

Abstract
More than 40 antimicrobial peptides and proteins (AMPs) are expressed in the oral cavity. These AMPs have been organized into 6 functional groups, 1 of which, cationic AMPs, has received extensive attention in recent years for their promise as potential antibiotics. The goal of this review is to describe recent advances in our understanding of the diverse mechanisms of action of cationic AMPs and the bacterial resistance against these peptides. The recently developed peptide GL13K is used as an example to illustrate many of the discussed concepts. Cationic AMPs typically exhibit an amphipathic conformation, which allows increased interaction with negatively charged bacterial membranes. Peptides undergo changes in conformation and aggregation state in the presence of membranes; conversely, lipid conformation and packing can adapt to the presence of peptides. As a consequence, a single peptide can act through several mechanisms depending on the peptide’s structure, the peptide:lipid ratio, and the properties of the lipid membrane. Accumulating evidence shows that in addition to acting at the cell membrane, AMPs may act on the cell wall, inhibit protein folding or enzyme activity, or act intracellularly. Therefore, once a peptide has reached the cell wall, cell membrane, or its internal target, the difference in mechanism of action on gram-negative and gram-positive bacteria may be less pronounced than formerly assumed. While AMPs should not cause widespread resistance due to their preferential attack on the cell membrane, in cases where specific protein targets are involved, the possibility exists for genetic mutations and bacterial resistance. Indeed, the potential clinical use of AMPs has raised the concern that resistance to therapeutic AMPs could be associated with resistance to endogenous host-defense peptides. Current evidence suggests that this is a rare event that can be overcome by subtle structural modifications of an AMP.
Keywords: antibacterial agents, antibiotic resistance bacterial, gram negative bacteria, gram positive bacteria, cell wall, cell membrane
Introduction
Antimicrobial peptides and proteins (AMPs) are ubiquitous and have been identified in vertebrates, invertebrates, plants, and bacteria. Several online databases catalog AMPs: The APD3 database maintained at the University of Nebraska (Wang et al. 2016) contains >2,500 peptides, and a recently launched data repository of antimicrobial peptides (accessed 10/2016) contains 17,360 sequences, including 4,582 general AMPs, 12,704 patented sequences, and 74 peptides in drug development (Fan et al. 2016). An AMP database restricted to peptides with antibiofilm activity contains about 200 entries (Di Luca et al. 2015). In addition to information about each AMP, these databases provide information on the target organisms for each peptide. The goal of this review is to describe recent advances in our understanding of the diverse mechanisms of action of cationic AMPs and the bacterial resistance against these peptides. The recently developed peptide GL13K is used as an example to illustrate many of the discussed concepts.
AMPs in the Oral Cavity
We previously reviewed >40 AMPs in the oral cavity and organized these into 6 functional categories, based on their reported antimicrobial activities (Gorr 2009; Gorr and Abdolhosseini 2011). We hypothesized that the diverse functional classes—which include cationic peptides, bacterial agglutination and adhesion, metal ion chelators, peroxidase activity, cell wall activity, and proteolytic inhibitors (Gorr 2009; Gorr and Abdolhosseini 2011)—enhance the antibacterial activity of oral fluids and limit the development of bacterial resistance to endogenous AMPs, although it should be noted that the in vivo function of individual AMPs is not entirely clear and a single AMP may exhibit multiple functions. Several AMPs are found in the oral cavity in concentrations below the experimentally determined minimal inhibitory concentration (MIC), casting some doubt on their antibacterial activity in the oral cavity (Gorr and Abdolhosseini 2011). As an example, ß-defensins are found in saliva at concentrations that are 1 to 2 orders of magnitude below the MIC for several oral bacteria, including Porphyromonas gingivalis, Aggregatibacter actinomycetemcomitans, and Streptococcus mutans (Gorr and Abdolhosseini 2011). However, the absence of functional LL-37 in Morbus-Kostmann disease is associated with a dramatic increase in periodontal disease, suggesting that this AMP has a direct effect on the survival of, or susceptibility to, periodontal pathogens (reviewed in Gorr and Abdolhosseini 2011). Indeed, LL-37 is active against several oral bacteria, including P. gingivalis, S. mutans, Treponema denticola, and Fusobacterium nucleatum.
Furthermore, synergistic interactions have been demonstrated for a number of antimicrobials, including AMPs, and could restore activity even below the MIC of the individual components (reviewed in Gorr and Abdolhosseini 2011; Bechinger 2015). Despite the abundance of AMPs, the oral environment permits the growth of oral bacteria, which quickly form biofilms in the absence of oral hygiene. Conversely, invading bacteria rarely cause infection even in the case of oral wounds or tooth extractions. Thus, it has long been known that the growth of bacteria found in air and water is inhibited by saliva, while oral bacteria are relatively resistant to saliva; that is, they are adapted to the oral environment (Bibby et al. 1938).
Design of AMPs
The rich mixture of AMPs found in the oral cavity, including at least 20 cationic AMPs (Gorr and Abdolhosseini 2011), provides a basis for the design of novel AMPs with potential therapeutic function. Such AMPs include P113, which was derived from histatin 5 (Rothstein et al. 2001); hLf1-11, from lactoferrin (Godoy-Gallardo et al. 2014); and GL13K, from parotid secretory protein (BPIFA2; Abdolhosseini et al. 2012; Balhara et al. 2013; Hirt and Gorr 2013; Table 1). The goal for these peptides is to achieve strong antibacterial activity with low toxicity to mammalian cells and low ability to induce resistance in bacteria.
