

Abstract
The mechanisms underlying disparate roles of the canonical Wnt signaling pathway in maintaining self‐renewal or inducing differentiation and lineage specification in embryonic stem cells (ESCs) are not clear. In this study, we provide the first demonstration that self‐renewal versus differentiation of human ESCs (hESCs) in response to Wnt signaling is predominantly determined by a two‐layer regulatory circuit involving β‐catenin, E‐cadherin, PI3K/Akt, and Slug in a time‐dependent manner. Short‐term upregulation of β‐catenin does not lead to the activation of T‐cell factor (TCF)‐eGFP Wnt reporter in hESCs. Instead, it enhances E‐cadherin expression on the cell membrane, thereby enhancing hESC self‐renewal through E‐cadherin‐associated PI3K/Akt signaling. Conversely, long‐term Wnt activation or loss of E‐cadherin intracellular β‐catenin binding domain induces TCF‐eGFP activity and promotes hESC differentiation through β‐catenin‐induced upregulation of Slug. Enhanced expression of Slug leads to a further reduction of E‐cadherin that serves as a β‐catenin “sink” sequestering free cytoplasmic β‐catenin. The formation of such a framework reinforces hESCs to switch from a state of temporal self‐renewal associated with short‐term Wnt/β‐catenin activation to definitive differentiation. Stem Cells 2015;33:1419–1433
Keywords: Wnt, β‐Catenin, E‐cadherin, Slug, Human embryonic stem cell, Self‐renewal, Differentiation
Introduction
Canonical Wnt/β‐catenin signaling has been suggested to facilitate the self‐renewal of mouse and human embryonic stem cells (ESCs) 1, 2, 3, 4, 5, 6, 7, 8. It has also been shown to enhance reprogramming of somatic cells to pluripotent stem cells 9, 10. However, the specific role of Wnt/β‐catenin in ESCs remains unclear. A number of studies have yielded opposing results in that Wnt signaling stimulates ESC differentiation and lineage specification 11, 12, 13, 14, 15, 16. How does one reconcile these seemingly disparate outcomes of one signaling pathway in ESCs?
The canonical Wnt signaling pathway is activated when a Wnt ligand protein binds to its receptors Frizzled and Lrp5/6, which leads to the inhibition of the GSK3β (Ser‐Thr kinase glycogen synthase kinase 3β)‐mediated degradation of β‐catenin. This allows the cytoplasmic β‐catenin accumulate and translocate into the nucleus where it interacts with the lymphoid enhancer factor and T‐cell factor (LEF/TCF) transcriptional complexes 17, 18, 19. Subsequently, this interaction induces or suppresses the expression of many specific downstream target genes, such as pluripotency gene Sox2, Nanog, Oct3/4, mesoderm gene Brachyury, cell adhesion molecule E‐cadherin, and E‐cadherin suppressors Snail and Slug 1, 20, 21. The gene expression profiles and subsequent biological functions, however, seem to be cell type‐ and/or signaling network dependent 19.
In addition to its pivotal role as a downstream effector of canonical Wnt signaling, β‐catenin is also an essential structure adaptor. It was first discovered as a component of a mammalian cell adhesion complex linking to the cytoplasmic tail of E‐cadherin to mediate cell‐cell adhesion 22. E‐cadherin is a transmembrane glycoprotein that mediates calcium‐dependent, homophilic cell‐cell adhesion in all epithelial tissues including ESCs 18. With the exception of being a supplementary marker for undifferentiated hESCs 23, the active role of E‐cadherin in ESC self‐renewal was not recognized until recently. A plausible explanation may be from a clear demonstration in 1996 that E‐cadherin was dispensable for mouse ESC maintenance 24. However, a recent study has shown that the loss of E‐cadherin results in rapid differentiation of mouse ESCs, suggesting a supporting role of E‐cadherin in mouse ESC self‐renewal 25. In addition, several groups including ours have reported that E‐cadherin contributes to hESC self‐renewal 26, 27, 28, 29, 30 and reprogramming of adult cells into pluripotent stem cells 31, 32, 33, 34. Furthermore, a recent study showed prominent E‐cadherin expression in naïve hESC colonies that was associated with enhanced single‐cell survival 35. Nonetheless, the function and mechanism underlying cell‐cell adhesion and Wnt signaling pathway in ESCs are not yet clearly understood 36.
We here demonstrate that crosstalk between β‐catenin, E‐cadherin intracellular β‐catenin binding domain, PI3K/Akt, and E‐cadherin suppressor Slug is key to Wnt/β‐catenin signaling in hESCs. The apparently disparate effects of Wnt/β‐catenin signal in self‐renewal and differentiation of hESCs are time‐dependent and regulated by a two‐layer circuit. In the first layer, short‐term Wnt/β‐catenin activation augments E‐cadherin‐mediated cell‐cell adhesion that associates with PI3K/Akt signaling and enhances hESC self‐renewal. Conversely, the second layer is characterized by long‐term Wnt/β‐catenin activation that promotes hESC differentiation definitively due to the increased free cytoplasmic β‐catenin exceeding the binding capacity of E‐cadherin and translocating to nuclei to functionally interact with TCF. Upregulation of Slug but not Snail gene expression after prolonged Wnt/β‐catenin activation serves as a turning point that switches hESCs from temporal self‐renewal to definitive differentiation. A time lag between the upregulation of E‐cadherin and its repressor Slug transcripts during the switch from short‐ to long‐term Wnt activation points to an intricate network underlying the seemingly opposing functions of Wnt signaling in ESCs. Our results highlight a delicate time‐dependent balance within Wnt signaling and will lead to more effective manipulation of human pluripotent stem cells for potential applications. Integration of the aforementioned regulatory circuit with Wnt/β‐catenin and other signaling pathways may also provide broad implications for stem cell and cancer research.
Materials and Methods
hESCs Culture, GSK3 Inhibitors, Wnt3a Treatments, and Clonogenic Assays
H1 and H9 hESC lines were obtained from Wicell and CA1 cell line was a gift from Dr. Nagy (Mount Sinai hospital, Toronto, Canada). All cell lines were approved for use by the local ethics board and the Stem Cell Oversight Committee of the Canadian Institutes of Health Research. Undifferentiated hESC were maintained under feeder‐free conditions in mouse embryonic fibroblast‐conditioned medium (MEFCM) supplemented with 12 ng/ml human recombinant basic fibroblast growth factor (bFGF, BD) as described previously 27, 28. Chemical defined medium (CDM) supplemented with 1xB27, 1xITS‐G, 1xNEAA, and 40 ng/ml bFGF 37 was used in the majority of experiments. The dosages of GSK3 inhibitor 6‐bromoindirubin‐3′‐oxime BIO (Calbiochem, Darmstadt, Germany, http://www.emdmillipore.com/CA/en) used, after serial dilution, in MEFCM and CDM were 2.5 and 0.83 μM, respectively; CHIR99021 (Tocris Bioscience, Bristol, UK, http://www.tocris.com), 3–6 μM; Wnt3a (R&D Systems, Minneapolis, MN, http://www.rndsystems.com), 100–200 ng/ml; PI3K inhibitor LY294002 (Cell signaling, Beverly, MA, http://www.cellsignal.com), 5 μM; Akt activator II SC79 (EMD Millipore, Etobicoke, Canada, http://www.emdmillipore.com/CA/en) 38, 4–6 μg/ml; Akt inhibitor VIII (Cayman Chemical, Ann Arbor, MI, https://www.caymanchem.com), 3–6 μM; IWP2 (Cayman Chemical), 2 μM. Clonogenic and self‐renewal assays were performed as described previously 27. See Supporting Information for detailed procedures.
Plasmids Transfection and siRNA Knockdown
Plasmid hE‐cadherin/Δβ‐catenin‐pcDNA3 (EcadΔβ), obtained from Addgene (Cambridge, MA, https://www.addgene.org) (#45772), was a kind gift from Dr. Barry Gumbiner 39. This mutant does not contain a β‐catenin binding region due to deletion of the last 35 amino acids of the E‐cadherin cytoplasmic domain, as defined by Stappert and Kemler 40. Plasmid pcDNA3‐βS33Y Beta‐catenin (βS33Y, Addgene #19286) 41 contains a tyrosine to serine missense mutation at codon 33 (S33Y, presumptive GSK3β phosphorylation site), leading to cellular accumulation and an ability to activate TCF transcription 41. Other plasmids obtained from Addgene include hE‐cadherin‐pcDNA3 (#45769) 39, pLKO‐siSlug3 (#10905) 42, and pLKO‐scrambled (#1864) 43. See Supporting Information for siRNA targeted Slug and E‐cadherin knockdown.
