

SUMMARY
The Akt pathway is a central regulator that promotes cell survival in response to extracellular signals. Depletion of SIRT7, a NAD+-dependent deacetylase which is the least studied sirtuin, is known to significantly increase Akt activity in mice through unknown mechanisms. In this study, we demonstrate that SIRT7 depletion in breast cancer cells results in Akt hyper-phosphorylation and increases cell survival following genotoxic stress. Mechanistically, SIRT7 specifically interacts with and deacetylates FKBP51 at residue Lysines 28 and 155 (K28 and K155), resulting in enhanced interactions among FKBP51/Akt/PHLPP, and Akt dephosphorylation. Mutating both lysines to arginines abolishes the effect of SIRT7 on Akt activity through FKBP51 deacetylation. Finally, energy stress strengthens SIRT7 mediated effects on Akt dephosphorylation through FKBP51, thus, sensitizes cancer cells to cytotoxic agents. These results reveal a direct role of SIRT7 in Akt regulation and raise the possibility of using the glucose analog 2-Deoxy-D-glucose (2DG) as a chemo-sensitizing agent.
Keywords: SIRT7, Akt signaling pathway, Breast Cancer, Chemo-sensitivity
Graphical abstract
INTRODUCTION
Sirtuins belong to a conserved family of nicotinamide adenine dinucleotide (NAD+) dependent class III histone deacetylases, which share sequence homology with yeast silent information regulator 2 (Sir2). Sir2 silences specific chromatin regions within the yeast genome (Smith and Boeke, 1997). The requirement of NAD+ for sirtuin activity couples sirtuin-dependent posttranslational modifications with cellular energy levels, thereby, affecting a broad ranges of processes including cell metabolism, cell proliferation, gene transcription, cell division, cellular stress response and tumor development (Finkel et al., 2009). Sirtuins are well-sought-after drug targets as their enzymatic activities are amenable to manipulation (Baur et al., 2012).
Mammals have seven sirtuins, SIRT1–7, most of which deacetylate both histone and non-histone proteins. SIRT7 is a lysine deacetylase that selectively removes an acetyl group from histone H3 lysine 18 (H3K18) (Barber et al., 2012). Hypoacetylation of H3K18 has been associated with high grade tumors and poor patient prognosis (Mosashvilli et al., 2010; Seligson et al., 2009). Evidence indicates that SIRT7 may act as an oncogene as its expression was elevated in several human cancers (Ashraf et al., 2006; De Nigris et al., 2002; Kim et al., 2013), although other studies also indicate that SIRT7 mRNA and protein level are down-regulated in cancers such as head and neck squamous cell carcinomas (Lai et al., 2013). Furthermore, SIRT7 is also shown to be an activator that stimulates ribosomal biogenesis in dividing cells (Tsai et al., 2014) by activating rDNA transcription through multiple mechanisms (Chen et al., 2016; Ford et al., 2006; Tsai et al., 2012), thereby supporting the high biosynthetic and metabolic demands of cancer cells. Recent studies also show that SIRT7 can modulate DNA repair and maintain genome integrity (Li et al., 2016; Vazquez et al., 2016). In addition to its role in cancer, SIRT7 also plays an important role in the regulation of cell death and damage by inhibiting p53, Ras, and Akt signaling pathways in the heart (Vakhrusheva et al., 2008a; Vakhrusheva et al., 2008b). However, its function in the regulation of cancer cells in response to genotoxic stress is less well studied.