Table 1.
Sequences of Peptides Discussed in This Review.
| Peptides | Sequence |
|---|---|
| Antimicrobial | |
| LL-37 | LLGDF FRKSK EKIGK EFKRI VQRIK DFLRN LVPRT ES |
| Magainin 2 | GIGKF LHSAK KFGKA FVGEI MNS-NH2 |
| Indolicidin | ILPWK WPWWP WRR-NH2 |
| hDefensin 5 | ATC*YC# RTGRC^ ATRES LSGVC# EISGR LYRLC^ C*R-NH2 |
| P113 | AKRHH GYKRK FH–NH2 |
| hLf1-11 | GRRRR SVQWC A |
| BAR | LEAAP KKVQD LLKKA NITVK GAFQL FS |
| 1018 | VRLIV AVRIW RR–NH2 |
| GL13NH2 | GQIIN LKASL DLL-NH2 |
| GL13K | GKIIK LKASL KLL-NH2 |
| Gramicidin S | cyclo[LdFPV-Orn-LdFPV-Orn] |
| MP196 | RWRWR W-NH2 |
| Cell-penetrating peptides with AMP activitya | |
| MAP | KLALK LALKA LKAAL KLA |
| Tat | GRKKR RQRRR PPQ |
| Bactenecin 7 | RRIRP RPPRL PRPRP RP(LPFPRPGPRPIPRP)3 |
Superscript symbols show the location of disulfide bridges. dD-amino acid. See text for references.
Cell-penetrating peptides are small cationic peptides that are able to translocate the plasma membrane (protein transduction) and deliver molecular cargo to the cell interior. This property overlaps with antimicrobial activity in some cases, as illustrated by the examples in the table.
Although it has long been known that AMPs contain positively charged and hydrophobic amino acid residues, a correlation of peptide sequence with biological activity has been elusive. However, recent progress in this area can now aid in the de novo design of AMPs. Thus, short synthetic AMPs require a balance of positive charges (R and K) and hydrophobicity (I, V, F, Y, W), which led to a prediction model with an almost 98% success rate (Mikut et al. 2016). A further step away from naturally occurring peptides is the use of peptide mimetics that are based on the structure of AMPs. Such mimetics can be less costly to produce and may not be as sensitive to biological inactivation or degradation as natural peptides (Beckloff et al. 2007; Laursen et al. 2015).
Models for AMP Function
Cationic AMPs have received intense interest as possible models for new antibacterial therapeutics. Thus, for the purposes of this review, we focus on recent developments in the mechanisms of antibacterial activity of cationic AMPs (Bechinger 2015) and the development of bacterial resistance to these peptides. As appropriate, the review discusses naturally occurring AMPs and modified cationic peptide designs, as exemplified by our recent development of the peptide GL13K (Abdolhosseini et al. 2012).
Crossing the Outer Membrane of Gram-Negative Bacteria
The cationic charge of the peptides leads to their several-fold accumulation next to negatively charged surfaces of gram-negative (outer membrane) or gram-positive (cell wall) bacteria, which present very different outer surfaces to attacking AMPs. On one hand, the cell wall of gram-positive bacteria represents a porous 40- to 80-nm-thick mesh that many AMPs seem to pass with relative ease (Malanovic and Lohner 2016). On the other, gram-negative bacteria present an outer membrane that some AMPs can quickly cross by a charge-exchange mechanism where the cationic peptides compete with Ca2+ and Mg2+ bound to lipopolysaccharide, possibly promoted by binding to outer membrane proteins (self-promoted uptake hypothesis; e.g., Anunthawan et al. 2015). These peptides then have access to the cell wall in gram-negative bacteria as well as the cell membrane and intracellular targets. Indeed, lysozyme is active against the cell wall of gram-negative bacteria if it is administered with EDTA to remove the lipopolysaccharide-bound divalent cations (Vaara 1992).
Action on the Bacterial Cell Wall
Recent single-cell experiments using fluorescent probes have revealed that AMPs may not be generally distributed on the cell surface but rather are restricted to foci associated with cell division, cell wall remodeling, or secretion (Choi et al. 2016; Rashid et al. 2016) and thereby interfere with these processes or cause cell lysis. We recently noted that GL13K, which was covalently immobilized on titanium surfaces, causes cell wall damage that is reminiscent of autolysis in the oral gram-positive bacteria Streptococcus gordonii (Chen et al. 2014). Since short immobilized peptides are unlikely to penetrate the cell membrane or enter the cell, such peptides offer an opportunity to analyze antimicrobial mechanisms that are limited to the cell surface. In unpublished work, we have now found that the L-enantiomer of GL13K enhances autolysis of gram-positive bacteria, while an all D-amino acid enantiomer of this peptide, D-GL13K, completely inhibits autolysis but not bactericidal activity (Hirt and Gorr, in preparation). Negatively charged cell wall components, including teichoic acid and lipoteichoic acid, are targets for AMP action and bacterial resistance to AMPs, as discussed in turn.
Action on the Bacterial Cell Membrane
Once AMPs have crossed the outer barriers—represented by the outer membrane and cell wall, respectively—their interaction with the plasma membrane and internal targets may follow similar mechanisms (Koprivnjak and Peschel 2011; Malanovic and Lohner 2016).
