

Abstract
This two‐sequence, three‐period crossover study is the first pharmacokinetic (PK) study to compare all three innovator formulations of tacrolimus (twice‐daily immediate‐release tacrolimus capsules [IR‐Tac]; once‐daily extended‐release tacrolimus capsules [ER‐Tac]; novel once‐daily tacrolimus tablets [LCPT]). Stable renal transplant patients were dosed with each drug for 7 days, and blood samples were obtained over 24 h. Thirty subjects were included in the PK analysis set. A conversion factor of 1:1:0.80 for IR‐Tac:ER‐Tac:LCPT was used; no dose adjustments were permitted during the study. The median (interquartile range) total daily dose was 6.0 (4.0–8.0) mg for IR‐Tac and ER‐Tac and 4.8 (3.3–6.3) for LCPT. Significantly higher exposure on a per milligram basis, lower intraday fluctuation and prolonged time (Tmax) to peak concentration (Cmax) were found for LCPT versus IR‐Tac or ER‐Tac. ER‐Tac showed no differences versus IR‐Tac in exposure, Cmax, Tmax or fluctuation. The observed exposure of IR‐Tac was used to normalize exposure for LCPT and ER‐Tac, resulting in the following recommended total daily dose conversion rates: IR‐Tac:ER‐Tac, +8%; IR‐Tac:LCPT, −30%; ER‐Tac:LCPT, −36%. After exposure normalization, Cmax was ~17% lower for LCPT than for IR‐Tac or ER‐Tac; Cmin was ~6% lower for LCPT compared with IR‐Tac and 3% higher compared with ER‐Tac.
Keywords: clinical research/practice, kidney transplantation/nephrology, clinical trial, immunosuppressant, calcineurin inhibitor: tacrolimus, pharmacokinetics/pharmacodynamics
Short abstract
This study evaluates the pharmacokinetic profile of three tacrolimus formulations and shows significant differences, highlighting the higher per mg exposure and a lower peak and delayed peak of extended‐release tacrolimus tablets when compared to the two other capsule formulations.
Abbreviations
- AE
adverse event
- ANCOVA
analysis of covariance
- ASTCOFF
a steady‐state pharmacokinetic comparison of all FK‐506 formulations
- AUC
area under the curve
- BMI
body mass index
- BPAR
biopsy‐proven acute rejection
- C0
predose concentration
- Cavg
average concentration
- CI
confidence interval
- Cmax
peak concentration
- Cmin
minimum concentration
- DBS
dried blood spot
- eGFR
estimated glomerular filtration rate
- ER‐Tac
extended‐release tacrolimus
- GI
gastrointestinal
- IQR
interquartile range
- IR‐Tac
immediate‐release tacrolimus
- LC‐MS/MS
liquid chromatography–mass spectrometry/mass spectrometry
- LCPT
once‐daily MeltDose tacrolimus tablets
- LSM
least square means
- MDRD‐4
Modification of Diet in Renal Disease
- PK
pharmacokinetics
- RGM
ratio of geometric means
- SAE
serious adverse event
- SD
standard deviation
- TDD
total daily dose
- TEAE
treatment‐emergent adverse event
- Tmax
time to maximum observed concentration
Introduction
Tacrolimus is an integral part of most kidney transplant patients’ immunosuppression drug therapy, with over 90% of kidney transplant recipients being discharged with a tacrolimus‐containing regimen 1. Three innovator formulations are currently available: traditional twice‐daily immediate‐release tacrolimus capsules (IR‐Tac; Prograf, Astellas Pharma US, Inc., Northbrook, IL), and two once‐daily formulations: a once‐daily MeltDose tablet formulation (LCPT; Envarsus XR [Envarsus in Europe]; Veloxis Pharmaceuticals, Inc., Edison, NJ) and a once‐daily extended‐release tacrolimus capsule (ER‐Tac; Astagraf XL; Astellas Pharma US, Inc., Northbrook, IL).
The pharmacokinetics (PK), efficacy and safety of both once‐daily formulations have been compared individually with those of IR‐Tac in clinical studies in de novo and conversion studies of renal transplant recipients 2, 3, 4, 5, 6. Comparisons of ER‐Tac with IR‐Tac have shown variable effects of formulation on peak concentration (Cmax) and time of peak concentration (Tmax) but have generally shown that the minimum concentration (Cmin) and 24‐h area under the curve (AUC24) are lower for ER‐Tac on a milligram to milligram basis 7, 8. Clinical studies of de novo and stable transplant patients have consistently shown that ER‐Tac requires a higher dose than IR‐Tac to achieve similar trough and exposure levels 7, 9, 10, 11, 12.
Although both LCPT and ER‐Tac have once‐daily dosing, important differences in their formulations affect their PK. The extended release of tacrolimus offered by ER‐Tac is a result of adding ethylcellulose, which acts to slow down the diffusion rate of tacrolimus, leading to a prolonged release 13. In contrast, LCPT's development was focused on MeltDose technology (Veloxis Pharmaceuticals, Inc., Edison, NJ), which improves the solubility of tacrolimus, and therefore bioavailability, by dispersing tacrolimus in a polymeric matrix 14. This results in a more distal distribution of tacrolimus in the gut 15. This formulation has also been shown to promote a more rapid attainment of therapeutic tacrolimus systemic exposure compared with that of IR‐Tac and requires a lower total daily dose to achieve therapeutic exposure levels 16, 17. LCPT is also associated with less fluctuation between maximum (“peak”) exposure and trough and with a lower peak exposure level 18. From a clinical perspective, LCPT is noninferior in terms of efficacy (composite endpoint, including graft loss, death, biopsy‐proven acute rejection [BPAR] and loss to follow‐up) when compared to IR‐Tac with a similar safety profile 17.
However, a direct head‐to‐head PK comparison of all three formulations has not yet been conducted. Therefore, the objective of this study was to compare the steady‐state PK profile of LCPT with IR‐Tac and ER‐Tac in stable kidney transplant recipients to provide clinicians with dose conversion strategies between formulations.
Materials and Methods
Study design
This steady‐state pharmacokinetic comparison of all FK‐506 formulations (ASTCOFF) study was an open‐label, randomized, two‐sequence, three‐period crossover trial (Figure 1; ClinicalTrials.gov NCT02339246). The primary objective of the study was to evaluate the PK profile of LCPT in stable renal transplant patients compared with the profiles of IR‐Tac and ER‐Tac, which in turn allows for the generation of clinical recommendations for converting patients from one formulation to another.
Figure 1.
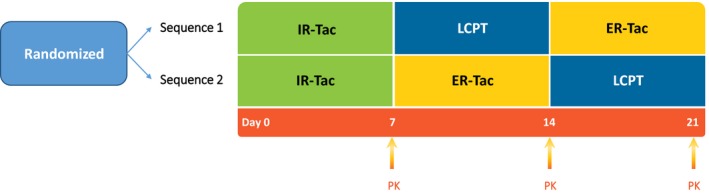
Study design. ER‐Tac, extended‐release tacrolimus; IR‐Tac, immediate‐release tacrolimus; LCPT, once‐daily, MeltDose tacrolimus; PK, pharmacokinetic profiling.
