in sham rats shown in Fig. 2. Quantitative analysis showed 24.6 % increase in tracer uptake in the EAE rat lumbar spinal cord, compared with the sham group (SUVR summed from 35 to 60 min, 1.47 ± 0.18 vs. 1.18 ± 0.04, P < 0.05, n = 4, Table 1). The increased percentage was 37.0 % when directly using lumbar spinal cord SUV values for comparison (SUV summed from 35 to 60 min, 1.00 ± 0.18 vs. 0.73 ± 0.14, P < 0.05, n = 4, Table 1). The difference in tracer uptake between the two groups was also estimated by DVR, calculated by LoganREF model with the abdominal aorta (blood) as the reference region. The resulting DVR was 1.41 ± 0.11 for EAE rats and 1.15 ± 0.08 for shams, with a 22.5 % increase (P < 0.01, n = 4, Table 1).
Fig. 1.
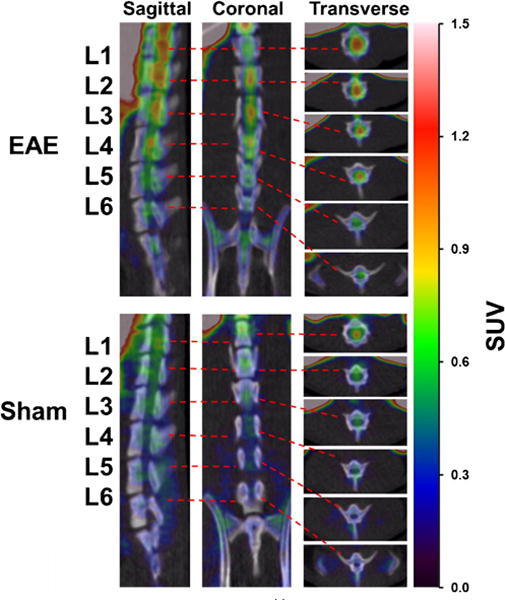
MicroPET/CT imaging of [11C]TZ3321 in EAE rat spinal cord. Representative images from sagittal, coronal and transverse views, summed from a 60-min microPET scan, showed high tracer uptake in EAE rat lumbar spinal cord (L1–L6).
Fig. 2.
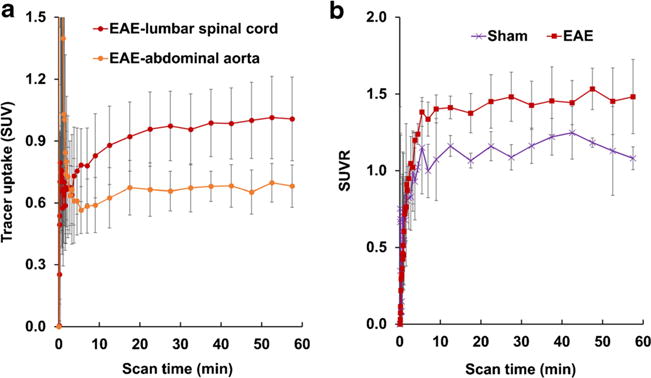
Time tissue–activity curves (TACs) of [11C]TZ3321 in rat lumbar spinal cord and abdominal aorta (n = 4). a TACs of EAE rats revealed that the tracer uptake in lumbar spinal cord was about 1.5-fold higher than the abdominal aorta region. b Normalized standardized uptake ratio (SUVR) curves of lumbar spinal cord, using abdominal aorta as the reference, showing increased tracer uptake in EAE lumbar spinal cord than shams.
Table 1.