Cationic AMPs typically exhibit a balance between positively charged and hydrophobic amino acid residues that permits them to adopt an amphipathic conformation (Fig. 1), which allows increased interaction with negatively charged surfaces and/or insertion into bacterial membranes (Fig. 2). A higher inside-negative transmembrane potential in bacteria further enhances electrostatic attraction. In contrast to bacterial membranes, the outer monolayers of eukaryotic membranes are composed of zwitterionic (overall neutral) lipids, thereby partly explaining selectivity between eukaryotic and prokaryotic cells. Notably, cancer cells lose some of the membrane asymmetry between the inner and outer monolayer, thereby making their outside more negative, which may explain why some cationic peptides are also active against cancer cells (Wakabayashi et al. 2016).
Figure 1.
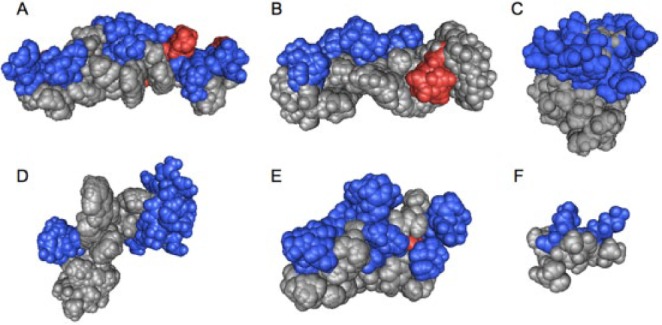
Amphipathic 3-dimensional structures of cationic antimicrobial peptides: (A) cathelicidin LL-37 (residues 2 to 30; pdb 2K6O), (B) magainin 2 (2MAG), (C) lactoferrin (residues 1 to 11; 1XV4), (D) indolicidin (1G89), (E) human defensin 5 (2LXZ), (F) helical model of GL13K (residues 5 to 13). The positively (blue) and negatively (red) charged residues are highlighted. The structures, except that of defensin 5, were obtained in the presence of detergent micelles by solution nuclear magnetic resonance spectroscopy. All low-energy conformations of the PDB files are included in the image to obtain a better view of the full conformational space. Notably, the peptides exhibit different degrees of hydrophobicity, hydrophobic moment, and amphipathicity and consequently differ in their membrane interactions. Space-filling models of cationic amphipathic peptides were created with the Cn3D software (Wang et al. 2000).
Figure 2.
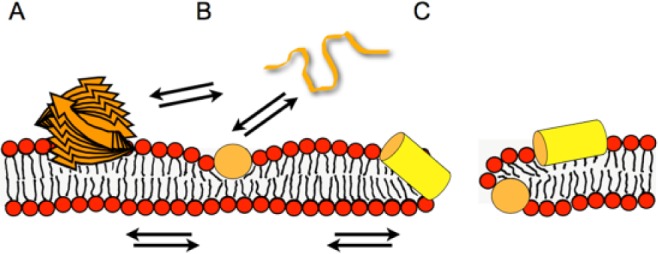
Models illustrating the membrane interactions of antimicrobial peptides. The peptide (orange and yellow) is illustrated as a stack of arrows (beta-sheet aggregate) (A), a random coil string (B), or a cylinder representing a helical structure (yellow, side view; orange, end-on view; B, C). Whereas at low peptide density, the soft membranes adjust to maintain the membrane integrity (B), at higher local peptide concentrations, the peptide-imposed curvature strain on the lipid bilayer causes transient openings (C). Multiple equilibria govern the membrane-association processes, including peptide in aqueous solution (random coil; B) ⇌ amphipathic monomers at the membrane surface ⇌ peptide-lipid supramolecular assemblies causing membrane openings. Additionally, depending on the peptide, beta-sheet membrane oligomers (A) or aggregated peptide structures form in solution. Panel B represents the adaptation of soft membranes to external stimuli (SMART model: Soft Membranes Adapt and Respond, also Transiently, in the presence of antimicrobial peptides), the alignment along the surface being a preliminary state to the carpet model, where a high peptide density causes membrane lysis. At intermediate peptide concentrations, transient openings form that have a toroidal shape made of lipids and peptide. To add another layer of complexity, cationic amphipathic AMPs have recently been shown to arrange in mesophase structures along the surface of charged lipid bilayers (Aisenbrey and Bechinger 2014).
The 3 models typically associated with cationic AMP-membrane interaction are barrel-stave, carpet, or toroidal-pore (Sani and Separovic 2016). The conformation of an AMP can be modified by the specific membrane environment. Thus, GL13K assumes a ß-turn conformation in the presence of negatively charged bacterial model membranes but not in the presence of eukaryotic model membranes (Balhara et al. 2013). The peptide removes membrane lipids by partial micellization (i.e., not a complete carpet mechanism; Balhara et al. 2013). Note that a single peptide can probably act through several mechanisms because the membrane structure, topology, aggregation, and lipid interactions of AMPs depend on the peptide structure, the peptide:lipid ratio, and the properties of the lipid membrane. These observations led to the proposal of the SMART model (Soft Membranes Adapt and Respond, also Transiently, in the presence of antimicrobial peptides; Bechinger 2015).
Many amphipathic peptides partition into the membrane interface of bacteria, where they orient along the membrane surface (Fig. 2). In this configuration, the peptides do not fill the membrane monolayer completely; therefore, this topology exerts considerable curvature strain on the lipid bilayer. Curvature strain is particularly high during the initial phases of membrane association when only the outer monolayer expands until membrane openings and transport allow for the peptide concentration between inner and outer lipid layer to equilibrate. At lower peptide concentrations, the soft lipid membranes adjust; however, membrane deformations and transient openings occur when more peptide associates with the lipid bilayer (Bechinger 2015). At even higher peptide concentrations, the membranes are disrupted, locally or globally. This view of amphipathic peptides interacting at the level of the membrane interface, rather than adopting a transmembrane alignment, has resulted in the design of some ultrashort amphipathic compounds (Arnusch et al. 2012; Ghosh and Haldar 2015) and peptide mimetics (Beckloff et al. 2007; Laursen et al. 2015).