Generation of Tet‐On Stable hESC Cell Lines
Stable E‐cadherin‐Tet‐On and vector‐Tet‐On hESCs were generated from H9 and H1 hESCs as reported previously 28. Stable EcadΔβ‐Tet‐On and βS33Y‐Tet‐On hESCs were generated using Lenti‐X Tet‐On advanced inducible expression system (Clontech, Mountain View, CA, http://www.clontech.com) according to the manufacturer's instructions 28. EcadΔβ and βS33Y genes were amplified by PCR from hEcadΔβ‐pcDNA3 and pcDNA3‐βS33Y Beta‐catenin, respectively. The amplified fragments were cloned into pLVX‐Tight‐Puro vector and verified by DNA sequencing (see Supporting Information for details). Stable transduced hESC colonies were selected and maintained as previously described 28. To upregulate E‐cadherin expression, the selected colonies were treated with 1 μg/ml of doxycycline. To express βS33Y and EcadΔβ, the hESCs were treated with 2 μg/ml of doxycycline unless otherwise specified.
Generation of Transgenic Wnt Reporter 7xTCF‐eGFP hESCs
Four Wnt reporter hESC sublines (7xTCF‐eGFP‐EcadΔβ‐Tet‐On, 7xTCF‐eGFP‐βS33Y‐Tet‐On, 7xTCF‐eGFP‐vector‐Tet‐On, and 7xTCF‐eGFP) were generated by transduction of the established EcadΔβ‐, βS33Y‐, and vector‐Tet‐On or wild‐type hESCs with a 7×TCF‐eGFP//SV40‐PuroR lentiviral vector containing seven Tcf/Lef‐binding sites and a puromycin resistance gene (Addgene #24305, a kind gift from Dr. Roel Nusse 44). Lentivirus was produced by transient transfection of 293T cells as described previously 45. Transduced hESCs were selected with puromycin (2 μg/ml) for 14 days. Wnt reporter Tet‐On‐hESC lines were maintained on drug‐resistant DR4 MEF feeder cells in hESC medium supplemented with bFGF (4 ng/ml), puromycin (1 μg/ml), and G418 (100 μg/ml). All Wnt reporter lines were passaged on Matrigel in MEFCM supplemented with bFGF (12 ng/ml) and puromycin (1 μg/ml) or G418 (100 μg/ml for Tet‐On lines) for two passages prior to the Wnt activation assay. The expression levels of TCF‐eGFP were determined by flow cytometry.
Flow Cytometry
To preserve membrane E‐cadherin expression, hESCs were dissociated with Cell Dissociation Buffer (Life Technologies, Waltham, MA, http://www.lifetechnologies.com) for 14 minutes, filtered through a 40‐μm cell strainer (BD), and immunolabeled with either Alexa‐Flour647 mouse anti‐human E‐cadherin antibody or isotype control (BD Biosciences, Mississauga, Canada, http://www.bdbiosciences.com) for 120 minutes on ice. Live cells identified by 7‐AAD exclusion were analyzed for E‐cadherin and TCF‐eGFP expression on a CyAn ADP 9 Analyzer (Beckman Coulter, Mississauga, Canada, https://www.beckmancoulter.com). Detailed procedures are described in Supporting Information.
Real‐Time Quantitative PCR and Western Blot
Total RNA extraction, quantitative PCR (Q‐PCR), and Western blot were performed as described previously 27. Forward and reverse primers used in Q‐PCR, antibodies used in Western blotting, and additional procedures are described in Supporting Information.
Immunofluorescence Staining, Fluorescence Microscopy, and Image Analysis
Experimental methods have been described previously 27, 28 and in Supporting Information.
Other Experimental Procedures
See Supporting Information.
Statistical Analysis
Results are expressed as mean ± SD. Statistical significance was determined using a Student's t test, ANOVA, or chi‐square wherever appropriate. Results were considered significant when p < .05 or <.01.
Results
Short‐Term GSK3 Inhibition Temporally Promotes E‐Cadherin Expression and hESC Self‐Renewal Whereas Long‐Term Inhibition Generates Opposing Results
Inhibition of GSK3 has been reported to either promote self‐renewal or induce differentiation of hESCs 2, 14, 46. We speculated that the dual role of GSK3 inhibition might be time‐dependent and controlled by undefined mechanisms. Consistent with our hypothesis, brief inhibition of GSK3 with BIO for 6 hours caused hESC colonies to change from thin and flat to thick and compact morphology with sharp boundaries and hard‐to‐distinguish individual cells (characteristics of undifferentiated colonies) when observed 24 hours later after the removal of BIO (Fig. 1A). Notably, gene and protein expression levels of E‐cadherin, Nanog, and Oct3/4 were elevated (Supporting Information Fig. S1A, S1B, and Fig. 1B–1E), and E‐cadherin protein on the cell membrane was enriched (Fig. 1B, 1C, immunocytochemistry; Fig. 1F, 1G, flow cytometry) after short‐term BIO treatment. Using clonogenic assays, a crucial method to assess ESC self‐renewal by reseeding an equal number of single hESCs in each group, we observed an approximately eightfold increase in total clonogenic capacity after short‐term BIO treatment (3‐fold increase in clonogenic assays × 2.6‐fold increase in total cell number after removal of BIO and continual culture for 4 days, Fig. 1H, 1I). It can be concluded that short‐term BIO treatment enhances E‐cadherin expression on the cell membrane and promotes hESC self‐renewal.
Figure 1.
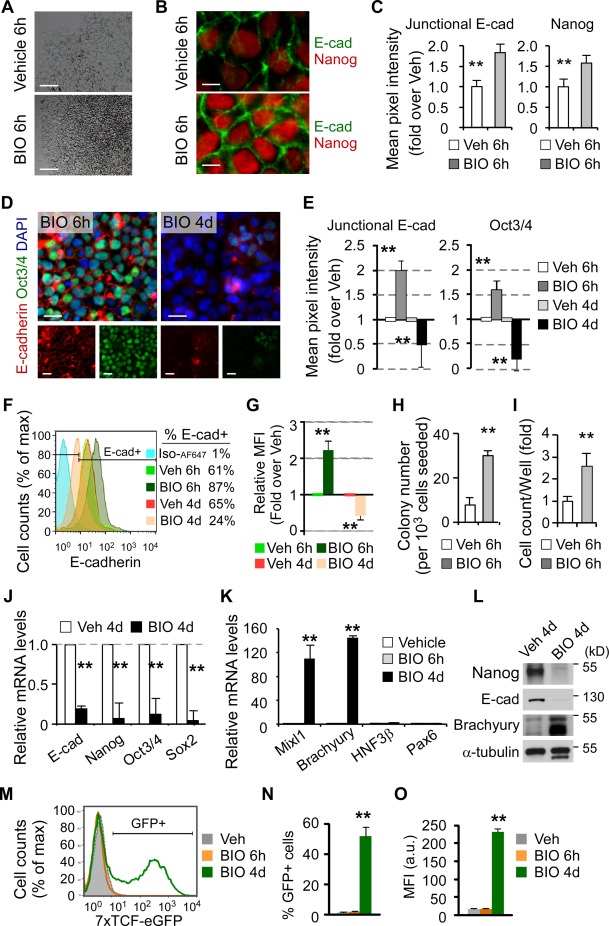
Short‐term GSK3 inhibition temporally promotes E‐cadherin expression and human embryonic stem cell (hESC) self‐renewal whereas long‐term inhibition generates opposite results. (A–L): hESCs were treated with either vehicle (Veh) or BIO for 6 hours. Twenty‐four hours after removal of BIO, hESCs exhibit thick and compact colonies (A, bright‐field images) that express higher levels of E‐cadherin (E‐cad), Nanog, and Oct3/4 (B and D, immunofluorescence staining; C and E, quantitative image analysis of fluorescence intensity, n > 100 from three independent experiments for each group; F and G, flow cytometry). They also show high cloning efficiency (H, clonogenic assays, n = 3). After removal of BIO and continual culture for 4 days, BIO treatment leads to a high cell yield (I). In contrast, long‐term BIO treatment leads to opposing results (D–G; J, Q‐PCR; L, Western blot), significantly enhancing the expression of mesoderm marker Mixl1 and Brachyury (K, Q‐PCR, HNF3β: endoderm, Pax6: ectoderm). Nuclei were counterstained with DAPI (D, blue). Scale bars = 100 µm (A), 10 µm (B), and 20 µm (D). (M–O): Representative flow cytometric histogram (M), percentage of live TCF‐eGFP‐positive cells (pregated on 7‐AAD‐negative live cells), and MFI after treatment with BIO for 6 hours or 4 days. All data are mean ± SD. **, p < .01 compared to vehicle control. Abbreviations: GFP, green fluorescent protein; MFI, median fluorescence intensity; TCF, T‐cell factor.
Next, to address whether prolonged inhibition of GSK3 suppresses E‐cadherin expression and promotes differentiation, we examined hESCs after treatment with BIO for 6 hours (short‐term) and 4 days (long‐term), respectively. In contrast to short‐term BIO treatment, long‐term BIO treatment markedly reduced Oct3/4 and E‐cadherin protein levels (Fig. 1D–1G), and significantly downregulated pluripotency transcripts Nanog, Oct3/4, and Sox2 (Fig. 1J).