PET measurements of radioactivity in EAE rats and sham control rats
| EAE, mean ± SD | Sham, mean ± SD | Pb | Increased percentage, % | |
|---|---|---|---|---|
| SUV-lumbar spinal cord | 1.00 ± 0.18 | 0.73 ± 0.14 | <0.05* | 37.0 |
| SUV-abdominal aorta | 0.68 ± 0.09 | 0.61 ± 0.09 | >0.05 | N/A |
| SUVRa | 1.47 ± 0.18 | 1.18 ± 0.04 | <0.05* | 24.6 |
| DVR-LoganREF | 1.41 ± 0.11 | 1.15 ± 0.08 | <0.01** | 22.5 |
n = 4
SUVR values were the ratio of summed SUV in lumbar spinal cord to abdominal aorta
P values were calculated by one-tailed t test
P < 0.05,
P < 0.01
Histological analysis was carried out to determine the expression of S1PR1 and its cellular localization in EAE rat lumbar spinal cord. Inflammatory cell infiltration and demyelination of the EAE rat spinal cord were confirmed by H&E and Luxol fast blue staining shown in Fig. 3. Compared with sham controls, increased S1PR1 expression as well as activation of microglia and astrocytes was observed in EAE rat spinal cord, primarily in the white matter shown in Fig. 4. Immunofluorescence staining revealed that S1PR1 was expressed in both activated microglia and astrocytes during EAE shown in Fig. 5. There were also a large number of IL-17-producing T cells in EAE rat spinal cord, which showed localization similar to S1PR1-positive cells in adjacent slices (Fig. 6). These data indicate that the increase in S1PR1 expression was associated with neuroinflammation in EAE lumbar spinal cord.
Fig. 3.
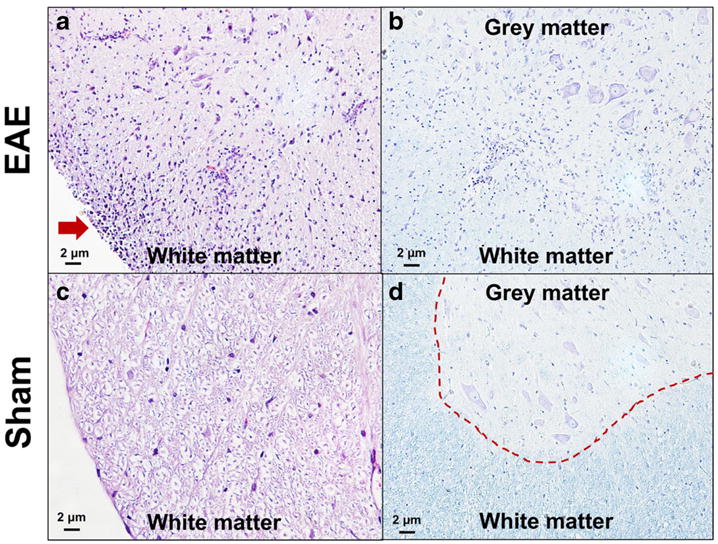
H&E staining and Luxol fast blue staining of lumbar spinal cords of EAE rats (a, b) and shams (c, d). H&E staining showed infiltration of inflammatory cells in EAE rat spinal cord (indicated by red arrow in a). Luxol fast blue staining of myelin (blue) revealed the borderline between gray matter and white matter (indicated by red dash line in d) became unclear (in b), indicating demyelination in EAE rat spinal cord.
Fig. 4.
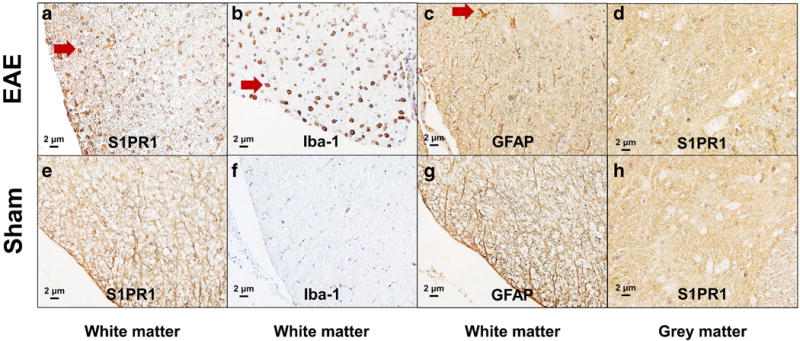
Immunohistochemical analysis of the lumbar spinal cord of EAE rats (a–d) and shams (e–h). Increased S1PR1 expression (red arrow in a), as well as activation of microglia (red arrow in b) and astrocytes (red arrow in c), was observed in the white matter of EAE rat spinal cord. No significant difference of S1PR1 expression in the gray matter of rat spinal cord in EAE (d) and sham (h) rats was observed.