Secondary study objectives included evaluating the daily tacrolimus trough level during each 7‐day crossover period, evaluating the labeled conversion factors when switching from the twice‐daily IR‐Tac formulation to once‐daily ER‐Tac or LCPT, and safety. The conversion factors used in the study were based on the Food and Drug Administration labeling for converting patients from IR‐Tac to LCPT and on the European Medicines Agency labeling for converting patients from IR‐Tac to ER‐Tac, because conversion labeling is not available in the United States. Sample size calculations determined that at least 14 subjects per sequence were required to have 85% power to show that, by following the labeled conversation rate, the ratio of geometric means (RGM) of AUC0–24 between LCPT and IR‐Tac would be bioequivalent based on the equivalence criteria of 0.80–1.25, inclusive, and based on two 1‐sided t‐tests, each at 5% significance level and assuming a standard deviation of 0.22 (ln scale). This was a single‐center study that received institutional review board approval (IRB# 2014–7906); recruitment took place at two centers: the Christ Hospital and the University of Cincinnati medical center. This study was conducted in accordance with the Declaration of Helsinki.
Eligible patients were randomized in a 1:1 fashion to one of the two treatment sequences. Study personnel involved in trial operations remained blinded to the randomization list.
Inclusion and exclusion criteria
Eligible patients had to be 18 years or older, have received a first or second renal transplant at least 6 months prior to study entry, be on a stable (no tacrolimus dose change in the 7 days prior to screening) immunosuppressive regimen consisting of any twice‐daily tacrolimus and mycophenolate with or without prednisone, have a BMI ≥19 kg/m2 and not be scheduled to start any new medications or agents that could interfere with tacrolimus blood levels during the study. Patients with an episode of rejection within 3 months of screening, having received another organ than a kidney, with an estimated glomerular filtration rate (eGFR) of ≤25 mL/min/1.73 m2, with severe gastroparesis or gastrointestinal conditions that could interfere with tacrolimus absorption, and pregnant or lactating women were excluded. Any patient omitting a tacrolimus dose within 48 h of PK measurements was also excluded from the PK analysis.
Intervention
After randomization, each patient received IR‐Tac (Prograf, the reference drug for both sequences, Astellas Pharma US, Inc., Northbrook, IL) followed by either LCPT followed by ER‐Tac, or ER‐Tac followed by LCPT, depending on the randomization sequence. At day 1 of each crossover period, patients were switched to a new formulation using a milligram to milligram total daily dose conversion factor of 1:1 for IR‐Tac to ER‐Tac, and 1:0.80 for IR‐Tac or ER‐Tac to LCPT, representing a 20% lower total daily dose. Doses were rounded to take into account the availability of dosage strengths. No immunosuppressant dose titrations (tacrolimus, mycophenolate or prednisone if present) were allowed during the study period.
Patients were required to remain fasting for at least 8 h prior to study drug administration and for 3 h following administration on the days of PK measurements. Dosing times were preassigned and remained constant throughout the study. To measure patient adherence, prior to taking their morning dose of tacrolimus, patients had to perform a fingerstick and apply their blood to a protein saver card with a predefined surface area; the protein saver card was returned to the investigators at every PK visit for trough tacrolimus measurement via dried blood spot (DBS) analysis by a central laboratory via tandem mass spectrometry 19, 20, 21. Patients were provided with a daily diary to record dosing times and daily trough tacrolimus measurement times and to document any delays or omissions in study doses. In addition, a whole blood sample for trough level at every PK visit was also obtained and analyzed by a local laboratory with an immunoassay (Abbott Architect platform, Abbott Laboratories, IL) for the purpose of safety assessment. All patients received study drug under the direct supervision of the investigators.
Safety laboratory assessments were obtained at screening and at every PK visit and were all analyzed at the clinical laboratory. Concomitant medications were reviewed at each study visit.
Pharmacokinetic profiles
Twenty‐four‐hour PK collections were performed at the end of each 1‐week period; a total of 17 or 21 time points were sampled over 24 h. Blood samples for tacrolimus were drawn as follows: IR‐Tac sampling strategy: predose concentration (C0) and then 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 12.5, 13, 13.5, 14, 14.5, 15, 16, 18, 20, and 24 h; LCPT and ER‐Tac: C0, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 14, 16, 18, 21, and 24 h. The additional time points for the IR‐Tac PK sampling were designed to better characterize the 12‐ to 24‐h PK profile of the evening dose.
The following PK parameters were a priori defined for analysis: area under the concentration–time curve from time 0 to 24 h (AUC0–24), maximum (peak) observed concentration (Cmax), time to maximum observed concentration (Tmax), minimum blood concentration observed over the 24‐h interval (Cmin; the value was taken from the observed concentration data at the 24‐h nominal time point), predose (nominal time = 0) concentration (C0), average concentration (Cavg) and percent peak‐to‐trough fluctuation of the drug concentration over the dosing interval (0–24) at the steady state (% fluctuation; calculated as 100*[(Cmax – Cmin)/Cavg]).
Bioanalytic methods
The central laboratory used (Lambda Therapeutics Research Inc., Toronto, Canada) conducted the tacrolimus whole blood level analyses according to principles of Good Laboratory Practice. The bioanalytical validated methods for assessing tacrolimus whole blood concentrations were analyzed by tandem mass spectrometry (liquid chromatography–mass spectrometry/mass spectrometry [LC‐MS/MS]). In brief, tacrolimus was extracted from whole blood and separated via high‐performance liquid chromatography and detected by using a TSQ Quantum tandem mass spectrometer (ThermoScientific, Waltham, MA).
Dried blood spot analysis was performed using previously validated and described technique 19, 20, 21, 22. Following extraction of tacrolimus from the DBS card, concentrations were evaluated by LC‐MS/MS on a platform consisting of Agilent components (Agilent, Santa Clara, CA) in combination with AB Sciex (AB Sciex, Foster City, CA) mass spectrometers at iC42 Clinical Research and Development (University of Colorado, CO).
Study drug
Study drugs were provided by the study sponsor, Veloxis Pharmaceuticals; both IR‐Tac and ER‐Tac were acquired from commercial supply. All acquired bottles were from a single lot for each product. All dosage strengths available were allowed during the study (0.5, 1, 5 mg for IR‐Tac and 1, 5 mg for ER‐Tac). For LCPT, tablets had to remain in their original packaging throughout the study, and dosage strengths included 0.75, 1, and 4 mg tablets. At every PK visit, patients had to return unused study drug and bottles for pill counting.
Safety assessments
Safety parameters included incidence of treatment emergent adverse events (TEAEs), serious adverse events (SAEs), graft failure, BPAR and death; changes in safety laboratory tests; changes in vital signs; and tacrolimus trough level determined by the local clinical laboratory for safety assessment. Patient eGFR was assessed by the Modification of Diet in Renal Disease (MDRD‐4) 23 and 24‐h urine collection at every PK visit to calculate creatinine clearance.
