

Abstract
The SATB2‐associated syndrome is a recently described syndrome characterized by developmental delay/intellectual disability with absent or limited speech development, craniofacial abnormalities, behavioral problems, dysmorphic features, and palatal and dental abnormalities. Alterations of the SATB2 gene can result from a variety of different mechanisms that include contiguous deletions, intragenic deletions and duplications, translocations with secondary gene disruption, and point mutations. The multisystemic nature of this syndrome demands a multisystemic approach and we propose evaluation and management guidelines. The SATB2‐associated syndrome registry has now been started and that will allow gathering further clinical information and refining the provided surveillance recommendations. © 2016 The Authors. American Journal of Medical Genetics Part A Published by Wiley Periodicals, Inc.
Keywords: SATB2, SATB2‐associated syndrome, Glass syndrome, 2q33.1 microdeletion syndrome, 2q32 deletion syndrome
INTRODUCTION
The SATB2‐associated syndrome (SAS) is a recently described syndrome characterized by developmental delay (DD)/intellectual disability (ID) with absent or limited speech development, craniofacial abnormalities including palatal and dental abnormalities, behavioral problems, and dysmorphic features [Docker et al., 2014; Zarate et al., 2015]. Skeletal anomalies and osteopenia were recently added to the list of distinctive features in SAS [Zarate et al., 2015]. Abnormalities of the SATB2 gene have also been described under the term Glass syndrome (OMIM 612313).
Alterations to the SATB2 Locus can result from a variety of different mechanisms that include contiguous deletions, intragenic deletions and duplications, translocations with secondary gene disruption, and point mutations. In this review, we will discuss the clinical features of SAS caused by the different mechanisms. It begins with a review of the function of the gene and how animal models have helped to elucidate its role in human disease, followed by a discussion on the current theories behind the molecular mechanisms that result from alterations in SATB2. Finally, after a brief discussion of the role of SATB2 in cancer, all the clinical information from reported cases with cytogenetic imbalances (including contiguous deletions, intragenic deletions and duplications, and translocations with gene disruption) and SATB2 point mutations is presented. Through reviewing all this information, we attempt to delineate the SAS phenotype, analyze for potential differences according to molecular mechanism, and comment on the current terminology being used when referring to alterations in the SATB2 gene. Recommendations for diagnosis and evaluation of these patients are also provided.
SATB2 STRUCTURE AND FUNCTION
SATB2 was originally identified as the causative gene on 2q32‐q33 associated with cleft palate, one of very few genomic regions where haploinsufficiency is significantly associated with isolated cleft palate [Brewer et al., 1999; FitzPatrick et al., 2003]. The SATB2 gene encodes a protein of 733 amino acids with two CUT domains and a homeodomain [FitzPatrick et al., 2003]. These functional domains are highly conserved across vertebrate taxa, where the human protein shares 100% identity with mouse, 98–100% identity with chicken, and up to 96% identity with zebrafish [FitzPatrick et al., 2003; Sheehan‐Rooney et al., 2010].
SATB2 functions as a transcription factor that binds to nuclear matrix‐attachment regions (MARs), where it activates transcription of multiple genes simultaneously [Dobreva et al., 2003; Gyorgy et al., 2008]. As such, SATB2 is a high‐level regulator of several gene regulatory networks (GRNs), and has critical roles in multiple developmental processes [Britanova et al., 2006; Dobreva et al., 2006].
SATB2 in Development
Studies in animal model systems have revealed diverse yet conserved roles for Satb2 during development. Satb2 is involved in jaw growth and patterning, upper layer neuron specification, and osteoblast differentiation [Britanova et al., 2006; Dobreva et al., 2006; Savarese et al., 2009]. Satb2 expression in the developing jaw, brain, and skeleton is conserved across a broad range of vertebrates, including zebrafish, Xenopus, chicken, and mouse [Sheehan‐Rooney et al., 2010; Fish et al., 2011]. These expression domains correspond to tissues that are affected by mutations in human patients with SAS [Zarate et al., 2015].
In mice, studies have found a critical role for Satb2 in brain development, particularly in the specification of cortical upper layer neurons [Alcamo et al., 2008; Britanova et al., 2008]. Satb2 is expressed in cortico‐cortical projection neurons that occupy the superficial layers of the cortex where they extend axons across the midline to form the corpus callosum [Alcamo et al., 2008; Britanova et al., 2008]. In the absence of Satb2, cortico‐cortical neurons are not properly specified and fail to extend axons laterally across the corpus callosum, and instead make subcortical projections [Britanova et al., 2006; Alcamo et al., 2008]. Thus, cognitive defects seen in SAS patients are likely associated with defects in the migration and axonal projections of cortical neurons.
The SAS phenotype also includes craniofacial anomalies that include micrognathia and cleft palate. Similarly, Satb2 knock‐out mice display hypoplasia of the distal jaw skeleton. At birth, mice lacking Satb2 have severely reduced jaw length and die from cleft palate [Britanova et al., 2006; Dobreva et al., 2006]. Interestingly, one quarter of mice heterozygous for Satb2 also die of cleft palate, while surviving heterozygotes exhibit a variable reduction in dentary length and facial asymmetry [Britanova et al., 2006; Fish et al., 2011]. These data mirror the variation in disease severity of patients affected by SAS, and suggest that Satb2 may be especially susceptible to perturbation during development [Fish, 2015].
Recently, the SAS phenotype was expanded to include skeletal anomalies and osteopenia [Zarate et al., 2015]. Similarly, in mice, loss of Satb2 leads to reduced levels of bone mineralization, resulting in short and brittle limb bones [Dobreva et al., 2006]. Studies have shown that Satb2 regulates osteogenesis by promoting the expression and function of osteoblast‐specific genes, including Runx2 and Atf4 [Dobreva et al., 2006; Gong et al., 2014].
MOLECULAR MECHANISMS BEHIND SATB2 ALTERATIONS
In both mice and humans, mutations in Satb2 are associated with variation in the severity of developmental defects. In mice, Satb2 acts in a dosage‐dependent manner. This is particularly evident in jaw size, where Satb2 +/− heterozygote mice exhibit significant variation in micrognathia and cleft palate. In humans, gene dosage effects (haploinsufficiency) with a resulting deficiency of functional SATB2 protein have been postulated to be the primary mechanism responsible for the clinical features seen in the SAS for those patients with SAS deriving from small interstitial deletions/duplications and those with chromosomal aberrations such as large deletions and translocations that directly disrupt SATB2 [Rosenfeld et al., 2009; Leoyklang et al., 2013; Lieden et al., 2014; Kaiser et al., 2015]. In addition, translocations with breakpoints that lie in the gene desert 3′ of SATB2 can also result in its functional haploinsufficiency and with clinical consequences that resemble those with heterozygous loss of function variants [Rainger et al., 2014].