Fig. 5.
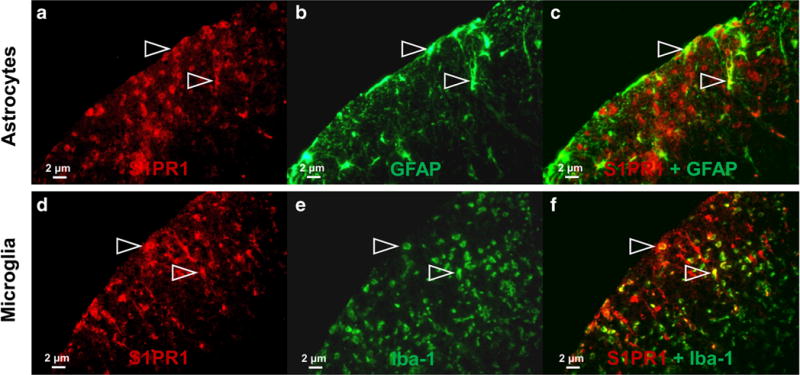
Double immunofluorescence staining for S1PR1 and GFAP (astrocytes)/Iba-1 (microglia) in the white matter of lumbar spinal cord of EAE rats. The colocalizations of S1PR1 (red) and GFAP (astrocytes, green)/Iba-1 (microglia, green) were indicated by white arrow.
Fig. 6.
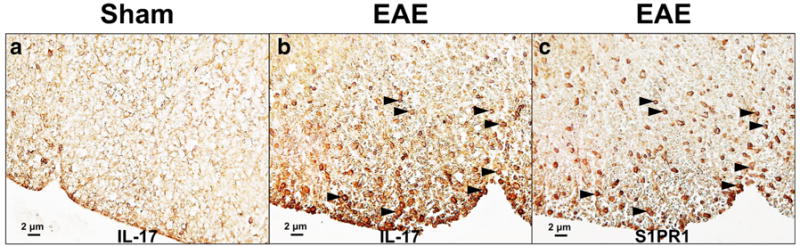
Immunohistochemical analysis of IL-17 in white matter of rat lumbar spinal cord. Increased IL-17-positive cells were found in EAE rat lumbar spinal cord (b, indicated by black arrow). The cellular location of IL-17 was similar as that of S1PR1-positive cells (c) as shown by the staining using adjacent slices.
Discussion
The present study demonstrated that microPET imaging with the S1PR1-specific radiotracer [11C]TZ3321 is able to detect the increase of S1PR1 expression in rat lumbar spinal cord in an EAE model of MS. Increased S1PR1 expression was further confirmed by immunohistochemical study in white matter of EAE rat lumbar spinal cord and colocalized within glial cells and infiltrating IL-17-producing T cells. Thus, [11C]TZ3321 PET imaging may provide a methodology to assess neuroinflammatory response in MS by quantitative measurement of S1PR1 expression.
In the female Lewis rat, active and passive EAE induced by MBP or by transfer of MBP-specific T cells typically produces severe CNS inflammation, with acute, modest demyelination (without remyelination) [26, 27]. Thus, MBP-induced EAE rat is a widely used animal model for the study of CNS inflammation. Using this animal model, Mattner et al. [28] showed that the degree of inflammation increases from rostral to caudal parts of the spinal cord and that the extent of lumbar/sacral inflammation was in good agreement with the rats’ clinical score. Notably, in this EAE model, lesions are found only in the spinal cord, not in the brain [26, 29].