Adherence assessment
Drug accountability and adherence evaluation were performed at each study visit by reviewing the patients’ daily diary and by pill counting. Adherence was primarily assessed by comparing the number of total doses taken with the total number of prescribed doses based on pill counts. Ad hoc analyses of adherence were performed by comparing the time when patients took their dose to the prescribed time, allowing for a 30‐min window on non‐PK days, and analysis of patient diaries.
Statistical analysis
All patients treated with study drug were included in the safety analysis; patients who successfully completed the 3‐week crossover periods were included in the PK analysis. The actual time of observed concentration‐time data was used to derive PK parameters via WinNonlin version 6.3 (Certara USA, Princeton, NJ) based on noncompartmental analysis and linear trapezoidal linear interpolation calculation methods. All PK parameters (except Tmax) were evaluated using a mixed effect analysis of covariance (ANCOVA) model in which period, sequence and formulation were fixed effects and patient within a sequence was a random effect. Natural logarithm transformation was performed for AUC, Cmax, Cavg and C0, and no transformation for other parameters was used for observed and dose‐corrected analyses. Time to maximal concentration was analyzed using the Wilcoxon signed‐rank test. Pearson linear correlation coefficients between AUC0–24 and Cmin were also estimated. Clinical safety parameters were tabulated for each treatment without statistical inferential testing.
One of the objectives of this study was to provide guidance to clinicians with regard to dose conversion rates when switching patients from one formulation to another to achieve similar exposure. As such, the exposure normalization analysis methodology examined the relative bioavailability of the employed dose conversion factor (80% for LCPT and 100% for ER‐Tac when converting from IR‐Tac) in this study based on the equal exposure principle (exposure is expressed as AUC0–24) before and after conversion from IR‐Tac to LCPT or ER‐Tac. The factors were applied to AUC0–24, Cmax and Cmin for each patient, and ANCOVA models were used to confirm whether the RGM for AUC0–24 was approximately 100% postnormalization.
All p‐values from inferential tests were reported as is without adjustment for multiple comparisons.
Results
Patient disposition
A total of 32 patients were screened for participation in the study between January 23, 2015, and February 27, 2015, and 31 were randomized, with 1 patient being a screen failure. Sixteen patients were randomized to the IR‐Tac/LCPT/ER‐Tac arm and 15 to the IR‐Tac/ER‐Tac/LCPT arm. All 31 randomized patients completed the study and are included in the safety analyses. One patient was not adherent with the study drug dosing requirement based on pill count while on LCPT treatment and was excluded from PK analysis for missing 5 doses (Figure 2). Baseline characteristics were similar across the groups, with a mean age of 50.1 versus 46.3 years; 56% versus 60% male; 81.3% versus 66.7% Caucasian; and with a mean time since transplant of 7 versus 5.2 years for the IR‐Tac/LCPT/ER‐Tac group compared to the IR‐Tac/ER‐Tac/LCPT group, respectively (Table 1). Based on the 30 subjects in the PK analysis, the median (interquartile range) total daily dose was 6.0 (4.0–8.0) mg for IR‐Tac and ER‐Tac and 4.8 (3.3–6.3) for LCPT.
Figure 2.
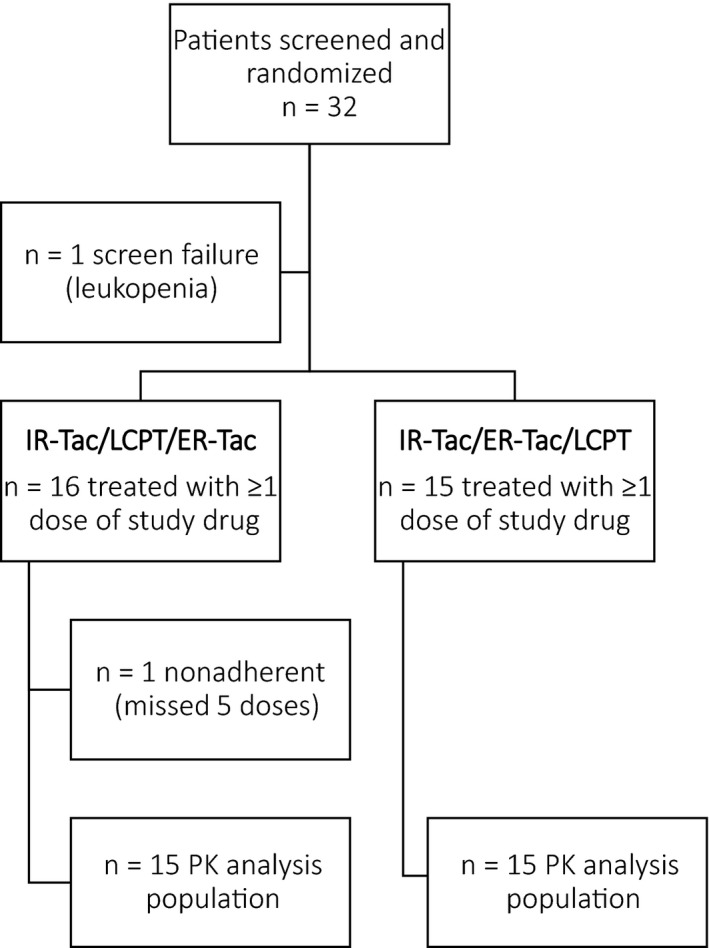
Patient attrition. ER‐Tac, extended‐release tacrolimus; IR‐Tac, immediate‐release tacrolimus; LCPT, once‐daily, MeltDose tacrolimus; PK, pharmacokinetics.
Table 1.
Patient characteristics
| All patients (n = 31) | IR‐Tac–LCPT–ER‐Tac (n = 16) | IR‐Tac–ER‐Tac–LCPT (n = 15) | |
|---|---|---|---|
| Age (years), mean (SD) | 48.3 (12.1) | 50.1 (11.0) | 46.3 (13.3) |
| Male sex, n (%) | 18 (58.1) | 9 (56.3) | 9 (60.0) |
| Race, n (%) | |||
| Caucasian | 23 (74.2) | 13 (81.3) | 10 (66.7) |
| African American | 7 (22.6) | 2 (12.5) | 5 (33.3) |
| Other | 1 (3.2) | 1 (6.3) | 0 (0) |
| Donor type, n (%) | |||
| Deceased | 3 (9.7) | 1 (6.3) | 2 (13.3) |
| Living | 28 (90.3%) | 15 (93.8%) | 13 (86.7%) |
| Years since transplant to study, mean (min–max) | 6.1 (0.7–14.2) | 7.0 (3.2–14.2) | 5.2 (0.7–9.2) |
| Baseline BMI (kg/m2), mean (SD) | 30.4 (4.9) | 30.9 (4.8) | 29.9 (5.1) |
BMI, body mass index; ER‐Tac, extended‐release tacrolimus; IR‐Tac, immediate‐release tacrolimus; LCPT, once‐daily, MeltDose tacrolimus; SD, standard deviation.