Akt is a serine/threonine kinase that is one of the most commonly upregulated oncoproteins in a variety of cancers. In addition, Akt hyper-activation is associated with tumorigenesis, and chemo-resistance. Akt phosphorylates a number of downstream proteins, all of which play important roles in regulation of cell survival, cell cycle progression, proliferation and metabolism. These processes are regulated, in part, by increased glucose uptake (Rathmell et al., 2003), glycolytic rate and lactate production (Elstrom et al., 2004) or by reciprocal control of growth signaling pathways such as the mammalian target of Rapamycin (mTOR) pathway. Full activation of Akt requires phosphorylation of both Thr308 and Ser473. Thr308 is phosphorylated by the 3-phosphoinosite-dependent protein kinase PDK1 (Alessi et al., 1997; Stephens et al., 1998), while Ser473 is phosphorylated by mTOR complex 2 (mTORC2) (Sarbassov et al., 2005). On the other hand, Akt Thr308 and Ser473 can be dephosphorylated by protein phosphatase 2A (PP2A) (Andjelković et al., 1996) and PH domain leucine-rich repeat protein phosphatase (PHLPP) (Brognard et al., 2007; Gao et al., 2005), respectively. Our laboratory is one of several groups firstly report that the interaction between Akt and the phosphatase PHLPP is facilitated by a scaffolding protein called FKBP51 (FK506 binding protein, also known as FKBP5), allowing for dephosphorylation of Akt at Ser473 and decreased Akt activity (Mulholland et al., 2011; Pei et al., 2009). Therefore, overexpression of FKBP51 results in sensitizing cells to different chemotherapeutics (Hou and Wang, 2012; Mulholland et al., 2011; Pei et al., 2009).
In the current study, we demonstrate that the NAD+-dependent protein deacetylase, SIRT7, affects cancer cell survival and responses to chemotherapy through the regulation of Akt pathway. Mechanistically, we find that SIRT7 removes acetyl groups from FKBP51 and enhances the formation of FKBP51-Akt-PHLPP complex. This subsequently results in suppression of the Akt pathway and increased chemo-sensitivity in breast cancer cells. Glucose deprivation or 2-Deoxy-D-glucose (2DG) treatment enhances interactions between SIRT7 and FKBP51 and the formation of FKBP51/Akt/PHLPP complex, resulting in Akt inactivation. Thus, our studies have identified an acetylation-dependent regulatory mechanism for the FKBP51-Akt-PHLPP axis, which can have a significant impact on cell proliferation and chemotherapy response.
RESULTS
Sirtuin Regulates FKBP51 Binding to Akt/PHLPP in Response to Energy Stress
We previously reported that FKBP51 was a scaffolding protein that bond to PHLPP and Akt, regulating Akt Ser473 phosphorylation (Pei et al., 2009). However, the mechanism involved in the regulation of FKBP51-mediated Akt activation remains unknown. To test which stimulus affects FKBP51 scaffolding function, we treated HEK293T cells stably expressing Flag-FKBP51 with different conditions: 1) AICAR, 2) glucose deprivation, 3) amino acid depletion, or growth factors 4) EGF, 5) PDGF, or 6) insulin (1–3, Fig. 1A; 4–6, data not shown). We found that only glucose deprivation significantly increased the interactions between FKBP51 and Akt/PHLPP (Fig. 1A). We then used 2-Deoxy-D-glucose (2DG), a glucose analog which targets glucose metabolism and mimics glucose deprivation, to confirm this finding. As shown in Fig. 1B, 2DG treatment also increased the binding of FKBP51 with Akt, similar to glucose deprivation.
Figure 1. Sirtuins are Involved in Regulating FKBP51 Binding with Akt/PHLPP in Response to Energy Stress.

(A) HEK293T cells stably expressing empty vector (EV) or Flag-FKBP51 were treated as indicated (NT, non-treated; AICAR, 5-Aminoimidazole-4-carboxamide ribonucleotide; -Glu, glucose-free media; -AA, amino acid deprivation) for 16 hours. The interactions between FKBP51 and Akt/PHLPP were determined by immunoprecipitation (IP) with anti-Flag beads and immunoblotted with the indicated antibodies.
(B and C) Cells stably expressing Flag-FKBP51were treated with either glucose-free medium or DMEM with 5mg/ml 2DG (B), or glucose-free in the presence or absence of Sirtinol (10μg/ml) (C) for 16 hours. The interactions between FKBP51 and Akt/PHLPP were determined by immunoprecipitation with anti-Flag and immunoblotted with the indicated antibodies.