S1PR1 signaling participates in pathology of different inflammatory diseases [30]. S1PR1 has been implicated in pathogenesis at multiple levels in MS, including immune-cell trafficking and activation, astrocyte proliferation, microglia activation, and potentially in altered blood–brain barrier function [31, 32]. This key role of S1PR1 in MS is underscored by the efficacy of fingolimod in treating remitting-relapsing MS patients [31]. Moreover, a strong increase of S1P1 expression was observed in human brain MS lesion by immunohistochemical analysis [10]. Colocalization analysis confirmed S1PR1 expression in both astrocytes and infiltrating immune cells [10], suggesting that the use of PET with a suitable S1PR1 radioligand should be able to assess the inflammatory response in MS. Recently, we reported that increased S1PR1 expression in response to vascular injury in a mouse wire injury model could be assessed by microPET imaging with the S1PR1-specific radiotracer [11C]TZ3321 [21]. Moreover, our previous biodistribution study in C57BL/6 mice showed high brain uptake of [11C]TZ3321, 7.09 ± 1.31%ID/g at 60 min post injection [21], which enables the application of the tracer in animal models of neuroinflammation. In the present study, PET imaging results clearly showed higher accumulation of [11C]TZ3321 in the spinal cord of EAE rats than that in sham controls. For quantitative assessment of the uptake of [11C]TZ3321 by PET, abdominal aorta (blood) was chosen as the reference region. The abdominal aorta is located parallel to the lumbar spinal cord and provides a convenient region for ROI delineation. Although there is S1PR1 expression on blood cells and endothelial cells [33], between EAE and sham rats, the change of radioactivity uptake of [11C]TZ3321 in abdominal aorta is negligible. Accordingly, the SUV ratio (lumbar spinal cord/abdominal aorta), as well as LoganREF model with abdominal aorta as the reference, revealed 20–30 % increase in EAE rats. Moreover, both SUV ratio and LoganREF model provide similar results, which ensure the application of reference-based analysis for [11C]TZ3321 PET imaging of spinal cord.
The cellular localization of S1PR1 in EAE rats versus sham control rats was further explored by post-PET immunohistochemistry and immunofluorescence. Increased expression of S1PR1 was detected mainly in the white matter, but not the gray matter, of EAE lumbar spinal cord. Immunohistological analysis focused on glial cells and immune cells, which are the main components of the white matter in EAE rat spinal cord. Our data revealed that EAE induced prominent activation of microglia and astrocytes, both of which showed S1PR1 expression. S1PR1 modulation of astrocytes has been found to be a key mechanism of fingolimod-induced direct CNS effects in MS, partially via the regulation of pro-inflammatory cytokine production by astrocyte S1PR1 signaling [7]. Concordantly, a recent study reported that S1PR1 activation induced production of IL-6, IL-8, and CCL2 in human astrocytic glioma which could be inhibited by fingolimod [34]. In contrast, the role of microglia in S1P/S1PR1 function and fingolimod treatment in MS has been poorly understood until recently. Noda and coworkers [12] found that fingolimod-induced S1PR1 activation downregulated activated microglial production of pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α), IL-1β, and IL-6. Fingolimod also upregulated microglial production of brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor (GDNF) and promoted the neuroprotective effects of microglia [12].
IL-17-producing T cells play a central role in the pathogenesis of MS [11, 35]. In our study, we found large amounts of IL-17-positive cells, with coexpression of S1PR1 in the EAE rat spinal cord. This observation was consistent with the findings in MS patients [36, 37]. It has been reported that S1P-S1PR1 signaling directly modulates the polarization of IL-17-producing T cells; impaired S1PR1 phosphorylation enhances the polarization of IL-17-producing T cells and exacerbates autoimmune neuroinflammation [11]. S1PR1 is also essential for the egress of IL-17-producing T cells from the lymph nodes under inflammatory conditions [16]. Moreover, fingolimod markedly decreases the infiltration of IL-17-producing T cells into the CNS [17].