Observed PK data
Figure 3(A) displays the whole blood concentrations of tacrolimus for each formulation, and Table 2 displays the observed PK parameters. When using labeled conversion rate (1:1 for IR‐Tac to ER‐Tac and 1:0.80 for IR‐Tac to LCPT), the AUC0–24 was significantly greater for LCPT compared with IR‐Tac (RGM: 117.0%; p = 0.002) and ER‐Tac (RGM: 125.7%; p < 0.001). The intraday peak‐to‐trough fluctuation was approximately 30% lower for LCPT compared with IR‐Tac (least square means [LSM] difference: −29.0%; p = 0.004) and ER‐Tac (LSM difference: −35.3%; p < 0.001). The Tmax was significantly longer at 5.9 h for LCPT compared with IR‐Tac and ER‐Tac (1.9 and 1.5 h, respectively; p < 0.001). Conversely, Tmax between IR‐Tac and ER‐Tac did not differ statistically (p = 0.669). The Cmin was significantly lower for ER‐Tac (5.1 ng/mL) compared with both LCPT (6.8 ng/mL, p < 0.001) and IR‐Tac (6.1 ng/mL, p = 0.001). There were no other statistically significant differences between IR‐Tac and ER‐Tac, nor were there significant period or sequence effects (Table 2). Overall, LCPT had a greater relative bioavailability with an increase of ~50% (p < 0.001) compared with both IR‐Tac and ER‐Tac on a milligram to milligram basis.
Figure 3.
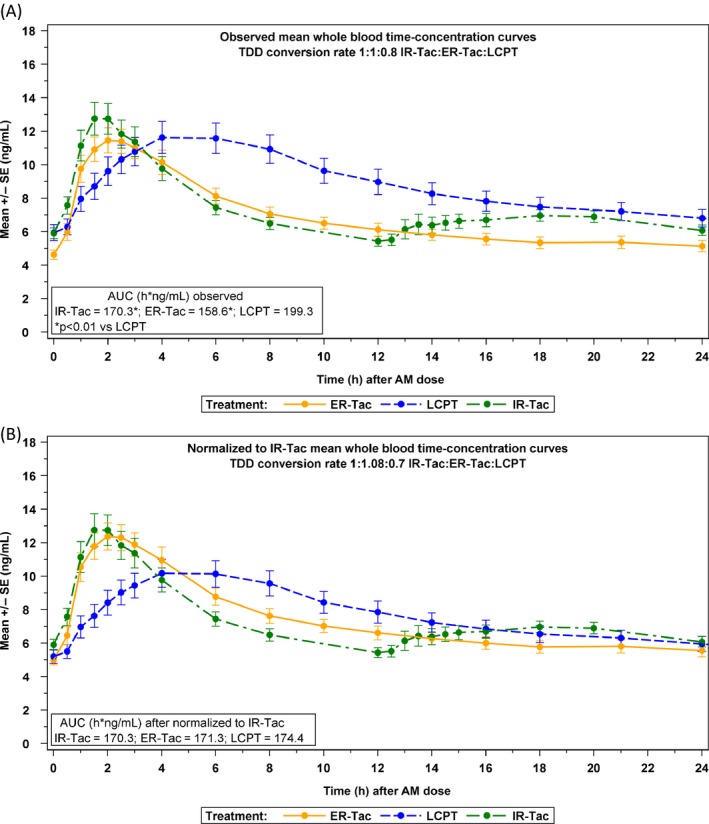
(A) Observed mean whole blood concentrations of tacrolimus based on conversion factors of 1:1:0.80 for IR‐Tac:ER‐Tac:LCPT (upper panel) versus (B) exposure (AUC)‐normalized mean whole blood concentrations of tacrolimus based on conversion factors of 1:1.08:0.70 (lower panel). AUC, area under the curve, ER‐Tac, extended‐release tacrolimus; IR‐Tac, immediate‐release tacrolimus; LCPT, once‐daily, MeltDose tacrolimus; SE, standard error of the mean; TDD, total daily dose.
Table 2.
Summary of observed pharmacokinetic parameters (n = 30)
| Observed PK parameter | Observed resultsa | Comparisonsb | ||||
|---|---|---|---|---|---|---|
| LCPT (L) | ER‐Tac (ER) | IR‐Tac (IR) | L versus IR (%) | ER versus IR (%) | L versus ER (%) | |
| TDD (mg/day) | 4.9 ± 2.3 | 6.1 ± 2.9 | 6.1 ± 2.9 | – | – | – |
| Median (IQR) | 4.8 (3.3–6.3) | 6.0 (4.0–8.0) | 6.0 (4.0–8.0) | – | – | – |
| AUC0–24 (h*ng/mL) |
213.4 ± 83.1 199.3 (38.6) |
165.0 ± 50.0 158.6 (29.1) |
176.5 ± 50.8 170.3 (27.2) |
117.0 (107.9, 127.0) p = 0.002 |
93.1 (85.8, 101.0) p = 0.149 |
125.7 (114.1, 138.5) p < 0.001 |
| Cmax (ng/mL) |
13.9 ± 5.3 12.9 (41.9) |
13.2 ± 4.4 12.5 (34.2) |
14.5 ± 5.5 13.6 (38.5) |
94.7 (85.8, 104.4) p = 0.354 |
91.8 (83.2, 101.3) p = 0.150 |
103.1 (92.4, 115.0) p = 0.638 |
| Cmin (ng/mL) |
6.8 ± 2.9 6.3 (42.4) |
5.1 ± 1.8 4.9 (33.3) |
6.1 ± 1.7 5.9 (27.0) |
107.0 (97.6, 117.2) p = 0.223 |
83.0 (75.7, 90.9) p = 0.001 |
128.9 (117.4, 141.6) p < 0.001 |
| Tmax (h) | 5.9 (1.5, 14.0) | 1.9 (0.9, 5.9) | 1.5 (0.9, 20.0) |
3.0 (1.6, 4.4) p < 0.001 |
0.1 (−0.4, 0.5) p = 0.670 |
3.0 (1.9, 4.0) p < 0.001 |
| Fluctuation (%) | 83.6 ± (51.7) | 118.9 ± (48.4) | 112.6 ± (53.1) |
−29.0 (−48.4, −9.6) p = 0.004 |
6.4 (−13.1, 25.7) p = 0.518 |
−35.3 (−53.4, −17.3) p < 0.001 |
Parameter fluctuation: Presented differences in LSM (95% CI) derived from ANCOVA models that included fixed effects of treatment, sequence, period (LCPT vs. ER‐Tac analyses) and random effect of subjects (sequence). p‐value was from 2‐sample t‐test.
Parameter Tmax: Presented median location shifts (95% CI) using Hodges–Lehmann estimation; the Hodges–Lehmann estimates of median location shift (“Comparisons” columns) may not match the difference in observed median values (“Observed Result” columns).
ANCOVA, analysis of covariance; AUC0–24, 24‐h area under the curve; CI, confidence interval; Cmax, maximal concentration; Cmin, minimal concentration; CV, coefficient of variation; ER‐Tac, extended‐release tacrolimus; IQR, interquartile range; IR‐Tac, immediate‐release tacrolimus; LCPT, once‐daily, MeltDose tacrolimus; LSM, least square means; PK, pharmacokinetics; RGM, ratio of geometric means; SD, standard deviation; TDD, total daily dose; Tmax, time to maximal concentration.