Consistent with the above observations, Mixl1 and Brachyury transcripts (primitive streak/mesoderm markers) were significantly upregulated after 4 days BIO treatment while remaining unchanged after 6 hours treatment (Fig. 1K). Western blot confirmed that long‐term BIO treatment resulted in differentiation of hESCs, as indicated by a significant downregulation of Nanog and E‐cadherin, and upregulation of Brachyury protein levels (Fig. 1L). Specifically, Wnt reporter TCF‐eGFP was not expressed after treatment with BIO for 6 hours but significantly upregulated after 4 days (Fig. 1M–1O). Thus, the opposing results in response to short‐ and long‐term BIO treatment indicate a time‐dependent manner of Wnt signaling pathway in the regulation of hESC self‐renewal versus differentiation. Similar results were also obtained by activating Wnt signaling pathway using GSK3 inhibitor CHIR99021 (Supporting Information Fig. S2). Collectively, these data illustrate that short‐term GSK3 inhibition enhances E‐cadherin expression on the cell membrane and hESC self‐renewal whereas long‐term inhibition results in opposite outcomes.
Dual Function of β‐Catenin in Canonical Wnt Signaling Pathway Is Responsible for hESC Self‐Renewal and Differentiation
Effects of GSK3 inhibition could be complicated by the diverse cellular activities of GSK3 (downstream of BIO and CHIR99021), which involves transcription, translation, cell cycle, metabolism, antiapoptosis, and signal transduction 47. To exclude possible nonspecific activity of GSK3, we used recombinant protein Wnt3a and obtained similar results (Supporting Information Figs. S3, S4). As β‐catenin is well recognized as the pivotal downstream effector of the Wnt signaling, we decided to examine β‐catenin to further study the discrepancy of Wnt signaling pathway on hESC self‐renewal and differentiation. To controllable over‐expression of β‐catenin in hESCs, we generated a stable hESC line using a tetracycline‐inducible lentiviral expression vector that expresses a nondegradable β‐catenin mutant βS33Y 41. A previous study has demonstrated that this mutant is functionally identical to β‐catenin, but undegradable due to the substitution of a specific residue that is normally phosphorylated by GSK3β, and therefore is able to activate TCF transcription 41.
Notably, after short‐term βS33Y expression, the increased levels of β‐catenin and Nanog proteins (data not shown), and an enhanced self‐renewal capacity (clonogenic assays), were observed (Fig. 2A). These results further confirm that β‐catenin signaling is involved in hESC self‐renewal. Conversely, long‐term (4–6 days) expression of βS33Y led to an increase in total cytoplasmic β‐catenin protein and a loss of Nanog expression in a doxycycline dosage‐dependent manner (Fig. 2B, 2C). Significantly, after long‐term expression of βS33Y with a high dosage of doxycycline, approximately 30% of colonies exhibited lower levels of Nanog and some cells completely lost Nanog expression together with accumulated β‐catenin in the nuclei (Fig. 2D, right panel), indicative of a differentiation property of hESCs due to the prolonged Wnt/β‐catenin activation. Consistently, after long‐term expression of βS33Y, clonogenic assays revealed a dose‐dependent impairment in hESC self‐renewal (Fig. 2E), and Western blotting showed diminished Nanog expression (Fig. 2F). Specifically, following an increase in TCF‐eGFP activity, E‐cadherin expression on the cell membrane was decreased in a doxycycline dosage‐dependent manner (Fig. 2G–2J). Together, these findings demonstrate that depending on the duration of Wnt activation, β‐catenin has dual function in the control of hESC self‐renewal versus differentiation through yet to be determined mechanisms.
Figure 2.
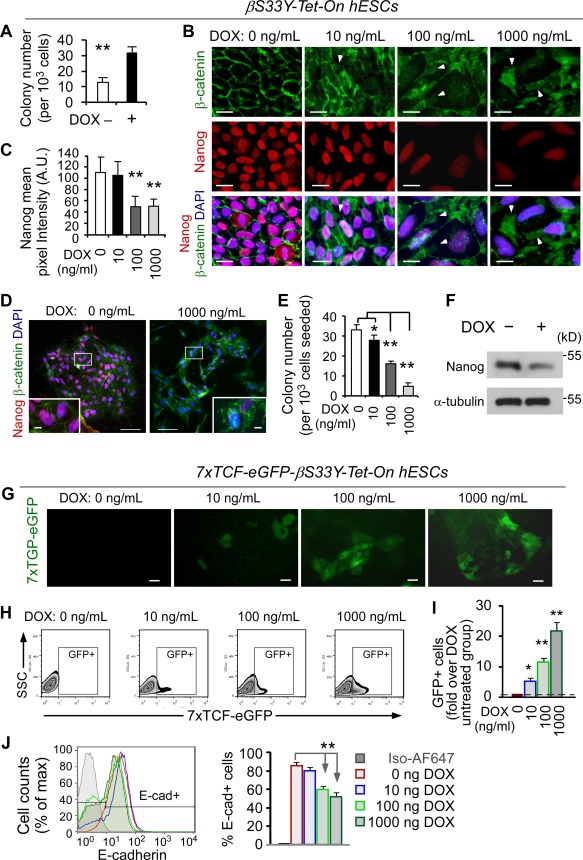
Dual function of β‐catenin in canonical Wnt signaling pathway is responsible for hESC self‐renewal and differentiation. (A): βS33Y‐Tet‐On hESCs were treated with doxycycline (DOX, 2 µg/ml) for 24 hours, followed by clonogenic assays (mean ± SD, n = 3, **, p < .01). (B–D): βS33Y‐Tet‐On hESCs were treated with different dosages of DOX for 4–6 days, followed by immunofluorescence staining of β‐catenin and Nanog (B), and quantification of mean pixel intensity of Nanog (C, n >100 cells/group). The nuclear translocation of β‐catenin was observed in 1,000 ng/ml of DOX group (D). Nuclei were counterstained with DAPI (blue). White arrows indicate cytoplasmic β‐catenin. Scale bars = 10 µm (insets in D: 1 µm). (E, F): Clonogenic assays (E, mean ± SD, n = 3, *, p < .05; **, p < .01) and Western blot analysis (F) for the same cells as described in (B)–(D). (G): TCF‐eGFP reporter activity remains invisible in the absence of DOX for 4–6 days but visible in the nuclear region in a DOX dose‐dependent manner, scale bars = 20 µm. (H, I): Flow cytometric analysis of 7xTCF‐eGFP hESCs as described in (G), pregated on single 7‐AAD‐negative live cells. The TCF‐eGFP reporter activity was measured by the percentage of GFP‐positive cells after treatment with different concentrations of DOX for 4–6 days (H). The expression of GFP‐positive cells is normalized to DOX‐untreated group that was arbitrarily set at 1 (I). (J): Flow cytometric analysis of the cells in (H) and (I) for E‐cadherin expression (E‐cad+) on the cell membrane indicates an inverse association with TCF‐eGFP activity as shown in (G)–(I), pregated on single 7‐AAD‐negative live cells. Data in (I) and (J) are means ± SD, n = 3, two independent experiments; **, p < .01 compared to DOX‐untreated group. Abbreviations: GFP, green fluorescent protein; hESC, human embryonic stem cell; TCF, T‐cell factor.
Short‐Term Upregulation of β‐Catenin Enhances hESC Self‐Renewal Through E‐Cadherin‐Associated PI3K/Akt Activation
To investigate how β‐catenin enhances hESC self‐renewal, we examined the relationship between β‐catenin and the adhesion molecule E‐cadherin. Several groups, including ours, have recently shown that E‐cadherin plays an essential role in the maintenance of hESCs 26, 27, 28, 29, 30, and in the reprogramming of somatic cells 31, 32, 33, 34. Our previous study also suggested that inadequate formation of E‐cadherin‐mediated cell‐cell contacts in hESCs hampered clonogenic capacity and self‐renewal of hESCs 27, 28. Since Wnt signaling has been shown to drive an epithelial‐to‐mesenchymal transition 48, one may speculate that even transient Wnt activation would repress E‐cadherin in hESCs.
To address this issue, we simultaneously assessed Wnt reporter TCF‐eGFP activity and E‐cadherin expression in hESCs using flow cytometry. Significantly, while the levels of E‐cadherin transcript and protein were increased, and E‐cadherin on the cell membrane was enriched (both percentage and median fluorescence intensity) after treatment with BIO, CHIR99021, or Wnt3a for 6 hours (Figs. 1, 4D‐scrambled + CHIR99021, Supporting Information Figs. S1–S4), the TCF‐eGFP expression was not elevated. This suggests that short‐term Wnt activation in hESCs leads to limited functional interaction of β‐catenin and TCF in the nucleus. In contrast, long‐term Wnt activation resulted in a significant increase in TCF‐eGFP reporter activity, which was inversely correlated with the E‐cadherin downregulation (Fig. 1, Supporting Information Figs. S2, S4). It is apparent that short‐term upregulation of β‐catenin in hESCs mainly promotes E‐cadherin expression on the cell membrane (as an intracellular adaptor protein for E‐cadherin) but not Wnt signaling in the nucleus.