The increased [11C]TZ3321 uptake in EAE rat lumbar spinal cord resulted from upregulation of S1PR1 expression in glial cells and infiltrated IL-17-producing T cells, as a reflection of the inflammatory status of EAE. Neuroimaging of inflammation in spinal cord lesions is currently a challenge. The radiotracers used in most studies target the translocator protein 18 kDa (TSPO), particularly the first-generation tracer [11C]PK11195 [38] and the second-generation tracer [18F]DPA-714 [22]. Clinical PET studies have revealed an increased TSPO signal in the white and gray matter of MS patients, which correlated with disease severity [39]. However, the application of TSPO radiotracers has been confounded by the expression of two different phenotypes of TSPO, with different binding affinities for all the TSPO ligands [40–42]. The existence of these two phenotypes of TSPO in human subjects thus adds variance to the clinical measurements [43]. Although the diameter of rat spinal cord is close to the resolution limit of the microPET scanner [22], which may affect the accuracy of ROI delineation, the promising microPET results from the EAE rat model of MS suggests that S1PR1 may provide a new approach imaging of neuroinflammation.
Conclusion
We reported here for the first time, imaging a key indicator of neuroinflammation, S1PR1, in spinal cord of EAE rats using the S1PR1-specific PET radiotracer [11C]TZ3321. The data clearly demonstrated that EAE-induced upregulation of S1PR1 expression in rat lumbar spinal cord can be assessed by PET imaging. Therefore, PET imaging with [11C]TZ3321 may provide a new method to detect neuroinflammatory response in patients with MS and other CNS diseases; PET tracers for this receptor may serve as a useful tool for drug evaluation and target engagement of S1PR1 modulation strategy.
Supplementary Material
Acknowledgments
This work was supported by DOE-Training in Techniques and Translation: Novel Nuclear Medicine Imaging Agents for Oncology and Neurology, DESC0008432, the Washington University School of Medicine Mallinckrodt Institute of Radiology (MIR) Cyclotron Facility Allotment #14-017, and NIH/NINDS, NS075527, and NIH/NIMH, MH092797. We thank Nicole Fettig, Margaret Morris, Amanda Roth, Lori Strong, and Ann Stroncek for their assistance with the microPET imaging studies, Marlene Scott and Bill Coleman in the Elvie L. Taylor Histology Core Facility of Washington University School of Medicine for sample embedding and H&E staining. The authors also like to thank Lynne A. Jones’ assistance in preparation of the manuscript.
Abbreviations
- 3D-OSEM
Three-dimensional ordered subset expectation maximization
- BDNF
Brain-derived neurotrophic factor
- CCL2
Chemokine (C-C motif) ligand 2
- CFA
Complete Freund’s adjuvant
- CNS
Central nervous system
- DAB
3,3-Diaminobenzidine
- EAE
Experimental autoimmune encephalomyelitis
- GDNF
Glial cell-derived neurotrophic factor
- GFAP
Glial fibrillary acidic protein
- HPLC
High-performance liquid chromatography
- HRP
Horseradish peroxidase
- Iba-1
Ionized calcium-binding adapter molecule 1
- IL
Interleukin
- MBP
Myelin basic protein
- MS
Multiple sclerosis
- PBS
Phosphate-buffered saline
- PET
Positron emission tomography
- S1PR1
Sphingosine-1-phosphate receptor 1
- SUV
Standardized uptake value
- TAC
Time–activity curve
- TNF-α
Tumor necrosis factor-α
- TSPO
Translocator protein 18 kDa
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11307-016-0944-y) contains supplementary material, which is available to authorized users.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14:183–193. doi: 10.1016/S1474-4422(14)70256-X. [DOI] [PubMed] [Google Scholar]
- 2.Ransohoff RM, Hafler DA, Lucchinetti CF. Multiple sclerosis—a quiet revolution. Nat Rev Neurol. 2015;11:134–142. doi: 10.1038/nrneurol.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Compston A, Confavreux C, Lassmann H, et al. McAlpine’s multiple sclerosis. 4th. Elsevier/Churchill Livingstone; Philadelphia: 2006. [Google Scholar]