Parameter AUC0–24, Cmax, Cmin: Presented arithmetic mean ± SD and geometric means (CV% of geometric means); Parameter TDD: Presented arithmetic mean ± SD; Tmax: Presented median (min, max).
Parameter AUC0–24, Cmax, Cmin: Presented RGM in percent (90% CI) derived from ANCOVA models that included fixed effects of treatment, sequence, period (LCPT vs. ER‐Tac analyses) and random effect of subjects (sequence). p‐value was from 2‐sample t‐test. Analysis was performed on natural log‐transformed data.
An ad hoc subgroup analysis to evaluate the differences in PK parameters among formulations between African Americans (n = 7) and non–African Americans (n = 23) was performed. Total median daily doses of all formulations were approximately twice as high for African Americans when compared with doses of non–African Americans (6.25, 8 and 8 mg vs. 3.25, 4 and 4 mg for LCPT, ER‐Tac and IR‐Tac, respectively). The overall differences in PK parameters among formulations for African Americans were similar to the main analysis, suggesting that LCPT provided a higher AUC0–24 (RGM 138.2, p < 0.01, and 149.1, p = 0.02), Cmax was similar despite a higher AUC0–24 (RGM 107.4, p = 0.54, and 109.8, p = 0.22), Cmin was higher (RGM 125.3, p = 0.02, and 151.0, p < 0.01), Tmax was delayed (RGM 2.48, p = 0.22, and 2.55, p < 0.01) and intraday peak‐to‐trough fluctuation was lower (−39.64, p = 0.07, and −54.08, p = 0.02) for LCPT versus IR‐Tac and LCPT versus ER‐Tac, respectively.
Exposure normalization and dose conversion results
When converting patients from IR‐Tac to LCPT, the study dose conversion rate applied resulted in significantly higher overall exposure for LCPT. This difference was not apparent when using the labeled conversion rate between IR‐Tac and ER‐Tac because the RGM of AUC0–24 was 93.1% (90% confidence interval [CI] 85.8–101.0).
For these reasons, the exposure normalization and dose conversion analysis was performed. A conversion factor (percentage of total daily dose of reference drug) of −30% yielded comparable AUC0–24 when converting from IR‐Tac to LCPT (AUC0–24 RGM of 102.4% [90% CI 94.4–111.1%, p = 0.627]. A conversion factor of −36% when converting from ER‐Tac to LCPT led to a RGM of 100.6% [90% CI 91.3–110.8%, p = 0.924]). A conversion factor of +8% was required to obtain similar exposures when converting from IR‐Tac to ER‐Tac (RGM of 100.6% [92.7–109.1%, p = 0.908]) (Figure 3B and Table 3). In addition, LCPT Cmax was reduced by 17% when compared with the Cmax of both IR‐Tac and ER‐Tac, with an RGM of approximately 82% (p = 0.002 and 0.006, respectively), whereas Cmax of ER‐Tac and IR‐Tac remained similar (p = 0.887).
Table 3.
Recommended dose conversions and resulting normalized PK parameters
| Dose conversion factor | LCPT (L) | IR‐Taca(IR) | RGMb and 90% CI | p‐value |
|---|---|---|---|---|
| −30% | Reference | L/IR | L versus IR | |
| −30% from IR‐Tac to LCPT based on normalized (AUC) exposure | ||||
| AUC0–24 (h*ng/mL) | 174.4 | 170.3 | 102.4 (94.4, 111.1) | 0.627 |
| Cmax (ng/mL) | 11.3 | 13.6 | 82.8 (75.1, 91.4) | 0.002 |
| Cmin (ng/mL) | 5.5 | 5.9 | 93.6 (85.4, 102.6) | 0.233 |
| Dose conversion rate | ER‐Tac (ER) | IR‐Taca(IR) | RGMb and 90% CI | p‐value |
|---|---|---|---|---|
| +8% | Reference | ER/IR | ER versus IR | |
| +8% from IR‐Tac to ER‐Tac based on normalized (AUC) exposure | ||||
| AUC24 (h*ng/mL) | 171.3 | 170.3 | 100.6 (92.7, 109.1) | 0.908 |
| Cmax (ng/mL) | 13.5 | 13.6 | 99.2 (89.9, 109.4) | 0.887 |
| Cmin (ng/mL) | 5.3 | 5.9 | 89.6 (81.8, 98.2) | 0.050 |
| Dose conversion rate | LCPT (L) | ER‐Taca(ER) | RGMb and 90% CI | p‐value |
|---|---|---|---|---|
| −36% | Reference | L/ER | L versus ER | |
| −36% from ER‐Tac to LCPT based on normalized (AUC) exposure | ||||
| AUC24 (h*ng/mL) | 159.5 | 158.6 | 100.6 (91.3, 110.8) | 0.924 |
| Cmax (ng/mL) | 10.3 | 12.5 | 82.5 (73.9, 92.0) | 0.006 |
| Cmin (ng/mL) | 5.0 | 4.9 | 103.1 (93.9, 113.2) | 0.579 |
AUC0–24, 24‐h area under the curve; CI, confidence interval; Cmax, maximal concentration; Cmin, minimal concentration; ER‐Tac, extended‐release tacrolimus; IR‐Tac, immediate‐release tacrolimus; LCPT, once‐daily, MeltDose tacrolimus; PK, pharmacokinetics; RGM, ratio of geometric means.
The AUC0–24 values used as reference were obtained from the observed data.
RGM is expressed as % value.
Correlation between AUC0–24 and Cmin
A robust correlation between AUC0–24 and Cmin was found for all three tacrolimus formulations across the observed range of Cmin and AUC0–24. Pearson's linear correlation coefficient between ln(AUC0–24) and ln(Cmin) was 0.92 (p < 0.001) for LCPT, 0.92 (p < 0.001) for ER‐Tac and 0.81 (p < 0.001) for IR‐Tac.
Adherence assessment
Overall mean (standard deviation [SD]) daily tacrolimus morning trough concentrations were assessed by DBS for days 1–6. Tacrolimus morning trough level assessed by DBS on each day within a period is also presented in Figure 4. To account for a potential carryover effect resulting from the change in tacrolimus formulations immediately following conversion, we removed the first 3 days of the 6‐day period from the following calculations. Morning tacrolimus trough concentrations for days 4–6 were 9.73 (3.61) ng/mL for LCPT, 8.13 (3.08) for ER‐Tac and 9.57 (3.46) for IR‐Tac. There were no statistically significant differences between LCPT and IR‐Tac (p = 0.812), but there were differences between LCPT and ER‐Tac (p = 0.021) and ER‐Tac and IR‐Tac (p = 0.036). Tacrolimus morning trough levels assessed by DBS on each day within a period are also presented in Figure 4.
Figure 4.