While the exact pathomechanism of point mutations in SATB2 remains unknown, the possibility of a dominant negative effect has been suggested [Leoyklang et al., 2007]. This theory was later explored further and the truncated SATB2 was, indeed, documented to interfere with the repressive function of the wild‐type SATB2 [Rosenfeld et al., 2009; Leoyklang et al., 2013]. These data suggest that variation in phenotypic penetrance in human patients may result from both differences in genetic background as well as differences in SATB2 function related to the specific genetic mechanism disrupting the SATB2 Locus. These differences are discussed in more detail below.
CLINICAL DATA
To obtain the clinical information presented here, an online literature search was conducted in PUBMED using the keywords: “SATB2,” “SATB2‐associated syndrome,” “Glass syndrome,” “2q32‐q33 deletion syndrome,” and “2q33.1 microdeletion syndrome.”
SATB2 and Isolated Clefting
The 2q32‐q35 area had been shown to be a cleft susceptibility locus in the past [Marazita et al., 2004]. However, studies looking at point mutations of SATB2 in patients with isolated orofacial clefts have failed to reveal alterations. An initial screen of 70 unrelated isolated cleft palate patients (including 23 patients with Pierre Robin sequence) found no pathogenic SATB2 mutations [FitzPatrick et al., 2003]. Similarly, targeted testing or full sequencing of SATB2 failed to reveal mutations in a combined population of over 350 patients with nonsyndromic cleft lip with or without cleft palate [Vieira et al., 2005; Gurramkonda et al., 2015]. The presence of other phenotypic features in addition to orofacial clefts is likely to increase the detection yield when looking for SATB2 alterations.
Cytogenetic Abnormalities Encompassing SATB2
Reports of individuals with large deletions, intragenic deletions and duplications, and rearrangements involving the SATB2 locus at chromosome 2q33.1 were reviewed. Only papers in which the alteration of SATB2 was well documented with supplemental molecular techniques (e.g., microarrays, fine mapping, FISH, PCR) were included. Similarly, several other cases of deletions that encompass SATB2 have been briefly mentioned in large studies although with no full phenotypic description and therefore, were excluded from this review [Talkowski et al., 2012; Conte et al., 2016]. The included cases are shown in Figure 1, while the phenotypic features are presented in Table I.
Figure 1.
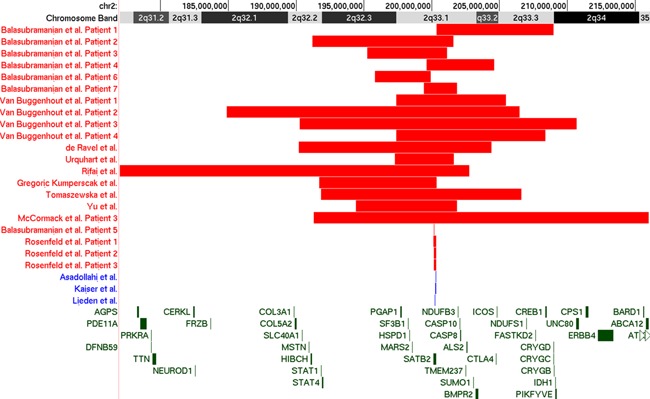
UCSC Genome Browser (GRCh37/hg19) assembly depiction of published deletions encompassing SATB2 included in this review. Deletions are represented in red while duplications are represented in blue. [Color figure can be viewed at wileyonlinelibrary.com].
Table I.
Frequencies of the Features Reported in More Than a Single Patient With Alterations of SATB2 According to Molecular Mechanism
| Large deletions | % | Intragenic duplication | % | Intragenic deletion | % | Gene disruptions | % | Point mutations | % | Total | % | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of patients (with feature/with data) | 17 | 3 | 4 | 6 | 11 | 41 | ||||||
| Males | 10 | 59 | 2 | 67 | 2 | 50 | 3 | 50 | 6 | 60 | 23 | 56 |
| Females | 7 | 41 | 1 | 33 | 2 | 50 | 2 | 33 | 4 | 40 | 16 | 39 |
| Age ranges in years (Median) | 0.5–37 (8.5) | 4–20 (10) | 3‐21 (7.75) | 0.1–33 (17.5) | 2.7–36 (4) | 0.1–37 (8) | ||||||
| DD/ID | 16/16 | 100 | 3/3 | 100 | 4/4 | 100 | 6/6 | 100 | 11/11 | 100 | 40/40 | 100 |
| Severe | 10 | 63 | 2 | 67 | 3 | 75 | 1 | 17 | 6 | 55 | 22 | 55 |
| Mild/Moderate | 3 | 19 | 0 | 0 | 1 | 25 | 2 | 33 | 4 | 36 | 10 | 25 |
| Speech delay | 16/16 | 100 | 3/3 | 100 | 4/4 | 50 | 5/6 | 83 | 10/11 | 91 | 38/40 | 95 |
| Absent | 6 | 38 | 0 | 0 | 2 | 50 | 2 | 33 | 9 | 82 | 19 | 48 |
| Limited | 8 | 50 | 3 | 100 | 2 | 50 | 2 | 33 | 1 | 9 | 16 | 40 |
| Dental | 15/16 | 94 | 3/3 | 100 | 3/4 | 75 | 2/2 | 100 | 7/9 | 78 | 30/34 | 88 |
| Abnormal shape/size of teeth | 7 | 44 | 1 | 33 | 0 | 0 | 1 | 50 | 7 | 78 | 16 | 47 |
| Crowding | 7 | 44 | 1 | 33 | 1 | 25 | 0 | 0 | 5 | 56 | 14 | 41 |
| Missing teeth | 5 | 31 | 1 | 33 | 0 | 0 | 2 | 100 | 0 | 0 | 8 | 24 |
| Delayed Primary dentition | 0 | 0 | 1 | 33 | 1 | 25 | 0 | 0 | 0 | 0 | 2 | 6 |
| Diastema | 2 | 13 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 6 |
| Behavior | 12/14 | 86 | 2/3 | 67 | 2/3 | 67 | 3/5 | 60 | 5/10 | 50 | 24/35 | 69 |
| Hyperactivity/distractibility | 6 | 43 | 1 | 33 | 0 | 0 | 0 | 0 | 1 | 10 | 8 | 23 |
| Autistic/repetitive behaviors | 2 | 14 | 1 | 33 | 1 | 33 | 1 | 20 | 2 | 20 | 7 | 20 |
| Agitation/Aggressive outbursts | 5 | 36 | 0 | 0 | 0 | 0 | 2 | 40 | 0 | 0 | 7 | 20 |
| Sleeping difficulties | 0 | 0 | 1 | 33 | 1 | 33 | 0 | 0 | 4 | 40 | 6 | 17 |
| Happy demeanor/inappropriate laughter/friendly | 2 | 14 | 2 | 67 | 1 | 33 | 0 | 0 | 1 | 10 | 6 | 17 |
| Difficult behavior | 3 | 21 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 10 | 4 | 11 |
| Sensory issues | 2 | 14 | 0 | 0 | 1 | 33 | 0 | 0 | 0 | 0 | 3 | 9 |
| Cleft palate | 8/17 | 47 | 2/3 | 67 | 2/4 | 50 | 4/6 | 67 | 8/11 | 73 | 24/41 | 59 |
| Brain MRI/CT performed | 11 | 65 | 2 | 67 | 1 | 25 | 2 | 33 | 10 | 91 | 26 | 63 |
| Normal | 6 | 55 | 1 | 50 | 1 | 100 | 0 | 0 | 5 | 50 | 13 | 50 |
| Abnormal | 5 | 45 | 1 | 50 | 0 | 0 | 2 | 100 | 5 | 50 | 13 | 50 |
| Enlarged ventricles | 2 | 12 | 0 | 0 | 0 | 0 | 2 | 33 | 1 | 9 | 5 | 19 |
| Abnormal myelination | 0 | 0 | 1 | 33 | 0 | 0 | 0 | 0 | 2 | 18 | 3 | 12 |