- 4.Marsolais D, Rosen H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat Rev Drug Discov. 2009;8:297–307. doi: 10.1038/nrd2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 6.Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. N Engl J Med. 2012;366:339–347. doi: 10.1056/NEJMct1101691. [DOI] [PubMed] [Google Scholar]
- 7.Choi JW, Gardell SE, Herr DR, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A. 2011;108:751–756. doi: 10.1073/pnas.1014154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galicia-Rosas G, Pikor N, Schwartz JA, et al. A sphingosine-1-phosphate receptor 1-directed agonist reduces central nervous system inflammation in a plasmacytoid dendritic cell-dependent manner. J Immunol. 2012;189:3700–3706. doi: 10.4049/jimmunol.1102261. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Cabrera PJ, Cahalan SM, Nguyen N, et al. S1P1 receptor modulation with cyclical recovery from lymphopenia ameliorates mouse model of multiple sclerosis. Mol Pharmacol. 2012;81:166–174. doi: 10.1124/mol.111.076109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Doorn R, Van Horssen J, Verzijl D, et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia. 2010;58:1465–1476. doi: 10.1002/glia.21021. [DOI] [PubMed] [Google Scholar]
- 11.Garris CS, Wu L, Acharya S, et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat Immunol. 2013;14:1166–1172. doi: 10.1038/ni.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noda H, Takeuchi H, Mizuno T, Suzumura A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J Neuroimmunol. 2013;256:13–18. doi: 10.1016/j.jneuroim.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Samoilova EB, Horton JL, Hilliard B, et al. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J Immunol. 1998;161:6480–6486. [PubMed] [Google Scholar]
- 14.Izikson L, Klein RS, Charo IF, et al. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiba K, Kataoka H, Seki N, et al. Fingolimod (FTY720), sphingosine 1-phosphate receptor modulator, shows superior efficacy as compared with interferon-beta in mouse experimental autoimmune encephalomyelitis. Int Immunopharmacol. 2011;11:366–372. doi: 10.1016/j.intimp.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Maeda Y, Seki N, Kataoka H, et al. IL-17-producing Vγ4+ γδ T cells require sphingosine 1-phosphate receptor 1 for their egress from the lymph nodes under homeostatic and inflammatory conditions. J Immunol. 2015;195:1408–1416. doi: 10.4049/jimmunol.1500599. [DOI] [PubMed] [Google Scholar]
- 17.Seki N, Maeda Y, Kataoka H, et al. Role of sphingosine 1-phosphate (S1P) receptor 1 in experimental autoimmune encephalomyelitis—I. S1P-S1P1 axis induces migration of Th1 and Th17 cells. Pharmacol Pharm. 2013;4:628–637. [Google Scholar]
- 18.Briard E, Orain D, Beerli C, et al. BZM055, an iodinated radiotracer candidate for PET and SPECT imaging of myelin and FTY720 brain distribution. Chemmedchem. 2011;6:667–677. doi: 10.1002/cmdc.201000477. [DOI] [PubMed] [Google Scholar]
- 19.Prasad VP, Wagner S, Keul P, et al. Synthesis of fluorinated analogues of sphingosine-1-phosphate antagonists as potential radiotracers for molecular imaging using positron emission tomography. Bioorg Med Chem. 2014;22:5168–5181. doi: 10.1016/j.bmc.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Shaikh RS, Schilson SS, Wagner S, et al. Synthesis and evaluation of fluorinated Fingolimod (FTY720) analogues for sphingosine-1-phosphate receptor molecular imaging by positron emission tomography. J Med Chem. 2015;58:3471–3484. doi: 10.1021/jm502021d. [DOI] [PubMed] [Google Scholar]
- 21.Jin H, Yang H, Liu H, et al. A promising carbon-11 labeled sphingosine-1-phosphate receptor 1 specific PET tracer for imaging vascular injury. J Nucl Cardiol. 2016 doi: 10.1007/s12350-015-0391-1. [DOI] [PubMed] [Google Scholar]