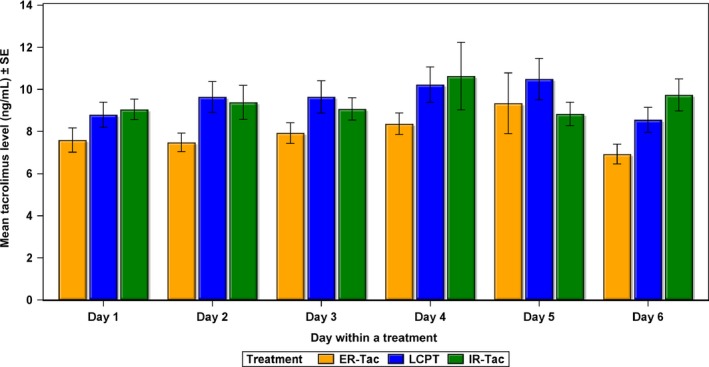
Group mean daily tacrolimus trough level (ng/ mL ) determined by the dried blood samples in each period. ER‐Tac, extended‐release tacrolimus; IR‐Tac, immediate‐release tacrolimus; LCPT, once‐daily, MeltDose tacrolimus; SE, standard error of the mean.
Overall adherence was excellent. An ad hoc analysis showed that when actual dosing time based on patient diaries was compared with prescribed dosing time, the proportion of doses taken within plus or minus 30 min of prescribed time was 91.9% (SD 12.8%), 87.1% (SD 14.2%) and 89.9% (SD 15.0%) for LCPT, ER‐Tac and IR‐Tac, respectively.
Safety
No patients discontinued study drug during the study because of adverse events. No deaths, BPARs, graft losses or SAEs occurred. A summary of the incidence of the most common TEAEs occurring in 5% or more of the patients overall by system organ class and preferred term for the safety set are presented in Table 4. The two most commonly reported system organ classes were “gastrointestinal disorders” and “general disorders and administration site conditions.” The most commonly reported adverse events (AEs) were diarrhea, peripheral edema and headache.
Table 4.
Overall summary of treatment‐emergent adverse events
| Total (n = 31) | LCPT (n = 31) | ER‐Tac (n = 31) | IR‐Tac (n = 31) | |||||
|---|---|---|---|---|---|---|---|---|
| Patients (%) | Events | Patients (%) | Events | Patients (%) | Events | Patients (%) | Events | |
| Patients with ≥1 TEAE, n (%) | 15 (48.4) | 29 | 6 (19.4) | 10 | 10 (32.3) | 16 | 3 (9.7) | 3 |
| TEAE severity, n (%) | ||||||||
| Mild | 14 (45.2) | 24 | 6 (19.4) | 7 | 9 (29.0) | 14 | 3 (9.7) | 3 |
| Moderate | 3 (9.7) | 5 | 1 (3.2) | 3 | 2 (6.5) | 2 | 0 | 0 |
| Number of TEAE per patient, median (min, max) | 0 (0.0, 5.0) | – | 0 (0.0, 4.0) | – | 0 (0.0, 3.0) | – | 0 (1.0) | – |
| Gastrointestinal disorders, n (%) | 6 (19.4) | 8 | 1 (3.2) | 1 | 4 (12.9) | 6 | 1 (3.2) | 1 |
| Diarrhea | 3 (9.7) | 3 | 0 | 0 | 2 (6.5) | 2 | 1 (3.2) | 1 |
| Vomiting | 2 (6.5) | 2 | 0 | 0 | 2 (6.5) | 2 | 0 | 0 |
| General disorders, n (%) | 6 (19.4) | 7 | 3 (9.7) | 4 | 3 (9.7) | 3 | 0 | 0 |
| Fatigue | 2 (6.5) | 2 | 2 (6.5) | 2 | 0 | 0 | 0 | 0 |
| Edema, peripheral | 3 (9.7) | 3 | 0 | 0 | 3 (9.7) | 3 | 0 | 0 |
| Infections and infestations, n (%) | 3 (9.7) | 3 | 1 (3.2) | 1 | 2 (6.5) | 2 | 0 | 0 |
| Nasopharyngitis | 2 (6.5) | 2 | 0 | 0 | 2 (6.5) | 2 | 0 | 0 |
| Nervous system disorders, n (%) | 4 (12.9) | 6 | 1 (3.2) | 1 | 2 (6.5) | 3 | 2 (6.5) | 2 |
| Headache | 3 (9.7) | 3 | 0 | 0 | 1 (3.2) | 1 | 2 (6.5) | 2 |
ER‐Tac, extended‐release tacrolimus; IR‐Tac, immediate‐release tacrolimus; LCPT, once‐daily, MeltDose tacrolimus; TEAE, treatment‐emergent adverse event.
Of the 31 patients treated with at least 1 dose of study drug, 15 (48.4%) unique patients had at least 1 TEAE: 6 (19.4%) patients in the LCPT treatment period, 10 (32.3%) patients in the ER‐Tac treatment period and 3 (9.7%) patients in the IR‐Tac treatment period.
Results of renal function assessments are presented in Table 5. There were no statistical differences between groups when assessed by MDRD‐4 or 24‐h urine collection.
Table 5.
Renal function assessments
| Parameter | IR‐Tac (N = 30) | ER‐Tac (N = 30)a | LCPT (N = 30)a | Treatment effect p‐valueb |
|---|---|---|---|---|
| Serum creatinine (mg/dL), mean (SD) | 1.28 (0.23) | 1.30 (0.29) | 1.28 (0.26) | 0.961 |
| 24 h urine total volume (mL), mean (SD) | 3019 (1312) | 3160 (2831) | 3335 (2616) | 0.873 |
| 24 h creatinine clearance, calculated (mL/min), mean (SD) | 77 (32) | 74 (33) | 76 (32) | 0.929 |
| eGFR (mL/min/1.73 m2) | ||||
| Non–African Americans, mean (SD) |
n = 23 61 (19.4) |
n = 23 61 (20.4) |
n = 22 63 (20.1) |
0.964 |
| African Americans, mean (SD) |
n = 7 72 (17.7) |
n = 6 71 (19.0) |
n = 7 71 (20.6) |
0.981 |
ER‐Tac, extended‐release tacrolimus; IR‐Tac, immediate‐release tacrolimus; LCPT, once‐daily, MeltDose tacrolimus; eGFR, glomerular filtration rate, estimated (mL/min/1.73 m); SD, standard deviation.
N = 29 for ER‐Tac group for 24‐h creatinine clearance; N = 29 for eGFR for LCPT.
p‐value from one‐way ANOVA with main effect of treatment.
Discussion
ASTCOFF is the first PK study to compare all three innovator tacrolimus formulations. The PK profile of LCPT differed significantly from the profiles of ER‐Tac and IR‐Tac and is consistent with previous studies 16, 17. When comparing the three formulations based on the study conversion factors, overall exposure was higher with LCPT, Tmax was delayed and Cmax was similar despite a higher exposure for LCPT. Given the importance of overall exposure to tacrolimus, dose conversion factors were derived on the basis of exposure normalization to IR‐Tac. This resulted in a lower and delayed peak with comparable Cmin for LCPT when compared with IR‐Tac and ER‐Tac to achieve comparable overall exposure. LCPT showed a higher tacrolimus systemic exposure, greater apparent bioavailability with lower drug dosing compared with the other two tacrolimus formulations. The characteristic high peak in tacrolimus PK following initial dosing of IR‐Tac was also observed for ER‐Tac, but not for LCPT, when normalizing the PK profiles for exposure. Furthermore, LCPT had less fluctuation between trough and peak exposures than did other formulations and a longer time to maximum concentration, as has been previously reported 18, 24. These observations are consistent with the “flatter” PK profile of LCPT as compared with the profiles of the other formulations. The clinical significance of this finding remains to be fully explored but may contribute in decreasing toxicity associated with tacrolimus peak levels, as recently suggested by a study comparing LCPT with IR‐Tac in which the severity of tremors was significantly reduced with LCPT with no correlation to Cmin levels 25.