To further elaborate whether E‐cadherin is involved in hESC self‐renewal, we subsequently over‐expressed E‐cadherin in hESCs using a tetracycline‐inducible lentiviral vector 28, and observed an increase in the expression of Nanog and Oct3/4 transcripts, formation of thick compact colonies with more than a fivefold increase in cell density, and enriched junctional E‐cadherin expression at cell‐cell contacts (Fig. 3A–3C). We then asked whether loss of E‐cadherin results in a decline of hESC self‐renewal. Indeed, siRNA targeted E‐cadherin knockdown significantly inhibited Nanog and Oct3/4 expression and impaired clonogenic capacity (Fig. 3D, 3E). Finally, we demonstrated that E‐cadherin knockdown also abrogated short‐term GSK3 inhibition‐induced upregulation of Nanog and Oct3/4 expression (Supporting Information Fig. S5). Collectively, the above results suggest that short‐term Wnt/β‐catenin activation enhances hESC self‐renewal through the augmentation of E‐cadherin expression.
Figure 3.
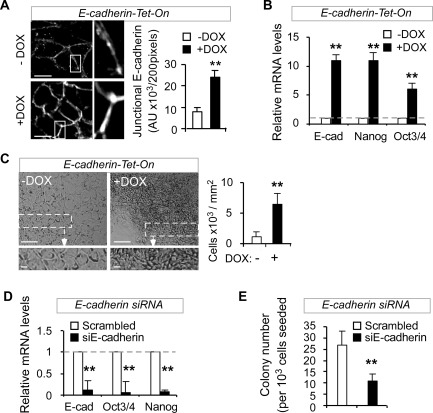
β‐Catenin enhances human embryonic stem cell (hESC) self‐renewal through upregulation of E‐cadherin. (A–C): Inducible over‐expression of E‐cadherin (E‐cad) in hESCs enhances E‐cadherin expression in cell‐cell junctions (A, immunofluorescence, left panel: enlarged images; DOX: 3 days), and increases the expression of E‐cadherin, Oct3/4, and Nanog (B, Q‐PCR, DOX: 24 hours) and cell density (C, bright‐field image and cell counting; low panel: enlarged images; DOX: 3 days). Scale bars = 10 µm (A), 20 µm (low panel of C), and 100 µm (upper panel of C); DOX, 2 µg/ml. (D, E): Reduced expression levels of E‐cadherin, Oct3/4, and Nanog (D, Q‐PCR) and diminished clonogenic capacity (E, clonogenic assays, n = 3, **, p < .01) in hESC 24 hours after E‐cadherin siRNA knockdown. H1, H9, or CA1 hESC lines were used in this figure.
To examine the underlying mechanism between E‐cadherin expression and hESC self‐renewal, we tested the effect of E‐cadherin‐targeted knockdown on PI3K/Akt activation. It has been documented that PI3K/Akt signaling enhances the expression of pluripotency genes and antagonizes hESC differentiation induced by over‐activation of Erk and Wnt signaling 14, 49. Significantly, E‐cadherin knockdown in hESCs resulted in a reduction of Akt activation via decreased Akt phosphorylation on Ser473, indicative of reduced PI3K/Akt signaling (Fig. 4A). Conversely, E‐cadherin upregulation led to an increase in PI3K/Akt activation (Fig. 4B) and clonogenic capacity in hESCs (Fig. 4C), while inhibition of PI3K/Akt abrogated the enhanced clonogenic capacity (Fig. 4C) and elevated Nanog and Oct3/4 expression induced by E‐cadherin upregulation (Supporting Information Fig. S5). Specifically, short‐term GSK3 inhibition‐induced upregulation of E‐cadherin protein, Akt phosphorylation, and Nanog and Oct3/4 transcripts was abolished by E‐cadherin knockdown, but restored by a specific Akt activator SC79 (Fig. 4D, Supporting Information Fig. S5). Furthermore, similar to E‐cadherin knockdown, Akt suppression blocked short‐term GSK3 inhibition‐induced upregulation of Nanog and Oct3/4 transcripts (Supporting Information Fig. S5). Taken together, these data demonstrate that short‐term Wnt/β‐catenin activation enhances hESCs self‐renewal through E‐cadherin upregulation that is associated with PI3K/Akt activation.
Figure 4.
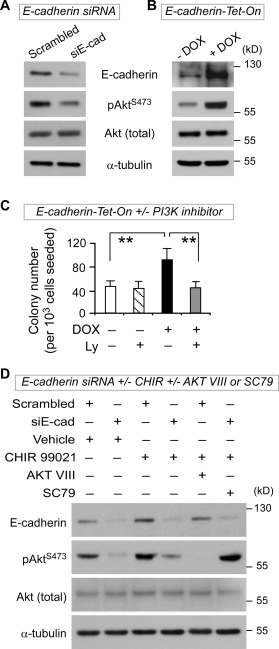
E‐cadherin associates with PI3K/Akt activation in response to short‐term Wnt/β‐catenin signaling‐enhanced human embryonic stem cell (hESC) self‐renewal. (A): Decreased phosphorylated AKT (pAKTS473) in hESCs after E‐cadherin knockdown for 48 hours (Western blot). (B): Increased phosphorylated AKT (pAKTS473) after E‐cadherin upregulation. E‐cadherin‐Tet‐On hESCs were treated with DOX (2 µg/ml) for 48 hours, followed by Western blot analysis. (C): Inhibition of PI3/Akt with LY294002 abrogates the enhanced clonogenic capacity induced by E‐cadherin upregulation. E‐cadherin‐Tet‐On hESCs were treated with or without DOX (2 µg/ml) in the presence or absence of LY294002 (5 µM) for 24 hours, followed by clonogenic assays (n = 3). (D): E‐cadherin knockdown abolishes short‐term GSK3 inhibition‐induced upregulation of E‐cadherin and phosphorylated Akt, while specific Akt activator SC79 counteracts the effect of E‐cadherin knockdown. After siRNA knockdown for 44 hours, hESCs (H9 and H1 lines) were treated with either CHIR99021 (6 µM) or vehicle (DMSO) for 6 hours, followed by Akt inhibitor VIII (6 µM), or Akt activator II SC79 (6 µg/ml), or vehicle for an additional 30 minutes. All data in this figure are mean ± SD and were generated from H1 and H9 lines. **, p < .01.
E‐Cadherin Sequesters β‐Catenin to Suppress Wnt‐Induced hESC Differentiation
While E‐cadherin has recently been revealed as an important component in hESC self‐renewal 26, 27, 28, 29, 30, its regulatory role in Wnt/β‐catenin signaling in ESCs remains undefined. It is known that cytoplasmic β‐catenin can either be phosphorylated by GSK3β which led to its degradation, or bound by E‐cadherin intracellular domain through direct protein‐protein interactions at the cell‐cell junctions. In either fate, accumulation of free cytoplasmic β‐catenin will be minimized, thus preventing translocation of β‐catenin into the nucleus to initiate expression of targeted downstream genes through interaction with the LEF/TCF complex 18, 50. The lack of nuclear shift of β‐catenin and TCF‐eGFP reporter activity in undifferentiated hESCs (Fig. 2B, 2D, 2G, 2H, DOX‐untreated group) prompted us to propose that, in addition to association with PI3K/Akt activation to regulate hESC self‐renewal and differentiation, E‐cadherin also influences Wnt signaling by sequestering β‐catenin.
To determine whether disruption of interaction between E‐cadherin and β‐catenin impairs hESC self‐renewal, we transfected hESCs with a truncated E‐cadherin that lacks the β‐catenin‐binding domain (EcadΔβ 39) in combination with other genes. Stappert and Kemler 40 have shown that EcadΔβ mutant allows more cytoplasmic β‐catenin to enter the nucleus and interact with LEF/TCF complex. Particularly, ectopic coexpression of βS33Y and EcadΔβ, but not coexpression of βS33Y and wild‐type E‐cadherin, led to a significant downregulation of pluripotency genes Nanog, Oct3/4, and Sox2 (Fig. 5A). However, this effect was rescued by coexpression of βS33Y with wild‐type E‐cadherin instead of EcadΔβ (Fig. 5A), demonstrating that the repressed expression of pluripotency genes after Wnt activation relies on the β‐catenin‐binding ability of E‐cadherin. Importantly, the Brachyury transcript was also significantly upregulated in comparison to controls after short‐term ectopic coexpression of βS33Y and EcadΔβ (Fig. 5B). These results suggest that mutating the β‐catenin binding domain of E‐cadherin abrogates self‐renewal enhanced by short‐term Wnt activation (as shown in Fig. 1) and sensitizes hESCs to Wnt‐induced differentiation.
Figure 5.