- 22.Abourbeh G, Theze B, Maroy R, et al. Imaging microglial/macrophage activation in spinal cords of experimental autoimmune encephalomyelitis rats by positron emission tomography using the mitochondrial 18 kDa translocator protein radioligand [18F]DPA-714. J Neurosci. 2012;32:5728–5736. doi: 10.1523/JNEUROSCI.2900-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Innis RB, Cunningham VJ, Delforge J, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 24.Logan J, Fowler JS, Volkow ND, et al. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Logan J, Fowler JS, Volkow ND, et al. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(−)-cocaine PET studies in human subjects. J Cereb Blood Flow Metab. 1990;10:740–747. doi: 10.1038/jcbfm.1990.127. [DOI] [PubMed] [Google Scholar]
- 26.Meeson AP, Piddlesden S, Morgan BP, Reynolds R. The distribution of inflammatory demyelinated lesions in the central nervous system of rats with antibody-augmented demyelinating experimental allergic encephalomyelitis. Exp Neurol. 1994;129:299–310. doi: 10.1006/exnr.1994.1172. [DOI] [PubMed] [Google Scholar]
- 27.Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS) Br J Pharmacol. 2011;164:1079–1106. doi: 10.1111/j.1476-5381.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattner F, Katsifis A, Staykova M, et al. Evaluation of a radiolabelled peripheral benzodiazepine receptor ligand in the central nervous system inflammation of experimental autoimmune encephalomyelitis: a possible probe for imaging multiple sclerosis. Eur J Nucl Med Mol Imaging. 2005;32:557–563. doi: 10.1007/s00259-004-1690-y. [DOI] [PubMed] [Google Scholar]
- 29.Linker RA, Lee DH. Models of autoimmune demyelination in the central nervous system: on the way to translational medicine. Exp Transl Stroke Med. 2009;1:5. doi: 10.1186/2040-7378-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kunkel GT, MacEyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12:688–702. doi: 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 32.Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33:91–101. doi: 10.1097/WNF.0b013e3181cbf825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res. 2014;55:1596–1608. doi: 10.1194/jlr.R046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seki N, Kataoka H, Sugahara K, et al. Role of sphingosine 1-phosphate (S1P) receptor 1 in experimental autoimmune encephalomyelitis—II. S1P-S1P1 axis induces pro-inflammatory cytokine production in astrocytes. Pharmacol Pharm. 2013;4:638–646. [Google Scholar]
- 35.Mehling M, Lindberg R, Raulf F, et al. Th17 central memory T cells are reduced by FTY720 in patients with multiple sclerosis. Neurology. 2010;75:403–410. doi: 10.1212/WNL.0b013e3181ebdd64. [DOI] [PubMed] [Google Scholar]
- 36.Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matusevicius D, Kivisakk P, He B, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 38.de Paula FD, Vlaming ML, Copray SC, et al. PET imaging of disease progression and treatment effects in the experimental autoimmune encephalomyelitis rat model. J Nucl Med. 2014;55:1330–1335. doi: 10.2967/jnumed.114.137216. [DOI] [PubMed] [Google Scholar]
- 39.Colasanti A, Piccini P. PET imaging in multiple sclerosis: focus on the translocator protein. PET and SPECT in neurology. Springer, Berlin Heidelberg. 2014:757–773. [Google Scholar]
- 40.Owen DR, Yeo AJ, Gunn RN, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1–5. doi: 10.1038/jcbfm.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kreisl WC, Fujita M, Fujimura Y, et al. Comparison of [C-11]-(R)-PK 11195 and [C-11]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage. 2010;49:2924–2932. doi: 10.1016/j.neuroimage.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Owen DR, Gunn RN, Rabiner EA, et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med. 2011;52:24–32. doi: 10.2967/jnumed.110.079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kreisl WC, Jenko KJ, Hines CS, et al. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab. 2013;33:53–58. doi: 10.1038/jcbfm.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.