In vitro model membrane experiments clearly show the interaction of AMPs with membranes and the disruption of lipid bilayers (e.g., Balhara et al. 2013), which correlates well with the killing/lysis of bacterial cells exposed to AMPs (Roversi et al. 2014). It is difficult to predict the exact nature of the AMP-lipid supramolecular assembly because peptides and lipids can adjust conformation, packing, and aggregation state (Bechinger 2015). However, the overall lipid geometry provides a guide to rationalize a number of experimental observations: Whereas the shape of some lipid molecules, including phosphatidylcholines, resembles cylinders, phosphatidylethanolamine exhibits a more cone-shaped structure (Bechinger 2015). Moreover, the small head group of phosphatidylethanolamine more easily accommodates the additional volume of the interfacial peptides. The more rigid membrane in the presence of cholesterol has been suggested to protect eukaryotic membranes from the action of AMPs. It is thus quite likely that a given peptide can cause different effects on phospholipid membranes, depending on lipid composition, temperature, and other environmental factors, and such interactions are best described by phase diagrams (Bechinger and Lohner 2006). The differences in membrane lipid composition of bacterial strains (Cheng et al. 2011; Malanovic and Lohner 2016) and the distinct environment that they need to propagate may thus contribute to the variable selectivity and activity of AMPs. A recent investigation showed that MP196, an arginine-rich minimalistic AMP, and gramicidin compete with the association of peripheral membrane proteins (Wenzel et al. 2014), an effect that is probably driven by electrostatic interactions and may further modulate peptide action at the membrane surface. Finally, it should be mentioned that the properties and sequences of many AMPs resemble those of cell-penetrating peptides (Pärn et al. 2015; Table 1) and that they may enter the cell before membrane rupture occurs. Once the peptides reach the cell interior, they may also interact with proteins, nucleic acids, and cellular organelles, which by itself constitutes a potential cell-killing mechanism (Scocchi et al. 2016).
Intracellular Targets
Accumulating evidence shows that, in addition to acting at the cell wall and cell membrane, AMPs may inhibit protein folding or enzyme activity or act intracellularly (e.g., on mitochondria, protein synthesis, DNA/RNA, or essential enzymes; reviewed in Scocchi et al. 2016). A recent proteome array analysis identified a range of intracellular protein targets for individual AMPs. Four tested AMPs—including bactenecin 7, a hybrid of pleurocidin and dermaseptin, a proline-arginine-rich peptide, and lactoferricin B—each showed 47 to 231 unique hits in Escherichia coli, while 30 proteins were common targets for all 4 peptides (Ho et al. 2016). The ability of individual AMPs to interact with multiple targets or multiple peptides to interact with a single target may limit the development of bacterial resistance. The use of drug-interaction clusters, as reported for the glycopeptide vancomycin and traditional antibiotics (Zhou et al. 2015), may further elucidate the synergistic and antagonistic actions of individual AMPs. Finally, the presence of an intracellular or periplasmic target may act in concert with membrane-permeabilizing activity. Thus, the recently discovered effect of GL13K on autolysis, as described earlier, presumably acts in addition to its reported interaction with lipid membranes (Balhara et al. 2013).
Bacterial Defenses and Resistance to AMPs
While it has been thought that AMPs would not cause widespread resistance due to their preferential attack on the cell membrane, the identification of specific protein targets, as reviewed here, opens the possibility for genetic mutations and bacterial resistance. Bacteria that inhabit the host microbiome and some invading bacteria clearly have the ability to coexist with or overcome the host AMPs. The corresponding bacterial defense mechanisms may also protect against therapeutic AMPs (Koprivnjak and Peschel 2011; Cole and Nizet 2016). Thus, a better understanding of these defense mechanisms is critical for further development of AMPs as therapeutic antimicrobials. The bacterial defense mechanisms discussed here are illustrated in Figure 3.
Figure 3.
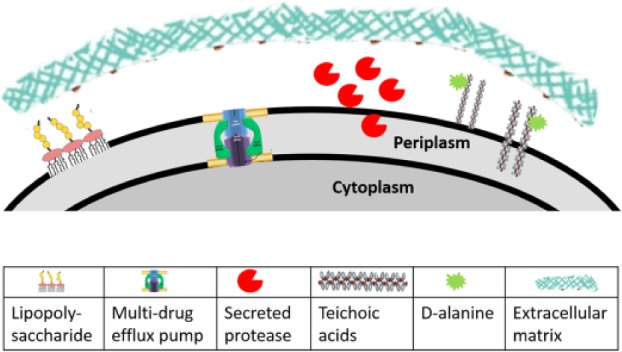
Bacterial resistance components against antimicrobial peptides as discussed in the text: secreted bacterial proteases (e.g., gingipains), lipopolysaccharides in the outer membrane of gram-negative bacteria (Rhee 2014), wall teichoic acid and lipoteichoic acid in the cell wall of gram-positive bacteria (Carvalho et al. 2014), D-alanine modification of teichoic and lipoteichoic acids, multidrug efflux pumps (Alvarez-Ortega et al. 2013), and extracellular biofilm matrix (Verma-Gaur et al. 2015). Symbols are from the references listed and used under Creative Commons licenses (https://creativecommons.org/licenses/); they have been modified for size and cropped to fit the figure.