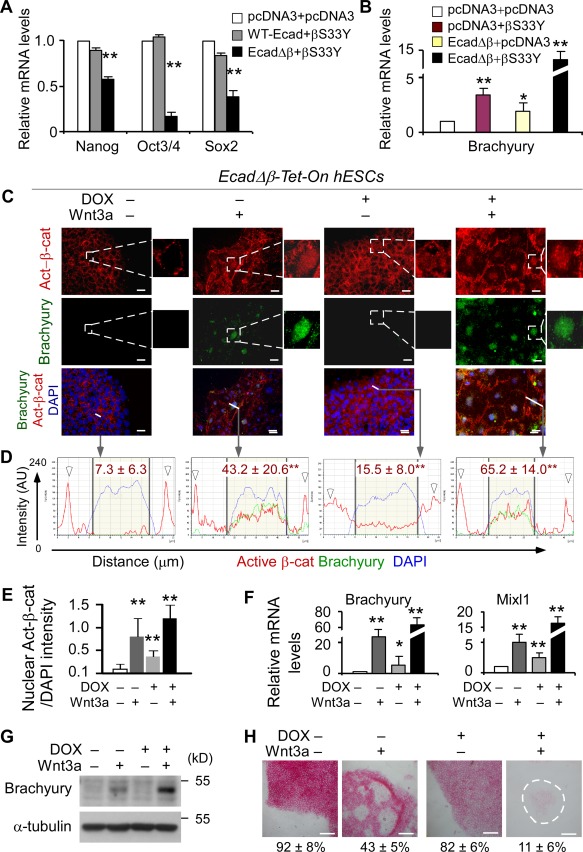
E‐cadherin sequesters β‐catenin to suppress Wnt‐induced hESC differentiation. (A): Q‐PCR analysis of hESCs cotransfected with βS33Y plasmids and either wild‐type E‐cadherin (WT‐Ecad) or EcadΔβ for 24 hours. pcDNA: control vector. (B): In comparison to control groups, co‐overexpression of EcadΔβ and βS33Y for 24 hours significantly increases Brachyury expression (Q‐PCR). (C): EcadΔβ‐Tet‐On hESCs were treated with or without doxycycline (DOX+, 2 µg/ml) or vehicle in the presence or absence of Wnt3a protein (100 ng/ml) for 4–6 days and analyzed by double‐immunostaining. Nuclei were counterstained with DAPI (blue). Scale bar = 20 µm. (D): Subcellular localization analysis of the expression of active β‐catenin and Brachyury as shown in (C). Plots of the intensity profile of active β‐catenin (red), Brachyury (green), and DAPI (blue) over a linear section of a whole cell (white lines in C) are representative of >100 cells/group analyzed. Data are expressed as AU versus length in microns. The arrowheads indicate the cell‐cell contact areas; the yellow area in the middle of each plot marks the nuclear region; mean ± SD in red on each plot represent the mean pixel intensity of the nuclear active β‐catenin from >100 cells/group. (E): Quantification of the ratio of active β‐catenin nuclear staining/DAPI intensity for the indicated groups in (C). Data are mean ± SD, four replicates from H9 line, **, p < .01 compared to DOX−Wnt3a− group. (F, G): Doxycycline‐induced EcadΔβ over‐expression promotes hESC differentiation in response to Wnt3a treatment as revealed by significant upregulation of Brachyury and Mixl1 (F, Q‐PCR; G, Western blot). (H): Alkaline phosphatase (AP) staining and quantitative analyses. At day 4 of treatments as indicated, cells were dissociated and reseeded. AP‐positive colonies were counted when >50% of the cells within the colony were positive for AP staining. The percentages of AP+ colonies were quantitatively assessed and are shown at the bottom. The dashed line indicates the boundary of the colony. Scale bar = 100 µm. The results represent three replicates ± SD from H1 and H9 lines. *, p <.05; **, p <.01. Abbreviations: AU, arbitrary unit; hESC, human embryonic stem cell.
To further confirm the above observation, we generated a stable hESC line using a tetracycline‐inducible lentiviral expression vector to control EcadΔβ expression using EcadΔβ plasmid 39. It is known that accumulation of β‐catenin that is specifically unphosphorylated at GSK3β sites is critical for β‐catenin/TCF‐mediated transcription 51. We therefore used a monoclonal antibody to detect the transcriptionally active form of β‐catenin (active β‐catenin), which recognizes the signaling form of β‐catenin specifically unphosphorylated at S37 and T41 51, 52. In comparison to untreated control, we observed an increase in nuclear‐localized active β‐catenin when EcadΔβ expression was induced by the addition of doxycycline alone for 4 days (Fig 5C–5E), indicative of a state of Wnt/β‐catenin signaling activation. Importantly, DOX‐induced EcadΔβ over‐expression plus Wnt3a stimulation led to a further increase in the nuclear accumulation of active β‐catenin and high‐level expressions of Brachyury and Mixl1 (Fig. 5C–5F). This suggests that hESCs expressing EcadΔβ without β‐catenin‐binding domain are more sensitive to Wnt/β‐catenin activation, resulting in early differentiation.
To quantify the extent by which hESC differentiation is dependent on functional interactions between E‐cadherin and β‐catenin, we cultured EcadΔβ‐Tet‐On hESCs in the presence or absence of Wnt3a for 4 days, then performed clonogenic assays and measured the numbers and intensity of alkaline phosphatase‐positive (AP+) colonies. Again, after addition of both Wnt3a (activation of Wnt signaling) and DOX (induction of EcadΔβ expression), differentiation was significantly enhanced based on the loss of AP staining and gain of Brachyury and Mixl1 expression (Fig. 5F–5H, AP+ colonies: Wnt3a alone vs. DOX+Wnt3a, 43% ± 5% vs. 11% ± 6%). As such, in addition to its association with PI3K/Akt activation, E‐cadherin serves as β‐catenin sink to delay hESC differentiation in response to prolonged Wnt activation.
We finally asked whether introducing EcadΔβ into hESCs enhances functional interaction of β‐catenin and TCF if E‐cadherin truly acts to sequester β‐catenin away from cytoplasm and TCF. Indeed, after being treated with DOX alone for 4 days, both percentage of TCF‐eGFP‐positive cells and median fluorescence intensity were increased up to five‐ to sevenfold in comparison to DOX‐untreated cells (Fig. 6A [fluorescence microscopy], 6B–6D [flow cytometry]), demonstrating that E‐cadherin is capable of minimizing nuclear β‐catenin‐TCF signaling. Consistently, EcadΔβ hESCs were also more sensitive to Wnt3a treatment, exhibiting an early TCF activation and greater TCF‐eGFP reporter activity than control groups (Fig. 6A–6D). Collectively, it can be concluded that E‐cadherin sequesters β‐catenin to suppress its nuclear localization and functional interaction with TCF.
Figure 6.
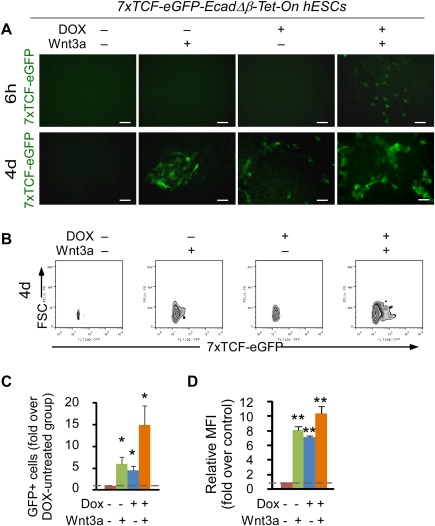
E‐cadherin sequesters β‐catenin and suppresses the functional interaction of β‐catenin and TCF in hESCs. Analysis of 7xTCF‐eGFP‐EcadΔβ‐Tet‐On hESCs after treatment with doxycycline (DOX+, 2 µg/ml) or vehicle in the presence or absence of Wnt3a protein (100 ng/ml) for the indicated time points. The cells were cultured on Matrigel‐coated plates. Media, DOX, and Wnt3a were changed daily. (A): TCF‐eGFP (green) expression pattern of hESCs in the absence or presence of DOX and/or Wnt3a for 6 hours or 4 days. Scale bar = 50 µm. (B–D): Representative flow cytometric plots (B), fold changes of TCF‐eGFP‐positive cells over DOX−Wnt3a− control (C) and MFI (D) in the absence or presence DOX and/or Wnt3a for 4 days, pregated on single 7‐AAD‐negative live cells. Mean ± SD from three independent experiments. *, p < .05; **, p < .01. Abbreviations: GFP, green fluorescent protein; hESC, human embryonic stem cell; MFI, median fluorescence intensity; TCF, T‐cell factor.
Interestingly, hESC differentiation progressed even after removing BIO at day 4 and cultivating in conventional conditions supporting self‐renewal, indicating that hESC differentiation induced by prolonged Wnt/β‐catenin activation is irreversible. This prompted us to examine the regulatory circuits associated with Wnt and E‐cadherin that switch hESCs from self‐renewal to definite differentiation.
β‐Catenin‐Induced Slug Upregulation Reinforces hESC Differentiation by Downregulating E‐Cadherin and Upregulating Brachyury
Nuclear transactivation of β‐catenin has been shown to upregulate expression of E‐cadherin suppressor genes Snail and Slug in other cell types to initiate epithelial‐mesenchymal transition 1, 21, 53. While β‐catenin/TCF transcription‐dependent and ‐independent in the control of ESC self‐renewal have been well studied, little is known about the crosstalk between Wnt/β‐catenin, Snail/Slug, and E‐cadherin in ESC fate decision. Because interaction between E‐cadherin and β‐catenin at cell‐cell junctions favors hESC self‐renewal (Figs. 3, 4, 5, 6), we propose the balance between self‐renewal and differentiation in response to Wnt/β‐catenin signaling is coordinated by a two‐layer circuit: (a) interaction between E‐cadherin and β‐catenin at cell membrane, and (b) crosstalk among E‐cadherin, Snail/Slug, and Wnt/β‐catenin pathway as determined below.