Contrary to the differences found in most PK parameters for LCPT as compared with ER‐Tac and IR‐Tac, ER‐Tac and IR‐Tac tended to be similar to each other in PK profiles. Results from these analyses showed a significantly lower trough for ER‐Tac compared with both LCPT and IR‐Tac. This observation is consistent with previous reports showing a decrease in overall exposure and Cmin when converting from IR‐Tac to ER‐Tac on a 1:1 ratio 12.
Race effects for LCPT have been previously published and are consistent with the ad hoc analysis that showed greater per milligram exposure in both African Americans and non–African Americans 18. In the subgroup ad hoc analysis, the LCPT followed the same pattern as the overall observed PK in that exposure; relative bioavailability and fluctuation differed for LCPT versus IR‐Tac and ER‐Tac. However, the point estimates and inferential statistics presented here have to be interpreted cautiously because this study was not designed nor initially powered to address this question. These findings may be explained by the increased solubility of LCPT or the delayed and more distal distribution in the gastrointestinal tract of LCPT in contrast with the other formulations 15, 26. This distal distribution may allow for partial bypass of presystemic metabolism since proximal and distal portions of the gastrointestinal (GI) tract express different levels of cytochrome p450 3A4 and p‐gylcoprotein 27.
Given the novel formulation that represents LCPT, Cmin to AUC0–24 correlations were also performed to allow for comparison of trough sampling across formulations as is currently done in most clinical settings. A robust correlation between AUC0–24 and Cmin was found for all three formulations. However, other limited sampling strategies could be explored in further studies 28.
Adherence as measured by diaries and DBS was excellent. We observed higher DBS trough levels on average (all days) for all formulations as compared to whole blood samples (PK days only). This may be explained by variability in DBS sampling moment, analytical steps required to perform extraction, effect of hematocrit, impact of food on tacrolimus absorption or other unobserved patient‐related factors. This observation is consistent with previous reports suggesting that DBS results tend to be higher than when measured by other methods relying on whole blood sampling 29.
Renal function was assessed at every PK visit and did not appear to be influenced by formulation, despite a higher exposure to LCPT when compared with IR‐Tac and ER‐Tac. This is contrary to other studies that found an association between tacrolimus exposure and renal function 30, 31. Given the short duration of exposure to each formulation, exposure time may not have been sufficient to observe changes in renal function. Nonetheless, the higher overall exposure with LCPT did not affect renal function in this study. Numerical differences in TEAEs among formulations were not considered clinically meaningful.
This trial has many strengths, including a robust randomized crossover design, adequate sample size, advanced analytical techniques, strict PK sampling protocols, predefined feeding schedules, strict adherence monitoring and, most importantly, included a sample of renal transplant recipients as opposed to healthy volunteers. Although sample size was adequate for the primary outcome, it did not allow for full assessment of the impact of recipient genotype on formulation PK. Investigators were not blinded to treatment groups and may therefore have unintentionally introduced biases. Finally, our sample constituted a group of stable renal transplant recipients who were mainly Caucasian males and who may not be representative of other populations. Generalizability to nonfasting conditions and uncontrolled conditions also may not be possible. These questions could be addressed in pragmatic trials.
Conclusion
Results from this comparative PK study of all three innovator tacrolimus formulations, conducted in stable renal transplant recipients, demonstrate that there are significant PK differences between LCPT and both IR‐Tac and ER‐Tac and that formulations are not interchangeable with LCPT. Based on the results of this study and exposure normalization analysis, a 36% total daily does (TDD) reduction is recommended when converting from ER‐Tac to LCPT and a 30% TDD reduction when converting from IR‐Tac to LCPT. The results also suggest an 8% TDD increase when converting from IR‐Tac to ER‐Tac; however, the 8% was not statistically significant and may be within the PK variability range. Although available dosage strengths may limit exact conversion, this information will facilitate achieving target tacrolimus exposure when converting between different formulations; in particular, at hospitals with limited formularies that may not provide all tacrolimus formulations.
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Acknowledgments
This study was funded by the sponsor, Veloxis Pharmaceuticals, Inc., Edison, NJ. The authors wish to sincerely acknowledge the work of Stefanie Young and Elizabeth Cole, for their help in conducting the PK sample collections, and Wei Du, PhD, for her statistical support. The authors also want to acknowledge all the patients for their time and dedication to this study. Medical writing support was provided by Kristin Kistler, PhD, Evidera. Portions of these data were presented at the European Society of Transplantation (ESOT) meeting, held September 13 – 16, 2015, in Brussels.
Tremblay S, Nigro V, Weinberg J, Woodle ES & Alloway RR. A Steady‐State Head‐to‐Head Pharmacokinetic Comparison of All FK‐506 (Tacrolimus) Formulations (ASTCOFF): An Open‐Label, Prospective, Randomized, Two‐Arm, Three‐Period Crossover Study. Am J Transplant 2017; 17: 432–442
Trial registration numbers: EudraCT/IND Identifier: IND 75,250, ClinicalTrials.gov NCT02339246.
[The copyright line for this article was changed on 07 October, 2016 after original online publication.]