Proteolytic Processing of AMPs
Proteolytic processing of AMPs is readily accomplished by many proteases secreted by bacteria, including the gingipains released by the oral pathogen P. gingivalis (Olsen and Potempa 2014). Substituting some or all amino acids in an AMP by D-amino acids has been used to increase resistance to proteolytic degradation of AMPs, including DJK-5, DJK-6, M33, and GL13K (Falciani et al. 2012; Hirt and Gorr 2013; de la Fuente-Núñez et al. 2015).
The D-amino acids are commonly assumed to be mirror images of the L-enantiomer. However, this assumption is not supported by recent simulations in pentapeptides that revealed that Ile and Thr, which have chiral side chains, can take on several conformations, depending on whether the side chain is in the same D- or L-form as the amino acid (Towse et al. 2014). Similarly, our studies with D-GL13K, which contains 2 Ile residues, revealed that the 2 peptides exhibit different propensity to form ß-turns and different effects on bacterial killing and resistance mechanisms (Pries et al. 2014), as described in detail next.
Surface Charge Modification
Cationic AMPs are attracted to the negatively charged outer membrane or cell wall of bacteria, but these, in turn, can reduce their surface charge and increase surface density to limit peptide adhesion. In gram-positive bacteria, D-alanylation of wall teichoic and lipoteichoic acids reduces the net negative charge and confers relative protection against AMPs (Koprivnjak and Peschel 2011; Simanski et al. 2013; Malanovic and Lohner 2016). Thus, dlt mutants, which are unable to D-alanylate teichoic acids, are relatively more susceptible to killing by cationic AMPs (Simanski et al. 2013). We found that a dlt mutant of Enterococcus faecalis is more susceptible to L-GL13K than wild-type bacteria, but, surprising, the D-GL13K enantiomer effectively kills both strains, suggesting that this peptide is able to bypass the D-alanylation resistance mechanism (Hirt and Gorr, in preparation). Since the 2 peptide enantiomers differ only in the overall chirality of their sequences, this result points to peptide chiral structure as a critical determinant of interaction with the cell wall.
Gram-negative bacteria can similarly regulate their surface charge by modification of the lipopolysaccharides that are part of the outer membrane by reduced phosphorylation, sugar substitution, or lipid addition. These modifications of lipopolysaccharide are thought to contribute to the relative resistance of the oral pathogen P. gingivalis to AMPs (Jain and Darveau 2010). Interestingly, we have found that L-GL13K does not kill P. gingivalis, whereas the bacteria are killed by D-GL13K. This difference is also seen in gingipain protease-negative strains, suggesting that it is not simply due to different proteolysis of the L- and D-peptide (Pries et al. 2014). Instead, these results suggest that D-GL13K can also bypass surface charge modification in gram-negative bacteria.
Active Efflux
AMPs that act intracellularly are susceptible to active efflux, similar to that used to resist traditional antibiotics (Koprivnjak and Peschel 2011; Alvarez-Ortega et al. 2013; Cole and Nizet 2016). Immobilized AMPs can be used to screen for surface active peptides that are not susceptible to efflux pumps (Chen et al. 2014). With this paradigm, it is possible to design AMPs that are immobilized on nano-/microparticles (Reinhardt and Neundorf 2016) that may avoid bacterial efflux.
Entrapment by Surface Proteins and Polysaccharides
A matrix of polysaccharide or DNA typically surrounds bacteria in a biofilm and limits AMP access to the cell surface. The polymer matrix may cause electrostatic repulsion of cationic peptides or trap the peptides to prevent them from reaching the embedded bacteria. As a result, only a minority of known AMPs, including GL13K (Hirt and Gorr 2013; Chen et al. 2014), are also known to be active against bacterial biofilms (Di Luca et al. 2015). Since biofilms are difficult to eradicate once they are established, recent strategies have focused on peptides that are specifically designed to prevent biofilm formation or eliminate existing biofilms (e.g., BAR and 1018; Daep et al. 2010; de la Fuente-Núñez et al. 2016). Some of these peptides specifically attack cellular mechanisms involved in biofilm formation (e.g., cell attachment) and thus may cause less bacterial resistance than bactericidal AMPs.
Cross-resistance with Host-Defense Peptides
A special concern for AMP resistance has been possible cross-resistance of bacteria to endogenous host-defense peptides (Dobson et al. 2014; Fleitas and Franco 2016). This concept could render bacteria resistant to our human host defenses after exposure to unrelated therapeutic AMPs. However, survival of resistant bacteria in vivo does not appear to vary predictably between AMP and traditional antibiotics (Dobson et al. 2014), and it appears that resistance to AMPs in general develops at low frequencies. Indeed, the long-standing use of nisin in foods and polymyxin in antibacterial wound ointments does not appear to have resulted in significant antibiotic resistance, and nisin-resistant strains may be more susceptible to other antibiotics (Martínez and Rodríguez 2005).