We first asked to what extent Snail or Slug was involved in Wnt/β‐catenin signaling in the control of hESC self‐renewal and differentiation. We found that Slug but not Snail transcript was increased significantly in a timely manner after 1 day BIO treatment and further upregulated in both transcript and protein levels after 4 days BIO treatment (Fig. 7A, 7B). Despite the initial increase of Snail protein in hESCs after short‐term Wnt activation, unexpectedly, neither Snail protein nor its transcript was significantly augmented in comparison to Slug after long‐term Wnt activation (Fig. 7A, 7B), suggesting that Slug is predominantly involved in the Wnt/β‐catenin pathway in hESCs.
Figure 7.
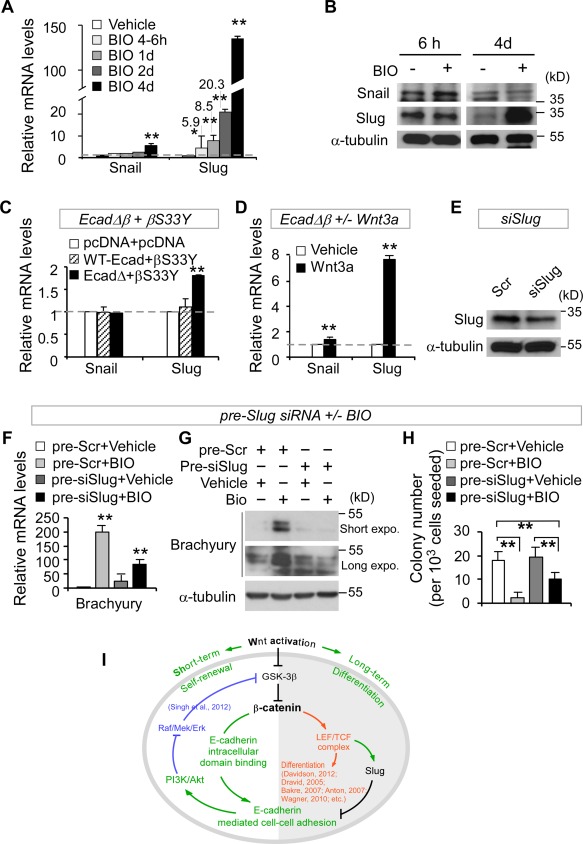
β‐Catenin‐induced Slug upregulation reinforces human embryonic stem cell (hESC) differentiation by downregulating E‐cadherin and upregulating Brachyury. (A, B): Long‐term but not short‐term BIO treatment enhances Slug expression in hESCs (A, Q‐PCR; B, Western blot). (C): hESCs cotransfected with EcadΔβ and βS33Y, but not with vectors (pcDNA) or with wild‐type E‐cadherin (WT‐Ecad) and βS33Y, enhances Slug expression (Q‐PCR). (D): Over‐expression of EcadΔβ together with short‐term Wnt3a treatment (100 ng/ml) promotes Slug expression in hESCs (Q‐PCR). (E): hESCs infected with pLKO‐siSlug plasmid (siSlug) for 2 days reduce Slug expression (Western blot). Scrambled: pLKO‐puro vector with a scrambled sequence that does not target any mRNA. (F–H): Slug knockdown (pre‐siSlug) followed by BIO treatment for 4 days results in a decrease of differentiation marker Brachyury (F, Q‐PCR; G, Western blot) and clonogenic capacity (H, clonogenic assays, n = 3) in hESCs. All data are mean ± SD, *, p <.05; **, p < .01. (I): The proposed working model (detailed in Supporting Information Fig. S7).
We next examined how a crosstalk between E‐cadherin and β‐catenin influences Slug expression in hESCs. Ectopic coexpression of βS33Y and EcadΔβ (which lacks the β‐catenin binding domain), but not coexpression of βS33Y and wild‐type E‐cadherin, led to upregulation of Slug expression (Fig. 7C) and downregulation of pluripotency genes Nanog, Oct3/4, and Sox2 in hESCs (Fig. 5A). This suggests that loss of a β‐catenin‐binding domain in E‐cadherin promotes Slug expression and impairs hESC self‐renewal. Consistently, ectopic expression of EcadΔβ mutant in response to short‐term Wnt3a treatment further augmented Slug expression in hESCs (Fig. 7D). Comparable results were observed in a Tet‐On stable hESC cell line expressing EcadΔβ after short‐term Wnt3a treatment as well (data not shown). These findings demonstrate that E‐cadherin and β‐catenin work together to regulate Slug expression after Wnt activation in hESCs.
We then asked whether Slug plays a potent role in the control of hESC self‐renewal and differentiation in response to Wnt signaling. To address this, we carried out transient knockdown of Slug using pLKO‐siSlug (Fig. 7E) 42. Suppression of Slug followed by long‐term BIO treatment for 4 days resulted in a significant decrease of differentiation marker Brachyury (Fig. 7F, 7G), and an increase in clonogenic capacity in comparison to scrambled control (Fig. 7H), indicating that Slug inhibition has an adverse effect on hESC differentiation after upregulation of Wnt signaling. Taken together, the above data provide the first evidence that loss of E‐cadherin β‐catenin‐binding domain or long‐term Wnt/β‐catenin activation upregulates Slug expression, which reinforces hESC differentiation.
Discussion
The disparate effects of Wnt/β‐catenin signaling pathway on ESC self‐renewal, differentiation, and lineage commitment are not fully understood. In this study, we demonstrate that the seemingly controversial biological roles of Wnt pathway in hESCs are largely time‐dependent and regulated by a two‐layer circuit. Short‐term activation of Wnt/β‐catenin signaling temporally enhances hESC self‐renewal via upregulating E‐cadherin expression at cell‐cell junctions (the first layer), leading to PI3K/Akt activation. Enhanced E‐cadherin expression also serves as a “sink” to reduce the accumulation of free cytoplasmic β‐catenin in favor of hESC self‐renewal. Conversely, long‐term activation of Wnt/β‐catenin signaling increases free cytoplasmic β‐catenin that exceeds the binding capacity of E‐cadherin, leading to translocation of β‐catenin into the nucleus and upregulation of E‐cadherin suppressor Slug expression. The upregulated Slug, but not Snail, in turn inhibits E‐cadherin expression and reinforces the accumulation of β‐catenin (the second layer), which leads to definitive differentiation of hESCs (working model, Fig. 7I).
The potential role of cell‐cell adhesion molecules in the regulation of Wnt/β‐catenin signaling pathway in ESCs has been speculated but not well documented, mainly due to the early demonstration of E‐cadherin as an unnecessary component in mouse ESC self‐renewal 24. As a result, the majority of studies regarding Wnt/β‐catenin signaling in ESCs are focused on its nuclear regulation via TCF transcriptional factors 1, 36, 54 or TCF‐independent functions through interaction with Oct3/4 13, 55. Current opposing observations regarding endogenous Wnt signaling in hESC self‐renewal versus differentiation further complicate this debate 8, 56, although we did not observe significant changes after inhibition of endogenous Wnt ligands' secretion with IWP2 (Supporting Information Fig. S2). Recent findings on the important role of E‐cadherin in ESCs 25, 26, 27, 28, 29, 30, 35 prompted us to investigate whether and how E‐cadherin and its associated molecules regulate Wnt/β‐catenin signaling pathway in ESCs.
Human ESCs provide a unique system to address this question because their self‐renewal is highly dependent on E‐cadherin expression. Interestingly, a recent report has suggested that retention of stabilized β‐catenin in the cytoplasm maintains self‐renewal of pluripotent stem cells 57. Here, we demonstrate that temporal hESC self‐renewal in response to short‐term β‐catenin activation is dependent on E‐cadherin upregulation, which associates with PI3K/Akt activation (Figs. 3, 4). Considerable studies have shown that PI3K/Akt pathway is specifically responsible for ESC self‐renewal 14, 49, 58, 59. Our findings provide the first evidence that links E‐cadherin‐mediated cell‐cell adhesion to PI3K/Akt activation in hESC self‐renewal.
In addition, we also provide the first demonstration that Wnt signaling‐induced hESC differentiation can be suppressed by cell‐cell adhesion molecule E‐cadherin. One underlying mechanism is via E‐cadherin intracellular domain that sequesters free cytoplasmic β‐catenin (Fig. 5). It is possible that other mechanisms may also come into play given the wide‐array of E‐cadherin and Wnt functions and cell type‐dependencies, which warrants further investigation in human pluripotent stem cells. For instance, in mouse ESCs, there is negligible transcriptional activation of β‐catenin during self‐renewal. β‐Catenin forms a complex with Oct3/4 and E‐cadherin in the membrane and it is this complex that is involved in regulating mESC self‐renewal and differentiation 60, 61. In cancer cells, E‐cadherin enhances the interaction between caveolin‐1 and β‐catenin near the cell membrane thereby reducing β‐catenin and TCF/LEF‐dependent transcription 62. E‐Cadherin‐mediated cell‐cell adhesion may also enhance the turnover of cytoplasmic β‐catenin by promoting the activity of a β‐catenin phospho‐destruction complex located near adhesion junctions 63. Conversely, unligated E‐cadherin initiates Wnt signaling since stabilization of E‐cadherin‐mediated cell‐cell junctions (i.e., formation of E‐cadherin‐p120‐catenin complex) inhibits the assembly of the Wnt signalosome 63.