| Agenesis of corpus callosum | 1 | 6 | 0 | 0 | 0 | 0 | 1 | 17 | 0 | 0 | 2 | 8 |
| Prominent perivascular spaces | 2 | 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 8 |
| Skeletal | 5/12 | 42 | 2/3 | 67 | 0/2 | 0 | 4/4 | 100 | 4/7 | 57 | 15/28 | 61 |
| Pectus excavatum | 2 | 17 | 1 | 33 | 0 | 0 | 0 | 0 | 1 | 14 | 4 | 14 |
| Kyphosis/lordosis | 1 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 14 | 2 | 7 |
| Tibiae bowing | 0 | 0 | 1 | 33 | 0 | 0 | 0 | 0 | 1 | 14 | 2 | 7 |
| Osteopenia/osteoporosis | 0 | 0 | 1 | 33 | 0 | 0 | 3 | 75 | 4 | 57 | 8 | 29 |
| Neurological (with data) | 17 | 3 | 4 | 3 | 9 | 36 | ||||||
| Feeding difficulties | 13 | 76 | 2 | 67 | 2 | 50 | 2 | 67 | 1 | 11 | 20 | 56 |
| Hypotonia | 9 | 53 | 1 | 33 | 0 | 0 | 0 | 0 | 5 | 56 | 15 | 42 |
| Clinical seizures | 3 | 18 | 0 | 0 | 0 | 0 | 2 | 67 | 3 | 33 | 8 | 22 |
| Abnormal gait | 5 | 29 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 11 | 6 | 17 |
| Hypertonicity/spasticity | 1 | 6 | 1 | 33 | 1 | 25 | 0 | 0 | 0 | 0 | 3 | 8 |
| Hyperreflexia | 0 | 0 | 1 | 33 | 1 | 25 | 0 | 0 | 0 | 0 | 2 | 6 |
| Dysmorphic features (with data) | 17 | 3 | 4 | 6 | 10 | 40 | ||||||
| High/prominent forehead/frontal bossing | 9 | 53 | 1 | 33 | 1 | 25 | 1 | 17 | 2 | 20 | 14 | 35 |
| Long face | 4 | 24 | 1 | 33 | 2 | 50 | 3 | 50 | 0 | 0 | 10 | 25 |
| Low‐set ears | 5 | 29 | 0 | 0 | 1 | 25 | 1 | 17 | 2 | 20 | 9 | 23 |
| Long philtrum | 4 | 24 | 1 | 33 | 0 | 0 | 0 | 0 | 3 | 30 | 8 | 20 |
| Down‐slanted palpebral fissures | 4 | 24 | 1 | 33 | 0 | 0 | 0 | 0 | 2 | 20 | 7 | 18 |
| High/broad nasal bridge | 3 | 18 | 1 | 33 | 1 | 25 | 2 | 33 | 0 | 0 | 7 | 18 |
| Smooth philtrum | 3 | 18 | 1 | 33 | 1 | 25 | 0 | 0 | 2 | 20 | 7 | 18 |
| Thin upper lip | 6 | 35 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 10 | 7 | 18 |
| Hypertelorism/telecanthus | 2 | 12 | 1 | 33 | 0 | 0 | 1 | 17 | 2 | 20 | 6 | 15 |
| Long nose | 0 | 0 | 0 | 0 | 1 | 25 | 5 | 83 | 0 | 0 | 6 | 15 |
| Small mouth | 1 | 6 | 0 | 0 | 1 | 25 | 3 | 50 | 1 | 10 | 6 | 15 |
| Upturned nose | 3 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 10 | 4 | 10 |
| Prominent nose | 2 | 12 | 1 | 33 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 8 |
| Short palpebral fissures | 1 | 6 | 1 | 33 | 0 | 0 | 0 | 0 | 1 | 10 | 3 | 8 |
| Short philtrum | 2 | 12 | 0 | 0 | 1 | 25 | 0 | 0 | 0 | 0 | 3 | 8 |
| Broad nose | 0 | 0 | 1 | 33 | 0 | 0 | 0 | 0 | 2 | 20 | 3 | 8 |
| Deeply set eyes | 2 | 12 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 10 | 3 | 8 |
| Anteverted nares | 0 | 0 | 1 | 33 | 0 | 0 | 0 | 0 | 1 | 10 | 2 | 5 |
| Flat face | 1 | 6 | 1 | 33 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 5 |
| Large ears | 0 | 0 | 1 | 33 | 1 | 25 | 0 | 0 | 0 | 0 | 2 | 5 |
| Low nasal bridge | 0 | 0 | 1 | 33 | 0 | 0 | 0 | 0 | 1 | 10 | 2 | 5 |
| Prominent ears | 1 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 10 | 2 | 5 |
| Thin/small nose | 1 | 6 | 0 | 0 | 0 | 0 | 1 | 17 | 0 | 0 | 2 | 5 |
| Triangular face | 1 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 10 | 2 | 5 |
| Craniofacial (with data) | 17 | 3 | 4 | 5 | 11 | 40 | ||||||
| Micrognathia | 6 | 35 | 1 | 33 | 3 | 75 | 3 | 60 | 7 | 64 | 20 | 50 |
| High (arched) palate | 8 | 47 | 1 | 33 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 23 |
| Flat occiput | 2 | 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 5 |
| Abnormal sinuses | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 18 | 2 | 5 |
| Extremities (with data) | 14 | 3 | 2 | 5 | 6 | 30 | ||||||
| Broad thumbs/halluces | 2 | 14 | 2 | 67 | 1 | 50 | 0 | 0 | 1 | 17 | 6 | 20 |
| Arachnodactyly | 1 | 7 | 0 | 0 | 0 | 0 | 4 | 80 | 0 | 0 | 5 | 17 |
| Clinodactyly | 2 | 14 | 0 | 0 | 0 | 0 | 1 | 20 | 1 | 17 | 4 | 13 |
| Small hands/feet | 2 | 14 | 1 | 33 | 0 | 0 | 0 | 0 | 1 | 17 | 4 | 13 |
| Contractures/camptodactyly | 2 | 14 | 1 | 33 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 10 |
| Club feet | 2 | 14 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 7 |
| Ophthalmologic (with data) | 14 | 0 | 4 | 4 | 6 | 28 | ||||||
| Strabismus | 1 | 7 | 0 | 0 | 1 | 25 | 1 | 25 | 3 | 50 | 6 | 21 |
| Refractive errors | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 50 | 3 | 11 |
| Tegumentary (with data) | 14 | 0 | 4 | 2 | 5 | 21 | ||||||
| Joint hypermobility/ligamentous laxity | 4 | 29 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 20 | 5 | 24 |
| Genitourinary (with data) | 11 | 1 | 4 | 0 | 0 | 16 | ||||||
| Small/undescended testicles | 4 | 36 | 0 | 0 | 1 | 25 | 0 | 0 | 0 | 0 | 5 | 31 |
| Inguinal hernia | 4 | 36 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 25 |
| Hypospadias | 2 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 13 |
| Cardiovascular (with data) | 9 | 1 | 3 | 0 | 0 | 13 | ||||||
| Atrial/ventricular septal defects | 2 | 22 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 15 |
| Pre‐ or postnatal growth retardation (with feature/with data) | 12/17 | 71 | 0/3 | 0 | 0/4 | 0 | 2/4 | 50 | 0/10 | 0 | 14/38 | 37 |
| Microcephaly (with feature/with data) | 6/17 | 35 | 1/3 | 33 | 1/4 | 25 | 1/2 | 50 | 1/8 | 9 | 10/34 | 29 |
| Ectodermal changes (with data) | 15 | 0 | 4 | 0 | 1 | 20 | ||||||
| Thin skin/reduced subcutaneous fat | 8 | 53 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 40 |
| Thin/fine/sparse hair | 6 | 40 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 30 |
Shaded rows represent the most common/proposed main clinical findings in this syndrome. All percentages have been adjusted to those with data.