As described above, cationic AMPs employ multiple modes of action (Scocchi et al. 2016), and bacteria that become resistant to 1 AMP may or may not become resistant to AMPs from a different functional class (Table 2). Moreover, we recently found that even subtle structural changes to an AMP can alter bacterial resistance. Thus, L-GL13K generates tolerance in Pseudomonas aeruginosa within a few days, while the D-amino acid version of this peptide does not cause tolerance (Larson et al. 2016). Nevertheless, it appears that good stewardship of these new drugs and combination treatments with traditional antibiotics is prudent, as suggested for existing antibiotics (Fleitas and Franco 2016). As an example, we found that GL13K in combination with tobramycin can eradicate biofilms of P. aeruginosa at concentrations where each drug alone is not effective (Hirt and Gorr 2013).
Table 2.
Bacterial Cross-resistance to AMPs.
| Treatmenta |
|||||||
|---|---|---|---|---|---|---|---|
| Selection | CSA-13b | Colistinb | Melittinc | Pexigananc | PGMLc | LL-37d | hNP-1d |
| Iseganan | Lower | Similar | Lower | ||||
| Pexiganan | Similar | Similar | |||||
| PGML | Similar | Similar | |||||
| Melittin | Similar | higher | |||||
| Streptomycin | Similar | Similar | Similar | ||||
| Vancomycin | Similar | Similar | Higher | Similar | Similar | ||
| Daptomycin | Higher | Higher | |||||
| CSA-13 | Highere | ||||||
Effect of selection for resistance to one antimicrobial peptide and protein (AMP) on subsequent sensitivity to treatment with another AMP. The initial selection to AMPs listed in the left column was followed by treatment with the AMP listed in the top row. The table indicates if the minimal inhibitory concentration to the second AMP treatment is lower, similar, or higher in the selected bacteria than wild-type control cells that were not selected with the first AMP.
Table cells refer to Staphylococcus aureus unless noted otherwise.
Pseudomonas aeruginosa.
Summary
The activities of cationic AMPs have moved beyond the membrane-penetrating models, and multiple bacterial proteins and/or nucleic acids may be targeted by a single AMP, while multiple AMPs may target a single protein. With the diverse functions of cellular targets, it is not surprising that AMPs have not yet generated widespread bacterial resistance, despite the long-term use of peptides such as nisin and polymyxin. Continued refinement of our understanding of AMP mechanisms of action and bacterial resistance promises to promote the design of new peptides with even better activity and safety profiles.
Author Contributions
B. Bechinger, contributed to conception, design, data acquisition, analysis, and interpretation, critically revised the manuscript; S.-U. Gorr, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
This review was made possible by a sabbatical stay for B.B. at the University of Minnesota School of Dentistry that was generously supported by the school’s Lasby Visiting Professor Fellowship and the University of Strasbourg. Funding by the Academic Consortium 21 allowed us to organize a workshop on the topic in October 2015 in Strasbourg, France. Research on GL13K was supported by the U.S. Public Health Service (grant 1R01DE017989) from the National Institute for Dental and Craniofacial Research and a University of Minnesota CTSI TPDF award supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (award UL1TR000114) and the University of Minnesota School of Dentistry (S.U.G.). B.B. is supported by the Agence Nationale de la Recherche (projects TRANSPEP 07-PCV-0018, membraneDNP 12-BSV5-0012, MemPepSyn 14-CE34-0001-01 and the LabEx Chemistry of Complex Systems 10-LABX-0026_CSC), the RTRA International Center of Frontier Research in Chemistry, the French Foundation for Medical Research, the University of Strasbourg, the CNRS, and the Région Alsace. The content is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. National Institutes of Health or the other funding organizations. U.S patent US 8,569,449 B2 issued to S.U.G. for the GL13K peptides. S.U.G. has no financial interests in the peptides.
Both authors declare that this work is free of conflict of interest.
References
- Abdolhosseini M, Nandula SR, Song J, Hirt H, Gorr SU. 2012. Lysine substitutions convert a bacterial-agglutinating peptide into a bactericidal peptide that retains anti-lipopolysaccharide activity and low hemolytic activity. Peptides. 35(2):231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aisenbrey C, Bechinger B. 2014. Molecular packing of amphipathic peptides on the surface of lipid membranes. Langmuir. 30(34):10374–10383. [DOI] [PubMed] [Google Scholar]
- Alvarez-Ortega C, Olivares J, Martinez JL. 2013. RND multidrug efflux pumps: what are they good for? Front Microbiol. 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anunthawan T, de la Fuente-Núñez C, Hancock RE, Klaynongsruang S. 2015. Cationic amphipathic peptides KT2 and RT2 are taken up into bacterial cells and kill planktonic and biofilm bacteria. Biochim Biophys Acta. 1848(6):1352–1358. [DOI] [PubMed] [Google Scholar]
- Arnusch CJ, Albada HB, van Vaardegem M, Liskamp RM, Sahl HG, Shadkchan Y, Osherov N, Shai Y. 2012. Trivalent ultrashort lipopeptides are potent pH dependent antifungal agents. J Med Chem. 55(3):1296–1302. [DOI] [PubMed] [Google Scholar]
- Balhara V, Schmidt R, Gorr SU, Dewolf C. 2013. Membrane selectivity and biophysical studies of the antimicrobial peptide GL13K. Biochim Biophys Acta. 1828(9):2193–2203. [DOI] [PubMed] [Google Scholar]