One unexpected finding in our study is the involvement of E‐cadherin suppressor Slug but not Snail in reinforcing the switch of hESCs from self‐renewal to differentiation in response to Wnt/β‐catenin signaling. Although Snail protein was temporarily increased after short‐term Wnt/β‐catenin activation (Fig. 7B), hESCs retained self‐renewal properties with increased clonogenicity (Fig. 1H). In contrast, Slug was upregulated after 1 day Wnt/β‐catenin activation and further increased thereafter (Fig. 7A, 7B). A similar trend was also observed when EcadΔβ mutant (lack of β‐catenin binding capacity) was over‐expressed after short‐term activation of Wnt signaling pathway (Fig. 7C, 7D). Furthermore, transient knockdown of Slug was capable of abrogating long‐term Wnt activation‐induced high level Brachyury expression, low level E‐cadherin expression, and poor clonogenic capacity (Fig. 7F–7H). These data suggest that increasing Slug expression represents a turning point in the fate decision of hESC self‐renewal versus differentiation. TCF1, TCF3, and TCF4 binding sites in Slug promoter have recently been revealed by Chip‐seq in several human cancer cell lines (ENCODE Consortium). Therefore, it is possible that E‐cadherin indirectly suppresses Slug expression by sequestering β‐catenin and reducing its effect on TCFs/Slug.
Although the direct connection between Slug and Brachyury is unclear, Brachyury has been shown to bind to the E‐cadherin promoter, and over‐expression of Brachyury resulted in downregulation of E‐cadherin and upregulation of Slug in epithelial tumor cells 64. It is possible that high levels of Brachyury and Slug expression after long‐term Wnt activation further suppress E‐cadherin expression, thereby skewing the balance from self‐renewal to differentiation.
Our proposed working model (Fig. 7I), which includes Wnt/β‐catenin, E‐cadherin, PI3K/Akt, and Slug, provides a mechanistic insight underlying the dynamic regulation between self‐renewal and differentiation in hESCs. To date, several signaling pathways have been shown to significantly influence ESC fate, including PI3K, Wnt, Activin A/Smad2,3, and Raf/Mek/Erk pathways 65. Recently, Singh et al. proposed the existence of a signaling network crosstalk in hESCs, which involves PI3K/Akt suppressing Erk and Wnt signaling to allow Smad2/3 to activate a specific subset of target genes required for self‐renewal 14. Our results further expand the current working model by revealing a new regulatory network comprising of β‐catenin, E‐cadherin, PI3K, and Slug in hESCs. Since β‐catenin collaborates with Smad2/3 to induce mesendoderm gene expression 14, it is possible that E‐cadherin could indirectly affect Smad2/3 pathway through reducing cytoplasmic‐free β‐catenin. As Mek/Erk signaling has been shown to be downregulated by E‐cadherin‐dependent PI3K/Akt pathway in differentiating intestinal epithelial cells 66, E‐cadherin‐associated PI3K activation may also contribute to the inhibition of Erk pathway to reduce hESC differentiation.
Conclusions
In summary, this study provides direct evidence, for the first time, that integrates β‐catenin, the cell‐cell adhesion molecule E‐cadherin, PI3K/Akt, and Slug into the current signaling network in the control of cell fate of pluripotent stem cells. Our work shows how E‐cadherin and Slug actively tunes Wnt signaling pathway and also provides a unique tool to controllably promote hESC self‐renewal by short‐term Wnt activation. In addition, these findings may shed light on the dual‐role of canonical Wnt signaling in developmental and cancer biology.
Author Contributions
T.‐S.H. and L.L.: conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing; L.M.‐N.: collection and assembly of data and data analysis and interpretation; D.J. and J.B.: collection of data; Z.Y., S.A.L.B., and D.F.: provision of study material; L.W.: conception and design, data interpretation, manuscript writing, financial support, and final approval of manuscript. T.‐S.H., L.L., and L.M.‐N. contributed equally to this study.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
Supporting information
Supplementary Information
Supplementary Information
Supplementary Information
Acknowledgments
We apologize to all those colleagues whose important work could not be cited owing to space limitations. We thank Andrew Sulaiman and Sarah Ooi for thorough reading of the manuscript. This work is supported by an operating grant from the Canadian Institutes of Health Research (CIHR) MOP‐111224 and Heart and Stroke Foundation of Ontario NA7186 to L.W.; a Canada Research Chair in Proteomics and Systems Biology and a NSERC strategic grant to D.F. (STPGP/396508‐2010); CIHR operating grant (MOP 62826) to S.A.L.B.
The copyright line for this article was changed on 2 February 2017 after original online publication
References
- 1. ten Berge D, Kurek D, Blauwkamp T et al. Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat Cell Biol 2011;13:1070–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sato N, Meijer L, Skaltsounis L et al. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK‐3‐specific inhibitor. Nat Med 2004;10:55–63. [DOI] [PubMed] [Google Scholar]
- 3. Singla DK, Schneider DJ, LeWinter MM et al. wnt3a but not wnt11 supports self‐renewal of embryonic stem cells. Biochem Biophys Res Commun 2006;345:789–795. [DOI] [PubMed] [Google Scholar]
- 4. Miki T, Yasuda SY, Kahn M. Wnt/beta‐catenin signaling in embryonic stem cell self‐renewal and somatic cell reprogramming. Stem Cell Rev 2011;7:836–846. [DOI] [PubMed] [Google Scholar]
- 5. Doble BW, Patel S, Wood GA et al. Functional redundancy of GSK‐3alpha and GSK‐3beta in Wnt/beta‐catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell 2007;12:957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hanna J, Markoulaki S, Mitalipova M et al. Metastable pluripotent states in NOD‐mouse‐derived ESCs. Cell Stem Cell 2009;4:513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ying QL, Wray J, Nichols J et al. The ground state of embryonic stem cell self‐renewal. Nature 2008;453:519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernandez A, Huggins IJ, Perna L et al. The WNT receptor FZD7 is required for maintenance of the pluripotent state in human embryonic stem cells. Proc Natl Acad Sci USA 2014;111:1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lluis F, Pedone E, Pepe S et al. Periodic activation of Wnt/beta‐catenin signaling enhances somatic cell reprogramming mediated by cell fusion. Cell Stem Cell 2008;3:493–507. [DOI] [PubMed] [Google Scholar]
- 10. Marson A, Foreman R, Chevalier B et al. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell 2008;3:132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bakre MM, Hoi A, Mong JC et al. Generation of multipotential mesendodermal progenitors from mouse embryonic stem cells via sustained Wnt pathway activation. J Biol Chem 2007;282:31703–31712. [DOI] [PubMed] [Google Scholar]
- 12. Dravid G, Ye Z, Hammond H et al. Defining the role of Wnt/beta‐catenin signaling in the survival, proliferation, and self‐renewal of human embryonic stem cells. Stem Cells 2005;23:1489–1501. [DOI] [PubMed] [Google Scholar]
- 13. Davidson KC, Adams AM, Goodson JM et al. Wnt/beta‐catenin signaling promotes differentiation, not self‐renewal, of human embryonic stem cells and is repressed by Oct4. Proc Natl Acad Sci USA 2012;109:4485–4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Singh AM, Reynolds D, Cliff T et al. Signaling network crosstalk in human pluripotent cells: A Smad2/3‐regulated switch that controls the balance between self‐renewal and differentiation. Cell Stem Cell 2012;10:312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anton R, Kestler HA, Kuhl M. Beta‐catenin signaling contributes to stemness and regulates early differentiation in murine embryonic stem cells. FEBS Lett 2007;581:5247–5254. [DOI] [PubMed] [Google Scholar]
- 16. Wagner RT, Xu X, Yi F et al. Canonical Wnt/beta‐catenin regulation of liver receptor homolog‐1 mediates pluripotency gene expression. Stem Cells 2010;28:1794–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clevers H, Nusse R. Wnt/beta‐catenin signaling and disease. Cell 2012;149:1192–1205. [DOI] [PubMed] [Google Scholar]
- 18. Nelson WJ, Nusse R. Convergence of Wnt, beta‐catenin, and cadherin pathways. Science 2004;303:1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol 2009;10:468–477. [DOI] [PubMed] [Google Scholar]
- 20. Cole MF, Johnstone SE, Newman JJ et al. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev 2008;22:746–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saegusa M, Hashimura M, Kuwata T et al. Requirement of the Akt/beta‐catenin pathway for uterine carcinosarcoma genesis, modulating E‐cadherin expression through the transactivation of slug. Am J Pathol 2009;174:2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCrea PD, Turck CW, Gumbiner B. A homolog of the armadillo protein in Drosophila (plakoglobin) associated with E‐cadherin. Science 1991;254:1359–1361. [DOI] [PubMed] [Google Scholar]