Large deletions
In 1989, a 16‐year‐old male with severe intellectual disability, epilepsy, microcephaly, cleft palate, short stature, and craniofacial dysmorphism was first described [Glass et al., 1989]. In this individual, a G banded karyotype of peripheral lymphocytes showed an interstitial deletion of the long arm of chromosome 2: del(2)(q32.2q33.1) [Glass et al., 1989]. Since this report, several other cases of 2q deletions have been reported but only 17 with sufficient molecular characterization to determine that the SATB2 gene was part of the deleted region [Van Buggenhout et al., 2005; de Ravel et al., 2009; Urquhart et al., 2009; Rifai et al., 2010; Balasubramanian et al., 2011; Mc Cormack et al., 2013; Tomaszewska et al., 2013; Yu et al., 2015; Gregoric Kumperscak et al., 2016]. The deletions range in size from 2.4 to 26.3 Mb.
The first report of patients with deletions at 2q33 with further molecular cytogenetic characterization was published in 2005 [Van Buggenhout et al., 2005]. In this report, four males with large deletions that included the SATB2 gene were described as having a consistent phenotype that included severe neurodevelopmental delay with significant speech impairment, craniofacial dysmorphism, dental and palatal anomalies, growth retardation, and behavioral issues. Most of these features were subsequently replicated in several case reports and a case series.
Overall, DD/ID is the most consistently reported finding in patients with large deletions (Table I). All cases described today with deletions that encompass the SATB2 gene have this feature independently of the deletion size in patients that were old enough to be assessed. When quantified, the degree of DD/ID has been reported as severe in over half the patients (55%). Verbal communication is the most affected area of development with speech delay in all patients that have been old enough to develop such expected skills and with absent speech in 38% of cases. Developmental regression and/or cognitive decline, on the other hand, had not been described until recently. Gregoric Kumperscak et al. [2016] described the case of an adult female with a history of a cleft palate and craniofacial dysmorphism with an 8.6 Mb deletion of 2q32.2q33.1 with 22 known OMIM genes including SATB2. The patient was documented to have cognitive decline between ages of 6 and 12 years of age and from mild ID (IQ 50–55) to severe ID (IQ of 25–30) and from poor to absent speech during the same timeframe. From the neurodevelopmental perspective, behavioral abnormalities are commonly described as well with a spectrum that goes from autistic behaviors and hyperactivity to aggression and difficult to control patterns of behavior. Other common features described in patients with large deletions include feeding difficulties (76%), pre‐ or postnatal growth retardation (71%), hypotonia (53%), thin skin or reduced subcutaneous fat (53%), cleft palate (47%), brain MRI abnormalities (45%), and dental crowding (44%).
Intragenic deletions and duplications
Intragenic deletions have only been reported in four patients thus far (Fig. 1). Rosenfeld et al. [2009] reported three patients (two females) with microdeletions of 2q33.1 that ranged from 173 to 185 kb in size and with only SATB2 included in the region. All three patients had severe developmental delay with little to no speech and dentofacial abnormalities, while two of the subjects also had unusual behavior [Rosenfeld et al., 2009]. In another case series, the smallest reported intragenic deletion thus far of only 35 kb was found in a 3‐year‐old male with a history of cleft palate, feeding difficulties, developmental delay with absent speech, facial dysmorphism, and minor skeletal anomalies [Balasubramanian et al., 2011].
Intragenic duplications seem to be equally rare with only three reported cases (Fig. 1). A 32 kb intragenic duplication that affects exon 4 was reported in a 4‐year‐old patient with a history of hypotonia, feeding difficulties, and a cleft palate. The patient also had DD with profoundly delayed speech, a double row of upper incisors, craniofacial dysmorphism, and behavior anomalies [Asadollahi et al., 2014]. In another case, a 10‐year‐old female with moderate to severe ID, nearly absent speech, sleeping problems, feeding difficulties, facial dysmorphic features, and dental anomalies was found to have a de novo 84‐kb duplication within chromosomal region 2q33.1 and encompassing exon 3 of the SATB2 gene [Kaiser et al., 2015]. Lastly, a 20‐year‐old male patient with moderate to severe ID, severe language impairment, cleft palate, dental anomalies, and facial dysmorphism was found to have a duplication of exons 5, 6, and 7 of SATB2 [Lieden et al., 2014].
Translocations with SATB2 disruption
Two children with de novo apparently balanced chromosomal rearrangements involving distal 2q32 were described with a combination of cleft palate, minor craniofacial and digital dysmorphisms, and delayed development, particularly of language skills [Brewer et al., 1999]. Through high‐resolution mapping of the 2q breakpoints in this pair of patients, disruption of the coding region of the SATB2 gene between exons 2 and 3 in one case (de novo 46,XY,t(2;3)(q33.1;q26.33)) and of the long‐range cis regulatory elements located in the centromeric gene desert 3′ area near SATB2 in the other (de novo 46,XX,t(2;11)(q32;p14)), were later documented [FitzPatrick et al., 2003; Rainger et al., 2014]. Four additional cases of SATB2 disruption secondary to chromosomal translocations were subsequently described [Baptista et al., 2008; Tegay et al., 2009; Talkowski et al., 2012; Rainger et al., 2014]. Developmental delay with significantly compromised speech development, facial dysmorphism, and craniofacial anomalies were described in all cases while bone mineralization abnormalities were present in three patients and seizures in two more (Table I).