- Bechinger B. 2015. The SMART model: soft membranes adapt and respond, also transiently, in the presence of antimicrobial peptides. J Pept Sci. 21(5):346–355. [DOI] [PubMed] [Google Scholar]
- Bechinger B, Lohner K. 2006. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim Biophys Acta. 1758(9):1529–1539. [DOI] [PubMed] [Google Scholar]
- Beckloff N, Laube D, Castro T, Furgang D, Park S, Perlin D, Clements D, Tang H, Scott RW, Tew GN, et al. 2007. Activity of an antimicrobial peptide mimetic against planktonic and biofilm cultures of oral pathogens. Antimicrob Agents Chemother. 51(11):4125–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibby BG, Hine MK, Clough OW. 1938. The antibacterial action of human saliva. J Am Dent Assoc. 25(8):1290–1302. [Google Scholar]
- Carvalho F, Sousa S, Cabanes D. 2014. How Listeria monocytogenes organizes its surface for virulence. Front Cell Infect Microbiol. 4:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Hirt H, Li Y, Gorr SU, Aparicio C. 2014. Antimicrobial GL13K peptide coatings killed and ruptured the wall of Streptococcus gordonii and prevented formation and growth of biofilms. PLoS One. 9(11):e111579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JT, Hale JD, Elliott M, Hancock RE, Straus SK. 2011. The importance of bacterial membrane composition in the structure and function of aurein 2.2 and selected variants. Biochim Biophys Acta. 1808(3):622–633. [DOI] [PubMed] [Google Scholar]
- Choi H, Rangarajan N, Weisshaar JC. 2016. Lights, camera, action! Antimicrobial peptide mechanisms imaged in space and time. Trends Microbiol. 24(2):111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JN, Nizet V. 2016. Bacterial evasion of host antimicrobial peptide defenses. Microbiol Spectr. 4(1):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daep CA, Novak EA, Lamont RJ, Demuth DR. 2010. Selective substitution of amino acids limits proteolytic cleavage and improves the bioactivity of an anti-biofilm peptide that targets the periodontal pathogen, Porphyromonas gingivalis. Peptides. 31(12):2173–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Núñez C, Cardoso MH, de Souza Cândido E, Franco OL, Hancock RE. 2016. Synthetic antibiofilm peptides. Biochim Biophys Acta. 1858(5):1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Núñez C, Reffuveille F, Mansour SC, Reckseidler-Zenteno SL, Hernández D, Brackman G, Coenye T, Hancock RE. 2015. D-enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem Biol. 22(2):196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Luca M, Maccari G, Maisetta G, Batoni G. 2015. BaAMPs: the database of biofilm-active antimicrobial peptides. Biofouling. 31(2):193–199. [DOI] [PubMed] [Google Scholar]
- Dobson AJ, Purves J, Rolff J. 2014. Increased survival of experimentally evolved antimicrobial peptide-resistant Staphylococcus aureus in an animal host. Evol Appl. 7(8):905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falciani C, Lozzi L, Pollini S, Luca V, Carnicelli V, Brunetti J, Lelli B, Bindi S, Scali S, Di Giulio A, et al. 2012. Isomerization of an antimicrobial peptide broadens antimicrobial spectrum to gram-positive bacterial pathogens. PLoS One. 7(10):e46259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Sun J, Zhou M, Zhou J, Lao X, Zheng H, Xu H. 2016. DRAMP: a comprehensive data repository of antimicrobial peptides. Sci Rep. 6:24482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleitas O, Franco OL. 2016. Induced bacterial cross-resistance toward host antimicrobial peptides: a worrying phenomenon. Front Microbiol. 7:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh C, Haldar J. 2015. Membrane-active small molecules: designs inspired by antimicrobial peptides. ChemMedChem. 10(10):1606–1624. [DOI] [PubMed] [Google Scholar]
- Godoy-Gallardo M, Mas-Moruno C, Fernández-Calderón MC, Pérez-Giraldo C, Manero JM, Albericio F, Gil FJ, Rodríguez D. 2014. Covalent immobilization of hLf1-11 peptide on a titanium surface reduces bacterial adhesion and biofilm formation. Acta Biomater. 10(8):3522–3534. [DOI] [PubMed] [Google Scholar]
- Gorr SU. 2009. Antimicrobial peptides of the oral cavity. Periodontol 2000. 51:152–180. [DOI] [PubMed] [Google Scholar]
- Gorr SU, Abdolhosseini M. 2011. Antimicrobial peptides and periodontal disease. J Clin Periodontol. 38 Suppl 11:126–141. [DOI] [PubMed] [Google Scholar]
- Hirt H, Gorr SU. 2013. Antimicrobial peptide GL13K is effective in reducing biofilms of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 57(10):4903–4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YH, Shah P, Chen YW, Chen CS. 2016. Systematic analysis of intracellular-targeting antimicrobial peptides, bactenecin 7, hybrid of pleurocidin and dermaseptin, proline–arginine-rich peptide, and lactoferricin B, by using Escherichia coli proteome microarrays. Mol Cell Proteomics. 15(6):1837–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Darveau RP. 2010. Contribution of Porphyromonas gingivalis lipopolysachharide to periodontitis. Periodontol 2000. 54(1):53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprivnjak T, Peschel A. 2011. Bacterial resistance mechanisms against host defense peptides. Cell Mol Life Sci. 68(13):2243–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson E, Hirt H, Gorr S. 2016. The antimicrobial peptide GL13K kills drug-resistant bacteria without causing resistance [abstract]. J Dent Res. 95:0428. [Google Scholar]