Nicotinamide adenine dinucleotide (NAD) and adenosine triphosphate (ATP) are two important energy-transfer molecules. Under energy stress conditions, the AMP/ATP ratio increases and AMP activates AMPK. However, AICAR (AMPK activator) treatment did not alter the interaction of FKBP51 with Akt/PHLPP (Fig. 1A), which indicated that glucose deprivation induced regulation of FKBP51 scaffolding function was independent of AMPK activation. Energy stress also increases the NAD+/NADH ratio in cells, activating NAD+ dependent sirtuins (Canto et al., 2009). Therefore, we hypothesized that sirtuins might be involved in regulation of the FKBP51-Akt-PHLPP complex and in turn, regulates Akt activity. To test this hypothesis, cells that stably overexpressing Flag-tagged FKBP51 were treated with a pan-sirtuin inhibitor, sirtinol, in the presence or absence of glucose or 2DG, and the interaction of FKBP51 and Akt was assessed. Treatment with sirtinol suppressed the binding of FKBP51 to Akt induced by glucose deprivation or 2DG treatment (Fig. 1C). These results raised the possibility that sirtuin may be involved in the regulation of FKBP51-Akt-PHLPP complex.
SIRT7 Specifically Interacts with FKBP51 and Regulates Akt Activation
In order to determine which sirtuin(s) might be involved in the regulation of FKBP51 and Akt/PHLPP binding, we transiently expressed 7 different constructs containing Flag-tagged sirtuins (SIRT1 to SIRT7) in HEK293T cells and performed immunoprecipitaion using beads conjugated to anti-Flag antibody. SIRT7, but not other sirtuins, showed an interaction with FKBP51 (Fig. 2A). The SIRT7-FKBP51 interaction was further confirmed by endogenous co-immunoprecipitation assays (Fig. 2B). Reciprocal immunoprecipitation with FKBP51 antibody also confirmed the interaction between FKBP51 and SIRT7 (Fig. 2B).
Figure 2. SIRT7 Specifically Interacts with FKBP51 and Regulates Phosphorylation of the Akt at Ser473.

(A) HEK293T cells were transfected with empty vector (EV) or full-length Flag-SIRT1-7 plasmids. Whole cell lysates were subjected to IP with anti-Flag beads. Immunoblotting was performed with anti-FKBP51 and anti-Flag antibodies.
(B) Endogenous interaction of FKBP51 and SIRT7 was detected in MDA-MB-231 cells by IP using SIRT7 antibody (top panel) or FKBP51 antibody (bottom panel) followed by immunoblotting with the anti-SIRT7 and anti-FKBP51 antibodies.
(C) MDA-MB-231 cells were transfected with indicated siRNAs for 72 hours and the phosphorylation levels of Akt, FoxO1 and GSK-3β in cell lysates were detected using the indicated antibodies by Western Blot. See also Figure S1.
(D) HEK293T cells were transfected with the indicated constructs and Akt phosphorylation was determined by Western Blot.
(E) The Akt phosphorylation in cell lysates isolated from sirt7+/+ or sirt7−/− cells was examined.
(F) sirt7−/− cells transfected with empty vector (EV), wide type SIRT7 and catalytic-dead mutant constructs for 48 hours, proteins were examined using the indicated antibodies.
The interaction between FKBP51 and SIRT7 prompted us to examine the potential role of SIRT7 in the regulation of Akt activation. In different breast cancer or prostate cancer cell lines, depletion of SIRT7 using both siRNA and shRNA significantly increased the phosphorylation of Akt at Serine 473, but not Threonine 308 (Fig. 2C and Fig. S1B & S1C). Consistent with the increased Akt activity, Akt downstream substrates, GSK-3β and FoxO1 also showed increased phosphorylation (pSer9 GSK-3β and pSer256 FoxO1) (Fig. 2C). We further confirmed this regulation using MEF cells derived from SIRT7 wild type (sirt7+/+) and knockout (sirt7−/−) embryos (Shin et al., 2013). Akt Serine 473 phosphorylation was stronger in sirt7−/− cells than that in sirt7+/+ cells (Fig. 2E). Conversely, overexpression of SIRT7 showed a reversed phenotype, i.e., decreased phosphorylation of Akt Serine 473 (Fig. 2D). In addition, we found that SIRT7 deacetylase activity was important for the regulation of Akt activation. Overexpressing WT SIRT7, but not the catalytic-dead SIRT7 H187Y mutant (HY mutant) in the sirt7−/− MEF cells reversed the increased Akt activity caused by SIRT7 deficiency (Fig. 2F). These results suggest that the negative regulation of Akt activity by SIRT7 is dependent on its deacetylase activity.