Point Mutations
Population data from the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) and the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/, accessed August, 2016), does not show any loss of function type variants present in the general population. The first report of an intragenic SATB2 point mutation came from a cohort of 59 unrelated patients with craniofacial dysmorphism, with or without intellectual disability that were screened for alterations in this gene. The only alteration in SATB2 was documented in a 36‐year‐old man with a history of cleft palate, generalized osteoporosis, profound intellectual disability, epilepsy, and a jovial personality [Leoyklang et al., 2013]. The same de novo nonsense variant (c.715C>T, p.R239*) in SATB2 identified in this patient was again found through trio‐exome sequencing in an unrelated 3‐year‐old girl with cleft palate, severely delayed speech development, mild hypotonia, facial dysmorphic features, and dental anomalies [Docker et al., 2014].
Nine additional patients with point mutations (eight de novo) in SATB2 have been described (four nonsense, three missense, one splice site, and one frameshift) (Fig. 2). In all cases, developmental delay/ID was present, while severe compromise of verbal communication was documented in the majority (Fig. 2, Table I). Of interest, two patients (one with a de novo missense variant and the other with a de novo nonsense variant) with Rett‐like phenotypes were recently described [Lee et al., 2016]. In addition to the history of developmental delay, dental anomalies, and craniofacial dysmorphism, both female patients were described to have limited purposeful hand movements with stereotyped repetitive movements, poor sleep at night, and bruxism.
Figure 2.
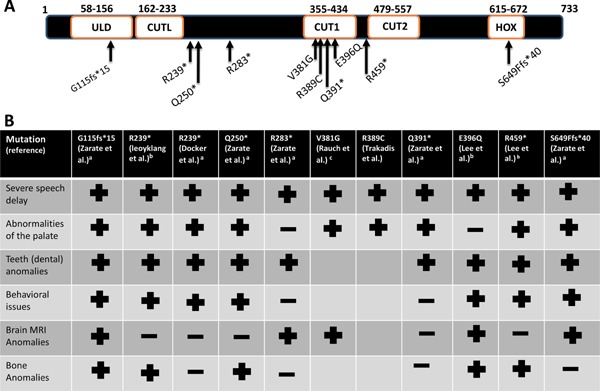
A. Schematic diagram of the SATB2 protein according to Uniprot and Pfam with location of point mutations depicted with arrows. In addition to the two CUT domains (CUT1 and CUT2) and a homeodomain (HOX), the SATB, ubiquitin‐like oligomerization domain (ULD) and the SATB, CUT1‐like DNA‐binding domain (CUTL) are depicted. B. Presence (+) or absence (−) of the main clinical features for those point mutations represented in A. Blank spaces represent no information provided for the given feature. aNM_001172517, accessed June 2016; bNM_15265, accessed June 2016; cNM_001172509, accessed June 2016. [Color figure can be viewed at wileyonlinelibrary.com].
SATB2 in Cancer
SATB2 is selectively expressed in glandular cells of the lower gastrointestinal tract with retained expression in the majority of primary and metastatic colorectal adenocarcinomas. This pattern of expression has contributed to the argument that SATB2 is a marker for colorectal adenocarcinomas and other hindgut well‐differentiated neuroendocrine tumors [Li et al., 2015; Kim et al., 2016]. SATB2 has been shown to be highly expressed in osteosarcoma cells and has been used as a sensitive marker for this type of bone malignancy [Seong et al., 2015; Davis and Horvai, 2016;]. In contrast, some reports suggest that SATB2 acts as a tumor suppressor, however no patients with documented SATB2 haploinsufficiency have been reported to have malignancies thus far [Brocato and Costa, 2015; Guo et al., 2015; Mansour et al., 2016; Wu et al., 2016]. Thus, the precise role of SATB2 in cancer remains to be fully elucidated.
CORE FEATURES OF SAS
Regardless of the underlying genetic mechanism that results in alteration of the SATB2 gene, some clinical features have been consistently described. Developmental delay/ID is almost universally reported and frequently in the severe range. Language development is the main area of development affected and often with very limited or absent speech. Behavioral abnormalities, facial dysmorphism, cleft palate, micrognathia, dental anomalies, and musculoskeletal changes have also been reported consistently in all etiological subgroups (Table I).
DIFFERENCES AMONG PATIENTS ACCORDING TO UNDERLYING SATB2 MECHANISM OF ALTERATION
Genitourinary anomalies such as hypospadias and inguinal hernias and cardiac defects such as atrial or ventricular septal defects have only been reported in patients with large deletions (Table I). Similarly, ectodermal changes (other than dental) seem to be particular prevalent in patients with large deletions but not with other mechanisms. In the report by Rifai et al. [2010] a 16‐year‐old female with a very large 2q31.2q33 deletion was described as having thin, atrophic skin and sparse, brittle, and slowly growing hair. Similar descriptions of hair sparseness and thin skin with reduced subcutaneous fat were present in a few additional cases [Van Buggenhout et al., 2005; de Ravel et al., 2009; Balasubramanian et al., 2011; Tomaszewska et al., 2013; Gregoric Kumperscak et al., 2016]. The issue of whether these ectodermal manifestations are the result of other deleted genes different than SATB2 remains to be elucidated.
CURRENT NOMENCLATURE: SAS, GLASS SYNDROME, OR 2q32‐q33/2q33.1 MICRODELETION SYNDROME?
Such as is the case for many genetic syndromes, alterations in SATB2 have been lumped together regardless of the mechanism in the Online Mendelian Inheritance in Man (OMIM) under the eponym “Glass syndrome” (OMIM 612313). This designation was made after the 1989 report by Glass et al. [1989] of a patient with a cytogenetically visible deletion in chromosome 2q32.2q33.1. As molecular cytogenetic techniques improved and better characterization of patients with deletions in this area became possible, other terms have been used based on the cytogenetic breakpoints with descriptions such as 2q33.1 microdeletion syndrome or 2q32 deletion syndrome [de Ravel et al., 2009; Rosenfeld et al., 2009; Rifai et al., 2010]. The designation of SATB2‐associated syndrome (SAS) was recently proposed by Docker et al. [2014] as a new clinically recognizable syndrome that should be considered in patients with ID and absent or severely impaired speech, cleft or highly arched palate, dental abnormalities, and skeletal anomalies.