- Laursen JS, Engel-Andreasen J, Olsen CA. 2015. β-Peptoid foldamers at last. AccChem Res. 48(10):2696–2704. [DOI] [PubMed] [Google Scholar]
- Malanovic N, Lohner K. 2016. Gram-positive bacterial cell envelopes: the impact on the activity of antimicrobial peptides. Biochim Biophys Acta. 1858(5):936–946. [DOI] [PubMed] [Google Scholar]
- Martínez B, Rodríguez A. 2005. Antimicrobial susceptibility of nisin resistant Listeria monocytogenes of dairy origin. FEMS Microbiol Lett. 252(1):67–72. [DOI] [PubMed] [Google Scholar]
- Mikut R, Ruden S, Reischl M, Breitling F, Volkmer R, Hilpert K. 2016. Improving short antimicrobial peptides despite elusive rules for activity. Biochim Biophys Acta. 1858(5):1024–1033. [DOI] [PubMed] [Google Scholar]
- Mishra NN, Bayer AS, Weidenmaier C, Grau T, Wanner S, Stefani S, Cafiso V, Bertuccio T, Yeaman MR, Nast CC, et al, et al. 2014. Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: relative roles of mprF and dlt operons. PLoS One. 9(9):e107426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen I, Potempa J. 2014. Strategies for the inhibition of gingipains for the potential treatment of periodontitis and associated systemic diseases. J Oral Microbiol. 6:24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pärn K, Eriste E, Langel Ü. 2015. The antimicrobial and antiviral applications of cell-penetrating peptides. In: Langel Ü, editor. Cell-penetrating peptides: methods and protocols. New York (NY): Springer New York; p. 223–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JE, Snarr J, Chaudhary V, Jennings JD, Shaw H, Christiansen B, Wright J, Jia W, Bishop RE, Savage PB. 2012. In vitro evaluation of the potential for resistance development to ceragenin CSA-13. J Antimicrob Chemother. 67(11):2665–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pries H, Gorr S, Hirt H. 2014. Gingipain-independent killing of Porphyromonas gingivalis by D-amino-acid antimicrobial peptide D-GL13K [abstract]. J Dent Res. 93:37. [Google Scholar]
- Rashid R, Veleba M, Kline KA. 2016. Focal targeting of the bacterial envelope by antimicrobial peptides. Front Cell Dev Biol. 4:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt A, Neundorf I. 2016. Design and application of antimicrobial peptide conjugates. Int J Mol Sci. 17(5)E701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SH. 2014. Lipopolysaccharide: basic biochemistry, intracellular signaling, and physiological impacts in the gut. Intest Res. 12(2):90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein DM, Spacciapoli P, Tran LT, Xu T, Roberts FD, Dalla Serra M, Buxton DK, Oppenheim FG, Friden P. 2001. Anticandida activity is retained in P-113, a 12-amino-acid fragment of histatin 5. Antimicrob Agents Chemother. 45(5):1367–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roversi D, Luca V, Aureli S, Park Y, Mangoni ML, Stella L. 2014. How many antimicrobial peptide molecules kill a bacterium? The case of pmap-23. ACS Chem Biol. 9(9):2003–2007. [DOI] [PubMed] [Google Scholar]
- Sani MA, Separovic F. 2016. How membrane-active peptides get into lipid membranes. Acc Chem Res. 49(6):1130–1138. [DOI] [PubMed] [Google Scholar]
- Scocchi M, Mardirossian M, Runti G, Benincasa M. 2016. Non-membrane permeabilizing modes of action of antimicrobial peptides on bacteria. Curr Top Med Chem. 16(1):76–88. [DOI] [PubMed] [Google Scholar]
- Simanski M, Glaser R, Koten B, Meyer-Hoffert U, Wanner S, Weidenmaier C, Peschel A, Harder J. 2013. Staphylococcus aureus subverts cutaneous defense by D-alanylation of teichoic acids. Exp Dermatol. 22(4):294–296. [DOI] [PubMed] [Google Scholar]
- Towse CL, Hopping G, Vulovic I, Daggett V. 2014. Nature versus design: the conformational propensities of D-amino acids and the importance of side chain chirality. Protein Eng Des Sel. 27(11):447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaara M. 1992. Agents that increase the permeability of the outer membrane. Microbiol Rev. 56(3):395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma-Gaur J, Qu Y, Harrison PF, Lo TL, Quenault T, Dagley MJ, Bellousoff M, Powell DR, Beilharz TH, Traven A. 2015. Integration of posttranscriptional gene networks into metabolic adaptation and biofilm maturation in candida albicans. PLoS Genet. 11(10):e1005590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Yano Y, Kawano K, Matsuzaki K. 2016. A pH dependent charge-reversal peptide for cancer targeting. Eur Biophys J [epub ahead of print 8 June 2016] in press. DOI: 10.1007/s00249-016-1145-y [DOI] [PubMed] [Google Scholar]
- Wang G, Li X, Wang Z. 2016. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44:D1087-D1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Geer LY, Chappey C, Kans JA, Bryant SH. 2000. Cn3d: sequence and structure views for Entrez. Trends Biochem Sci. 25(6):300–302. [DOI] [PubMed] [Google Scholar]
- Wenzel M, Chiriac AI, Otto A, Zweytick D, May C, Schumacher C, Gust R, Albada HB, Penkova M, Krämer U, et al. 2014. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc Natl Acad Sci U S A. 111(14):E1409–E1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, Kang TM, Yuan J, Beppler C, Nguyen C, Mao Z, Nguyen MQ, Yeh P, Miller JH. 2015. Synergistic interactions of vancomycin with different antibiotics against escherichia coli: trimethoprim and nitrofurantoin display strong synergies with vancomycin against wild-type E. coli. Antimicrob Agents Chemother. 59(1):276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]