References
- 1. Hart A, Smith JM, Skeans MA, et al. OPTN/SRTR annual data report 2014: Kidney. Am J Transplant 2016; 16(Suppl 2): 11–46. [Google Scholar]
- 2. Silva HT, Yang HC, Abouljoud M, et al. One‐year results with extended‐release tacrolimus/MMF, tacrolimus/MMF and cyclosporine/MMF in de novo kidney transplant recipients. Am J Transplant 2007; 7: 595–608. [DOI] [PubMed] [Google Scholar]
- 3. Krämer BK, Charpentier B, Bäckman L, et al. Tacrolimus once daily (ADVAGRAF) versus twice daily (PROGRAF) in de novo renal transplantation: A randomized phase III study. Am J Transplant 2010; 10: 2632–2643. [DOI] [PubMed] [Google Scholar]
- 4. Alloway R, Steinberg S, Khalil K, et al. Conversion of stable kidney transplant recipients from a twice daily Prograf‐based regimen to a once daily modified release tacrolimus‐based regimen. Transplant Proc 2005; 37: 867–870. [DOI] [PubMed] [Google Scholar]
- 5. Slatinska J, Rohal T, Wohlfahrtova M, Viklicky O. Long‐term follow‐up of stable kidney transplant recipients after conversion from tacrolimus twice daily immediate release to tacrolimus once‐daily prolonged release: A large single‐center experience. Transplant Proc 2013; 45: 1491–1496. [DOI] [PubMed] [Google Scholar]
- 6. de Jonge H, Kuypers DR, Verbeke K, Vanrenterghem Y. Reduced C0 concentrations and increased dose requirements in renal allograft recipients converted to the novel once‐daily tacrolimus formulation. Transplantation 2010; 90: 523–529. [DOI] [PubMed] [Google Scholar]
- 7. Barraclough K, Isbel N, Johnson D, Campbell S, Staatz C. Once‐ versus twice‐daily tacrolimus: Are the formulations truly equivalent? Drugs 2011; 71: 1561–1577. [DOI] [PubMed] [Google Scholar]
- 8. Niioka T, Satoh S, Kagaya H, et al. Comparison of pharmacokinetics and pharmacogenetics of once‐ and twice‐daily tacrolimus in the early stage after renal transplantation. Transplantation 2012; 94: 1013–1019. [DOI] [PubMed] [Google Scholar]
- 9. Wlodarczyk Z, Ostrowski M, Mourad M, et al. Tacrolimus pharmacokinetics of once‐ versus twice‐daily formulations in de novo kidney transplantation: A substudy of a randomized phase III trial. Ther Drug Monit 2012; 34: 143–147. [DOI] [PubMed] [Google Scholar]
- 10. Wlodarczyk Z, Squifflet JP, Ostrowski M, et al. Pharmacokinetics for once‐ versus twice‐daily tacrolimus formulations in de novo kidney transplantation: A randomized open‐label trial. Am J Transplant 2009; 9: 2505–2513. [DOI] [PubMed] [Google Scholar]
- 11. Crespo M, Mir M, Marin M, et al. De novo kidney transplant recipients need higher doses of Advagraf compared with Prograf to get therapeutic levels. Transplant Proc 2009; 41: 2115–2117. [DOI] [PubMed] [Google Scholar]
- 12. Hougardy J‐M, Broeders N, Kianda M, et al. Conversion from Prograf to Advagraf among kidney transplant recipients results in sustained decrease in tacrolimus exposure. Transplantation 2011; 91: 566–569. [DOI] [PubMed] [Google Scholar]
- 13. Caillard S, Moulin B, Buron F, et al. Advagraf, a once‐daily prolonged release tacrolimus formulation, in kidney transplantation: Literature review and guidelines from a panel of experts. Transpl Int 2015. doi: 10.1111/tri.12674. [DOI] [PubMed] [Google Scholar]
- 14. Van Arnum P. Formulation development forum: Controlled agglomeration for poorly soluble drugs. Pharm Technol 2011; 35: 46. [Google Scholar]
- 15. Nigro V, Glicklich A, Weinberg J. Improved bioavailability of MELTDOSE once‐daily formulation of tacrolimus (LCP‐Tacro) with controlled agglomeration allows for consistent absorption over 24 hrs: A scintigraphic and pharmacokinetic evaluation. American Transplant Congress, 2013. Abstract number: B1034.
- 16. Budde K, Bunnapradist S, Grinyo JM, et al. Once daily LCP‐Tacro MeltDose tacrolimus versus twice daily tacrolimus in de novo kidney transplants: One‐year results of phase 3, double‐blind, randomized trial. Am J Transplant 2014; 14: 2796–2806. [DOI] [PubMed] [Google Scholar]
- 17. Bunnapradist S, Ciechanowski K, West‐Thielke P, et al. Conversion from twice‐daily tacrolimus to once‐daily extended release tacrolimus (LCPT): The phase III randomized MELT trial. Am J Transplant 2013; 13: 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gaber AO, Alloway RR, Bodziak K, Kaplan B, Bunnapradist S. Conversion from twice‐daily tacrolimus capsules to once‐daily extended‐release tacrolimus (LCPT): A phase 2 trial of stable renal transplant recipients. Transplantation 2013; 96: 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Christians U, Jacobsen W, Serkova N, et al. Automated, fast and sensitive quantification of drugs in blood by liquid chromatography–mass spectrometry with on‐line extraction: Immunosuppressants. J Chromatogr B Biomed Sci Appl 2000; 748: 41–53. [DOI] [PubMed] [Google Scholar]
- 20. Clavijo C, Hoffman K, Thomas J, et al. A sensitive assay for the quantification of morphine and its active metabolites in human plasma and dried blood spots using high‐performance liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 2011; 400: 715–728. [DOI] [PubMed] [Google Scholar]
- 21. Clavijo CF, Thomas JJ, Cromie M, et al. A low blood volume LC‐MS/MS assay for the quantification of fentanyl and its major metabolites norfentanyl and despropionyl fentanyl in children. J Sep Sci 2011; 34: 3568–3577. [DOI] [PubMed] [Google Scholar]
- 22. Shokati T, Bodenberger N, Gadpaille H, et al. Quantification of the immunosuppressant tacrolimus on dried blood spots using LC‐MS/MS. J Vis Exp 2015; 105: e52424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levey AS, Coresh J, Greene T, et al. Using standardized serum creatinine values in the Modification of Diet in Renal Disease study equation for estimating glomerular filtration rate. Ann Intern Med 2006; 145: 247–254. [DOI] [PubMed] [Google Scholar]
- 24. Alloway RR, Eckhoff DE, Washburn WK, Teperman LW. Conversion from twice‐daily tacrolimus capsules to once‐daily extended‐release tacrolimus (LCPT): Phase II trial of stable liver transplant recipients. Liver Transpl 2014; 20: 564–575. [DOI] [PubMed] [Google Scholar]
- 25. Langone A, Steinberg SM, Gedaly R, et al. Switching study of kidney transplant patients with tremor to LCP‐TacrO (STRATO): An open‐label, multicenter, prospective phase 3b study. Clin Transplant 2015; 29: 796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gavhanea YN, Yadavb AV. Loss of orally administered drugs in GI tract. Saudi Pharm J 2012; 20: 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thörn M, Finnström N, Lundgren S, Rane A, Lööf L. Cytochromes P450 and MDR1 mRNA expression along the human gastrointestinal tract. Br J Clin Pharmacol 2005; 60: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm 1981; 9: 503–512. [DOI] [PubMed] [Google Scholar]
- 29. Koster RA, Alffenaar J‐WC, Greijdanus B, Uges DRA. Fast LC‐MS/MS analysis of tacrolimus, sirolimus, everolimus and cyclosporin A in dried blood spots and the influence of the hematocrit and immunosuppressant concentration on recovery. Talanta 2013; 115: 47–54. [DOI] [PubMed] [Google Scholar]
- 30. Kolonko A, Chudek J, Wie̢cek A. Improved kidney graft function after conversion from twice daily tacrolimus to a once daily prolonged‐release formulation. Transplant Proc 2011; 43: 2950–2953. [DOI] [PubMed] [Google Scholar]
- 31. Tinti F, Meçule A, Poli L, et al. Improvement of graft function after conversion to once daily tacrolimus of stable kidney transplant patients. Transplant Proc 2010; 42: 4047–4048. [DOI] [PubMed] [Google Scholar]