Having different mechanisms that result in alterations of a given gene and a recognizable phenotype is of common occurrence in genetics (EHMT1 and Kleefstra syndrome or ARID1B and Coffin–Siris syndrome, to name just a couple of examples). For SATB2 alterations, however, the multiple designations for a fairly consistent phenotype depending on the underlying pathomechanism has created some degree of confusion particularly for families of affected patients (personal communication) so that there are even separate social media support groups, one for deletions and another for point mutations. In this review, we have documented a consistent phenotype without major differences among the different SATB2 genetic alterations. The phenotypic differences among individuals appear to relate to differences in severity, rather than differences in the affected system, with few exceptions as mentioned above. Therefore, in an attempt to unify the nomenclature, we suggest the use of SATB2‐associated syndrome as the preferred term.
DIAGNOSTIC EVALUATION AND HEALTH SURVEILLANCE
The use of the following acronym of major features to help recognize this syndrome is proposed: S, Severe speech anomalies; A, Abnormalities of the palate; T, Teeth anomalies; B, Behavioral issues with or without Bone or Brain MRI anomalies, and age of onset before 2 years of age (S.A.T.B.2). In agreement with the proposal by Docker et al. [2014], SAS should be considered in patients that display early neurodevelopmental delay with severely compromised or absent speech and who also have palatal and dental abnormalities. The multisystemic nature of SAS requires ongoing multisystem evaluation. While the literature on SAS continues to expand, given the spectrum of documented abnormalities thus far, we outline a proposal for initial evaluation and subsequent surveillance in Table II.
Table II.
Suggested Evaluations and Health Surveillance/Treatment for SATB2‐Associated Syndrome
| System | Initial evaluation | Surveillance/treatment |
|---|---|---|
| Genetic | SATB2 sequencing with deletion/duplication analysis/array CGH | Provide genetic counseling |
| Neurological | Consider brain MRI and EEG at baseline | Treat seizures if present, Neurosurgery referral if enlarged ventricles present |
| Physical therapy evaluation | Physical and occupational therapies if needed | |
| Occupational therapy evaluation | Orthotics or mechanical aids | |
| Consider referral to habilitation | ||
| Psychological/psychiatric | Developmental evaluation | Treat behavioral issues if needed |
| Neuropsychological evaluation | ||
| Language | Speech evaluation | Aggressive speech/language therapy (3×/week) |
| Augmentative and alternative communication devices | ||
| Craniofacial | Evaluate for cleft palate/submucous cleft palate | Cleft palate/submucous cleft palate repair |
| Gastrointestinal | Assess feeding | Special nipples/bottle for cleft palate, feeding education |
| Musculoskeletal | Consider osteopenia evaluation (bone density) | Optimize bone mineralization as needed |
| Consider referral to orthopedics | ||
| Dental | Dental evaluation | Dental/orthodontic management |
Besides whole exome sequencing, point mutations of SATB2 will also be detected through the currently offered next generation sequencing panels that include this gene for the evaluation of neurodevelopmental phenotypes and/or intellectual disability. The addition of SATB2 to panels targeting craniofacial phenotypes including cleft palate could potentially help identify patients suspected to have a syndromic cause at an earlier age prior to the development of the more distinctive neurodevelopmental phenotype. Lastly, the addition of deletion and duplication analysis to the analysis of SATB2 is recommended because of the recently reported cases with these types of alterations. From the genetics perspective, there are no reports of parental transmission for point mutations or intragenic imbalances, suggesting a highly predominant de novo nature of the SAS.
Given that the greatest impact of SAS is on the neurological system with different structural and functional abnormalities of the central nervous system, a comprehensive neurodevelopmental evaluation at diagnosis seems warranted. With the high frequency of brain neuroimaging abnormalities documented in reported cases, brain MRI should be considered as part of the evaluation process. Enlarged ventricles have been documented before in a few cases and further neurosurgical care might be needed. While clinical seizures and gait abnormalities have been documented in a relatively small proportion of cases, neurological medical management could also be required. Of interest, subclinical seizures with EEG abnormalities have been reported in some patients [Leoyklang et al., 2007; Lee et al., 2016]. Physical and occupational therapies assessment is equally indicated, with therapy provided as necessary. From the neurodevelopmental perspective, medical management of behavioral issues could be required with further psychological mental health support. Lastly, with the almost universal presence of severely affected speech, an early evaluation for speech therapy is indicated. The authors are aware of at least one patient that started Prompts for Restructuring Oral Muscular Phonetic Targets (PROMPT) and aggressive speech therapy (3×/week) at 2 years with great speech improvement over time. Equally, augmentative and alternative communication devices such as Picture Exchange Communication System (PECS) may provide additional help in the affected patients.
With the majority of patients diagnosed later in life, it is very likely that craniofacial care would have already been established to repair potential cleft palate defects. For those without an obvious palatal defect, an otolaryngology evaluation could also be necessary to diagnose a submucous‐type cleft palate. Similarly, early feeding education is likely to be required with a high frequency of feeding difficulties (apparently larger than anticipated compared to patients with non‐syndromic cleft palate). The high frequency and the broad spectrum of dental anomalies that start with primary dentition are also likely to need early assessment and management.
Finally, while the overall frequency of musculoskeletal abnormalities does not appear to be very high, we call special attention to the relatively higher incidence of documented osteopenia and/or osteoporosis. While numbers are still very limited, this type of bone health abnormality appears to be particularly common in patients with point mutations. Based on the current information available evaluation for such problems especially in late childhood should be considered.
In summary, SAS is a relatively recently described syndrome caused by alterations in the SATB2 gene. Animal models have documented the role of this gene in craniofacial, bone, and brain development, organs that have been noticed to be affected in humans with alterations in this gene. The use of the acronym S.A.T.B.2 (S, Severe speech anomalies; A, Abnormalities of the palate; T, Teeth anomalies; B, Behavioral issues with or without Bone or Brain MRI anomalies, and age of onset before 2 years of age) should help identify patients affected with SAS that do not get diagnosed through expanded next generation sequencing panels and/or whole exome sequencing. Because there are still relatively few patients that have received this diagnosis and with many questions that remain unanswered, we have started a SAS patient database. Any clinician or laboratory specialist with knowledge of a patient with a SATB2 mutation is encouraged to visit a dedicated website for this syndrome www.SATB2gene.com. Similarly, parents and caregivers will be able to visit the website for further information and resources.
Zarate YA, Fish JL. 2017. SATB2‐associated syndrome: Mechanisms, phenotype, and practical recommendations. Am J Med Genet Part A 173A: 327–337.
Conflicts of interest: None.