- 23. Carpenter MK, Frey‐Vasconcells J, Rao MS. Developing safe therapies from human pluripotent stem cells. Nat Biotechnol 2009;27:606–613. [DOI] [PubMed] [Google Scholar]
- 24. Larue L, Antos C, Butz S et al. A role for cadherins in tissue formation. Development 1996;122:3185–3194. [DOI] [PubMed] [Google Scholar]
- 25. Chou YF, Chen HH, Eijpe M et al. The growth factor environment defines distinct pluripotent ground states in novel blastocyst‐derived stem cells. Cell 2008;135:449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu Y, Zhu X, Hahm HS et al. Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self‐renewal by small molecules. Proc Natl Acad Sci USA 2010;107:8129–8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li L, Wang S, Jezierski A et al. A unique interplay between Rap1 and E‐cadherin in the endocytic pathway regulates self‐renewal of human embryonic stem cells. Stem Cells 2010;28:247–257. [DOI] [PubMed] [Google Scholar]
- 28. Li L, Wang BH, Wang S et al. Individual cell movement, asymmetric colony expansion, Rho‐associated kinase and E‐cadherin impact the clonogenicity of human embryonic stem cells. Biophys J 2010;98:2442–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohgushi M, Matsumura M, Eiraku M et al. Molecular pathway and cell state responsible for dissociation‐induced apoptosis in human pluripotent stem cells. Cell Stem Cell 2010;7:225–239. [DOI] [PubMed] [Google Scholar]
- 30. Chen G, Hou Z, Gulbranson DR et al. Actin‐myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell 2010;7:240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen T, Yuan D, Wei B et al. E‐cadherin‐mediated cell‐cell contact is critical for induced pluripotent stem cell generation. Stem Cells 2010;28:1315–1325. [DOI] [PubMed] [Google Scholar]
- 32. Redmer T, Diecke S, Grigoryan T et al. E‐cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep 2011;12:720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Samavarchi‐Tehrani P, Golipour A, David L et al. Functional genomics reveals a BMP‐driven mesenchymal‐to‐epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010;7:64–77. [DOI] [PubMed] [Google Scholar]
- 34. Li R, Liang J, Ni S et al. A mesenchymal‐to‐epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010;7:51–63. [DOI] [PubMed] [Google Scholar]
- 35. Gafni O, Weinberger L, Mansour AA et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 2013;504:282–286. [DOI] [PubMed] [Google Scholar]
- 36. Watanabe K, Dai X. A WNTer revisit: New faces of beta‐catenin and TCFs in pluripotency. Sci Signal 2011;4:pe41. [DOI] [PubMed] [Google Scholar]
- 37. Wang S, Tian R, Li L et al. An enhanced chemically defined SILAC culture system for quantitative proteomics study of human embryonic stem cells. Proteomics 2011;11:4040–4046. [DOI] [PubMed] [Google Scholar]
- 38. Jo H, Mondal S, Tan D et al. Small molecule‐induced cytosolic activation of protein kinase Akt rescues ischemia‐elicited neuronal death. Proc Natl Acad Sci USA 2012;109:10581–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gottardi CJ, Wong E, Gumbiner BM. E‐cadherin suppresses cellular transformation by inhibiting beta‐catenin signaling in an adhesion‐independent manner. J Cell Biol 2001;153:1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stappert J, Kemler R. A short core region of E‐cadherin is essential for catenin binding and is highly phosphorylated. Cell Adhes Commun 1994;2:319–327. [DOI] [PubMed] [Google Scholar]
- 41. Kolligs FT, Hu G, Dang CV et al. Neoplastic transformation of RK3E by mutant beta‐catenin requires deregulation of Tcf/Lef transcription but not activation of c‐myc expression. Mol Cell Biol 1999;19:5696–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gupta PB, Kuperwasser C, Brunet JP et al. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet 2005;37:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sarbassov DD, Guertin DA, Ali SM et al. Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science 2005;307:1098–1101. [DOI] [PubMed] [Google Scholar]
- 44. Fuerer C, Nusse R. Lentiviral vectors to probe and manipulate the Wnt signaling pathway. PLoS One 2010;5:e9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jia D, Yang W, Li L et al. β‐Catenin and NF‐κB co‐activation triggered by TLR3 stimulation facilitates stem cell‐like phenotypes in breast cancer. Cell Death Differ. 2014. Sep 26. doi: 10.1038/cdd.2014.145. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsutsui H, Valamehr B, Hindoyan A et al. An optimized small molecule inhibitor cocktail supports long‐term maintenance of human embryonic stem cells. Nat Commun 2011;2:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wray J, Kalkan T, Gomez‐Lopez S et al. Inhibition of glycogen synthase kinase‐3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat Cell Biol 2011;13:838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kemler R, Hierholzer A, Kanzler B et al. Stabilization of beta‐catenin in the mouse zygote leads to premature epithelial‐mesenchymal transition in the epiblast. Development 2004;131:5817–5824. [DOI] [PubMed] [Google Scholar]
- 49. McLean AB, D'Amour KA, Jones KL et al. Activin a efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3‐kinase signaling is suppressed. Stem Cells 2007;25:29–38. [DOI] [PubMed] [Google Scholar]
- 50. Nusse R. Wnt signaling and stem cell control. Cell Res 2008;18:523–527. [DOI] [PubMed] [Google Scholar]
- 51. Staal FJ, Noort Mv M, Strous GJ et al. Wnt signals are transmitted through N‐terminally dephosphorylated beta‐catenin. EMBO Rep 2002;3:63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van Noort M, Meeldijk J, van der Zee R et al. Wnt signaling controls the phosphorylation status of beta‐catenin. J Biol Chem 2002;277:17901–17905. [DOI] [PubMed] [Google Scholar]
- 53. Sakai D, Tanaka Y, Endo Y et al. Regulation of Slug transcription in embryonic ectoderm by beta‐catenin‐Lef/Tcf and BMP‐Smad signaling. Dev Growth Differ 2005;47:471–482. [DOI] [PubMed] [Google Scholar]
- 54. Lyashenko N, Winter M, Migliorini D et al. Differential requirement for the dual functions of beta‐catenin in embryonic stem cell self‐renewal and germ layer formation. Nat Cell Biol 2011;13:753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kelly KF, Ng DY, Jayakumaran G et al. beta‐catenin enhances Oct‐4 activity and reinforces pluripotency through a TCF‐independent mechanism. Cell Stem Cell 2011;8:214–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blauwkamp TA, Nigam S, Ardehali R et al. Endogenous Wnt signalling in human embryonic stem cells generates an equilibrium of distinct lineage‐specified progenitors. Nat Commun 2012;3:1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim H, Wu J, Ye S et al. Modulation of beta‐catenin function maintains mouse epiblast stem cell and human embryonic stem cell self‐renewal. Nat Commun 2013;4:2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Storm MP, Bone HK, Beck CG et al. Regulation of Nanog expression by phosphoinositide 3‐kinase‐dependent signaling in murine embryonic stem cells. J Biol Chem 2007;282:6265–6273. [DOI] [PubMed] [Google Scholar]
- 59. Watanabe K, Ueno M, Kamiya D et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 2007;25:681–686. [DOI] [PubMed] [Google Scholar]
- 60. Faunes F, Hayward P, Descalzo SM et al. A membrane‐associated beta‐catenin/Oct4 complex correlates with ground‐state pluripotency in mouse embryonic stem cells. Development 2013;140:1171–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Livigni A, Peradziryi H, Sharov AA et al. A conserved Oct4/POUV‐dependent network links adhesion and migration to progenitor maintenance. Curr Biol 2013;23:2233–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Torres VA, Tapia JC, Rodriguez DA et al. E‐cadherin is required for caveolin‐1‐mediated down‐regulation of the inhibitor of apoptosis protein survivin via reduced beta‐catenin‐Tcf/Lef‐dependent transcription. Mol Cell Biol 2007;27:7703–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maher MT, Flozak AS, Stocker AM et al. Activity of the beta‐catenin phosphodestruction complex at cell‐cell contacts is enhanced by cadherin‐based adhesion. J Cell Biol 2009;186:219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fernando RI, Litzinger M, Trono P et al. The T‐box transcription factor Brachyury promotes epithelial‐mesenchymal transition in human tumor cells. J Clin Invest 2010;120:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pera MF, Tam PP. Extrinsic regulation of pluripotent stem cells. Nature 2010;465:713–720. [DOI] [PubMed] [Google Scholar]
- 66. Laprise P, Langlois MJ, Boucher MJ et al. Down‐regulation of MEK/ERK signaling by E‐cadherin‐dependent PI3K/Akt pathway in differentiating intestinal epithelial cells. J Cell Physiol 2004;199:32–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Information
Supplementary Information