REFERENCES
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Farinas I, Grosschedl R, McConnell SK. 2008. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 57:364–377. [DOI] [PubMed] [Google Scholar]
- Asadollahi R, Oneda B, Joset P, Azzarello‐Burri S, Bartholdi D, Steindl K, Vincent M, Cobilanschi J, Sticht H, Baldinger R, Reissmann R, Sudholt I, Thiel CT, Ekici AB, Reis A, Bijlsma EK, Andrieux J, Dieux A, FitzPatrick D, Ritter S, Baumer A, Latal B, Plecko B, Jenni OG, Rauch A. 2014. The clinical significance of small copy number variants in neurodevelopmental disorders. J Med Genet 51:677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian M, Smith K, Basel‐Vanagaite L, Feingold MF, Brock P, Gowans GC, Vasudevan PC, Cresswell L, Taylor EJ, Harris CJ, Friedman N, Moran R, Feret H, Zackai EH, Theisen A, Rosenfeld JA, Parker MJ. 2011. Case series: 2q33.1 microdeletion syndrome‐further delineation of the phenotype. J Med Genet 48:290–298. [DOI] [PubMed] [Google Scholar]
- Baptista J, Mercer C, Prigmore E, Gribble SM, Carter NP, Maloney V, Thomas NS, Jacobs PA, Crolla JA. 2008. Breakpoint mapping and array CGH in translocations: Comparison of a phenotypically normal and an abnormal cohort. Am J Hum Genet 82:927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer CM, Leek JP, Green AJ, Holloway S, Bonthron DT, Markham AF, FitzPatrick DR. 1999. A locus for isolated cleft palate, located on human chromosome 2q32. Am J Hum Genet 65:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, Sestan N, Molnar Z, Tarabykin V. 2008. Satb2 is a postmitotic determinant for upper‐layer neuron specification in the neocortex. Neuron 57:378–392. [DOI] [PubMed] [Google Scholar]
- Britanova O, Depew MJ, Schwark M, Thomas BL, Miletich I, Sharpe P, Tarabykin V. 2006. Satb2 haploinsufficiency phenocopies 2q32‐q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am J Hum Genet 79:668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocato J, Costa M. 2015. SATB1 and 2 in colorectal cancer. Carcinogenesis 36:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte F, Oti M, Dixon J, Carels CE, Rubini M, Zhou H. 2016. Systematic analysis of copy number variants of a large cohort of orofacial cleft patients identifies candidate genes for orofacial clefts. Hum Genet 135:41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JL, Horvai AE. 2016. SATB2 expression is sensitive but may not be specific for osteosarcoma compared to other high‐grade primary bone sarcomas. Histopathology 69:84–90. [DOI] [PubMed] [Google Scholar]
- de Ravel TJ, Balikova I, Thiry P, Vermeesch JR, Frijns JP. 2009. Another patient with a de novo deletion further delineates the 2q33.1 microdeletion syndrome. Eur J Med Genet 52:120–122. [DOI] [PubMed] [Google Scholar]
- Dobreva G, Chahrour M, Dautzenberg M, Chirivella L, Kanzler B, Farinas I, Karsenty G, Grosschedl R. 2006. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 125:971–986. [DOI] [PubMed] [Google Scholar]
- Dobreva G, Dambacher J, Grosschedl R. 2003. SUMO modification of a novel MAR‐binding protein, SATB2, modulates immunoglobulin mu gene expression. Genes Dev 17:3048–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docker D, Schubach M, Menzel M, Munz M, Spaich C, Biskup S, Bartholdi D. 2014. Further delineation of the SATB2 phenotype. Eur J Hum Genet 22:1034–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish JL. 2015. Developmental mechanisms underlying variation in craniofacial disease and evolution. Dev Biol 415:188–197. [DOI] [PubMed] [Google Scholar]
- Fish JL, Villmoare B, Kobernick K, Compagnucci C, Britanova O, Tarabykin V, Depew MJ. 2011. Satb2, modularity, and the evolvability of the vertebrate jaw. Evol Dev 13:549–564. [DOI] [PubMed] [Google Scholar]
- FitzPatrick DR, Carr IM, McLaren L, Leek JP, Wightman P, Williamson K, Gautier P, McGill N, Hayward C, Firth H, Markham AF, Fantes JA, Bonthron DT. 2003. Identification of SATB2 as the cleft palate gene on 2q32‐q33. Hum Mol Genet 12:2491–2501. [DOI] [PubMed] [Google Scholar]
- Glass IA, Swindlehurst CA, Aitken DA, McCrea W, Boyd E. 1989. Interstitial deletion of the long arm of chromosome 2 with normal levels of isocitrate dehydrogenase. J Med Genet 26:127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Qian Y, Yang F, Wang H, Yu Y. 2014. Lentiviral‐mediated expression of SATB2 promotes osteogenic differentiation of bone marrow stromal cells in vitro and in vivo. Eur J Oral Sci 122:190–197. [DOI] [PubMed] [Google Scholar]
- Gregoric Kumperscak H, Krgovic D, Vokac NK. 2016. Specific behavioural phenotype and secondary cognitive decline as a result of an 8.6 Mb deletion of 2q32.2q33.1. J Int Med Res 44:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Xiong D, Yao X, Gu W, Zhang H, Yang B, Peng B, Liu M, Zheng J. 2015. Decreased SATB2 expression is associated with metastasis and poor prognosis in human clear cell renal cell carcinoma. Int J Clin Exp Pathol 8:3710–3718. [PMC free article] [PubMed] [Google Scholar]
- Gurramkonda VB, Syed AH, Murthy J, Lakkakula BV. 2015. SATB2 gene variants in non‐syndromic cleft lip with or without cleft palate in Indian population. J Oral Biol Craniofac Res 5:161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorgy AB, Szemes M, de Juan Romero C, Tarabykin V, Agoston DV. 2008. SATB2 interacts with chromatin‐remodeling molecules in differentiating cortical neurons. Eur J Neurosci 27:865–873. [DOI] [PubMed] [Google Scholar]
- Kaiser AS, Maas B, Wolff A, Sutter C, Janssen JW, Hinderhofer K, Moog U. 2015. Characterization of the first intragenic SATB2 duplication in a girl with intellectual disability, nearly absent speech and suspected hypodontia. Eur J Hum Genet 23:704–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CJ, Baruch‐Oren T, Lin F, Fan XS, Yang XJ, Wang HL. 2016. Value of SATB2 immunostaining in the distinction between small intestinal and colorectal adenocarcinomas. J Clin Pathol, [epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Lee JS, Yoo Y, Lim BC, Kim KJ, Choi M, Chae JH. 2016. SATB2‐associated syndrome presenting with Rett‐like phenotypes. Clin Genet 89:728–732. [DOI] [PubMed] [Google Scholar]
- Leoyklang P, Suphapeetiporn K, Siriwan P, Desudchit T, Chaowanapanja P, Gahl WA, Shotelersuk V. 2007. Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum Mutat 28:732–738. [DOI] [PubMed] [Google Scholar]
- Leoyklang P, Suphapeetiporn K, Srichomthong C, Tongkobpetch S, Fietze S, Dorward H, Cullinane AR, Gahl WA, Huizing M, Shotelersuk V. 2013. Disorders with similar clinical phenotypes reveal underlying genetic interaction: SATB2 acts as an activator of the UPF3B gene. Hum Genet 132:1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Yuan J, Wei L, Zhou L, Mei K, Yue J, Gao H, Zhang M, Jia L, Kang Q, Huang X, Cao D. 2015. SATB2 is a sensitive marker for lower gastrointestinal well‐differentiated neuroendocrine tumors. Int J Clin Exp Pathol 8:7072–7082. [PMC free article] [PubMed] [Google Scholar]
- Lieden A, Kvarnung M, Nilssson D, Sahlin E, Lundberg ES. 2014. Intragenic duplication—A novel causative mechanism for SATB2‐associated syndrome. Am J Med Genet A 164A:3083–3087. [DOI] [PubMed] [Google Scholar]
- Mansour MA, Hyodo T, Akter KA, Kokuryo T, Uehara K, Nagino M, Senga T. 2016. SATB1 and SATB2 play opposing roles in c‐Myc expression and progression of colorectal cancer. Oncotarget 7:4993–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazita ML, Murray JC, Lidral AC, Arcos‐Burgos M, Cooper ME, Goldstein T, Maher BS, Daack‐Hirsch S, Schultz R, Mansilla MA, Field LL, Liu YE, Prescott N, Malcolm S, Winter R, Ray A, Moreno L, Valencia C, Neiswanger K, Wyszynski DF, Bailey‐Wilson JE, Albacha‐Hejazi H, Beaty TH, McIntosh I, Hetmanski JB, Tuncbilek G, Edwards M, Harkin L, Scott R, Roddick LG. 2004. Meta‐analysis of 13 genome scans reveals multiple cleft lip/palate genes with novel loci on 9q21 and 2q32‐35. Am J Hum Genet 75:161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Cormack A, Taylor J, Gregersen N, George AM, Love DR. 2013. Delineation of 2q32q 35 deletion phenotypes: Two apparent “proximal” and “distal” syndromes. Case Rep Genet 2013:823451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainger JK, Bhatia S, Bengani H, Gautier P, Rainger J, Pearson M, Ansari M, Crow J, Mehendale F, Palinkasova B, Dixon MJ, Thompson PJ, Matarin M, Sisodiya SM, Kleinjan DA, Fitzpatrick DR. 2014. Disruption of SATB2 or its long‐range cis‐regulation by SOX9 causes a syndromic form of Pierre Robin sequence. Hum Mol Genet 10:2569–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifai L, Port‐Lis M, Tabet AC, Bailleul‐Forestier I, Benzacken B, Drunat S, Kuzbari S, Passemard S, Verloes A, Aboura A. 2010. Ectodermal dysplasia‐like syndrome with mental retardation due to contiguous gene deletion: Further clinical and molecular delineation of del(2q32) syndrome. Am J Med Genet Part A 152A:111–117. [DOI] [PubMed] [Google Scholar]
- Rosenfeld JA, Ballif BC, Lucas A, Spence EJ, Powell C, Aylsworth AS, Torchia BA, Shaffer LG. 2009. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS ONE 4:e6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese F, Davila A, Nechanitzky R, De La Rosa‐Velazquez I, Pereira CF, Engelke R, Takahashi K, Jenuwein T, Kohwi‐Shigematsu T, Fisher AG, Grosschedl R. 2009. Satb1 and Satb2 regulate embryonic stem cell differentiation and Nanog expression. Genes Dev 23:2625–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong BK, Lau J, Adderley T, Kee L, Chaukos D, Pienkowska M, Malkin D, Thorner P, Irwin MS. 2015. SATB2 enhances migration and invasion in osteosarcoma by regulating genes involved in cytoskeletal organization. Oncogene 34:3582–3592. [DOI] [PubMed] [Google Scholar]
- Sheehan‐Rooney K, Palinkasova B, Eberhart JK, Dixon MJ. 2010. A cross‐species analysis of Satb2 expression suggests deep conservation across vertebrate lineages. Dev Dyn 239:3481–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM, Pereira S, Ruderfer D, Kirby A, Ripke S, Harris DJ, Lee JH, Ha K, Kim HG, Solomon BD, Gropman AL, Lucente D, Sims K, Ohsumi TK, Borowsky ML, Loranger S, Quade B, Lage K, Miles J, Wu BL, Shen Y, Neale B, Shaffer LG, Daly MJ, Morton CC, Gusella JF. 2012. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149:525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegay DH, Chan KK, Leung L, Wang C, Burkett S, Stone G, Stanyon R, Toriello HV, Hatchwell E. 2009. Toriello‐Carey syndrome in a patient with a de novo balanced translocation [46, XY, t(2;14)(q33;q22)] interrupting SATB2. Clin Genet 75:259–264. [DOI] [PubMed] [Google Scholar]
- Tomaszewska A, Podbiol‐Palenta A, Boter M, Geisler G, Wawrzkiewicz‐Witkowska A, Galjaard RJ, Zajaczek S, Srebniak MI. 2013. Deletion of 14.7 Mb 2q32.3q33.3 with a marfanoid phenotype and hypothyroidism. Am J Med Genet A 161A:2347–2351. [DOI] [PubMed] [Google Scholar]
- Urquhart J, Black GC, Clayton‐Smith J. 2009. 4.5 Mb microdeletion in chromosome band 2q33.1 associated with learning disability and cleft palate. Eur J Med Genet 52:454–457. [DOI] [PubMed] [Google Scholar]
- Van Buggenhout G, Van Ravenswaaij‐Arts C, Mc Maas N, Thoelen R, Vogels A, Smeets D, Salden I, Matthijs G, Fryns JP, Vermeesch JR. 2005. The del(2)(q32. 2q33) deletion syndrome defined by clinical and molecular characterization of four patients. Eur J Med Genet 48:276–289. [DOI] [PubMed] [Google Scholar]
- Vieira AR, Avila JR, Daack‐Hirsch S, Dragan E, Felix TM, Rahimov F, Harrington J, Schultz RR, Watanabe Y, Johnson M, Fang J, O'Brien SE, Orioli IM, Castilla EE, Fitzpatrick DR, Jiang R, Marazita ML, Murray JC. 2005. Medical sequencing of candidate genes for nonsyndromic cleft lip and palate. PLoS Genet 1:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Chen J, Qin Y, Mo X, Huang M, Ru H, Yang Y, Liu J, Lin Y. 2016. SATB2 suppresses gastric cancer cell proliferation and migration. Tumour Biol 37:4597–4602. [DOI] [PubMed] [Google Scholar]
- Yu N, Shin S, Lee KA. 2015. First korean case of SATB2‐associated 2q32‐q33 microdeletion syndrome. Ann Lab Med 35:275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate YA, Perry H, Ben‐Omran T, Sellars EA, Stein Q, Almureikhi M, Simmons K, Klein O, Fish J, Feingold M, Douglas J, Kruer MC, Si Y, Mao R, McKnight D, Gibellini F, Retterer K, Slavotinek A. 2015. Further supporting evidence for the SATB2‐associated syndrome found through whole exome sequencing. Am J Med Genet Part A 167A:1026–1032. [DOI] [PubMed] [Google Scholar]