Downstream effects of Akt phosphorylation include phosphorylation of forkhead family transcription factors such as FKHR (FoxO1) (Guo et al., 1999) and its subsequent cytoplasmic localization (Brunet et al., 1999; Zhang et al., 2002). Additionally, Akt activation also increases glucose uptake (Hajduch et al., 2001). To further confirm the effect of SIRT7 on Akt activity, FoxO1 immunofluorescence and glucose uptake assays were performed in breast cancer cells as well as sirt7+/+ and sirt7−/− MEF cells (Fig. S1D & S1E). Consistent with increased Akt activity, FoxO1 translocated from the nucleus to cytoplasm in both SIRT7 depleted breast cancer cells and sirt7 knockout MEFs (Fig. S1D). Glucose uptake also increased in SIRT7 depleted breast cancer cell and sirt7 knockout MEFs (Fig. S1E), which was likely due to Akt activation (Fig. 2C & 2E).
SIRT7 Regulates Cellular Response to Different Chemotherapeutics
FKBP51 mediated suppression of the Akt pathway plays an important role in chemo-sensitivity (Mulholland et al., 2011; Pei et al., 2009). Because SIRT7 negatively regulates Akt activity, we next set out to determine whether SIRT7 affects cellular responses to various chemotherapeutic drugs. We treated a pancreatic cancer cell line, Su86, and breast cancer cell lines, MCF7 and MDA-MB-231 with a series of commonly used chemotherapeutic agents, including microtubule stabilizers, nucleoside analogs, anthracyclines and topoisomerase inhibitors. Compared with control cells, down-regulation of SIRT7 resulted in dramatically increased resistance to these treatments in both breast cancer (Fig. 3A & 3B) and pancreatic cancer cells (Fig. S2A). Similarly, loss of SIRT7 expression resulted in increased resistance of MEF cells to these treatments (Fig. 3C and Fig. S2B). In contrast, overexpression of SIRT7 in the sirt7−/− MEF cells resulted in hypersensitivity to paclitaxel (Fig. 3D), gemcitabine and epirubicin (Fig. S3A & S3B). However, overexpressing the catalytic-inactive SIRT7 mutant, SIRT7 HY, did not lead to hypersensitivity to paclitaxel treatment, which indicated that intact deacetylase activity of SIRT7 is essential for SIRT7 mediated regulation of chemo-sensitivity (Fig. 3D, Fig. S3A & S3B). Overall, these results have established an important role of SIRT7 in the regulation of cellular responses to chemotherapeutic drugs.
Figure 3. SIRT7 Depletion Sensitizes Cancer Cells to Different Genotoxic Agents.

(A) MCF7 cells were transfected with negative siRNA or SIRT7 siRNA and were then treated with the indicated drugs for 72 hours. Cell survival was determined by MTS assay as described in Methods.
(B) MDA-MB-231 cells were transfected with negative siRNA or SIRT7 siRNA. Cells were treated with the indicated drugs and cell survival was determined as in (A). See also Figure S2.
(C) sirt7+/+ or sirt7−/− cells were treated with indicated drugs. Cell survival was determined as in (A). See also Figure S3.
(D) sirt7−/− MEF cells transfected with empty vector (EV), SIRT7 WT and SIRT7 H187Y mutants were treated with paclitaxel. Cell survival was determined as previously stated. (A–D) Each point indicates a mean value for three independent experiments. Error bars represent standard error of the mean (n=3).
SIRT7 Regulates Akt Activity through its Regulation of the Scaffolding Function of FKBP51
Our previous study suggested that FKBP51 functioned as a scaffolding protein to facilitate PHLPP binding to Akt and promote dephosphorylation of Akt at Serine 473 (Pei et al., 2009). Because SIRT7 specifically interacts with FKBP51 (Fig. 2A & 2B), and negatively regulates Akt activation, we then asked whether SIRT7 regulated Akt activity through modulating FKBP51 scaffolding functions, in other words, regulating FKBP51 binding to Akt/PHLPP. To test this hypothesis, we knocked down SIRT7 in MDA-MB-231, followed by immunoprecipitation to determine the interactions between endogenous FKBP51 and Akt/PHLPP. As shown in Fig. 4A, we found that FKBP51 interacted with both Akt and PHLPP in control cells. However, a substantial decrease in binding of FKBP51 with both Akt and PHLPP was observed when SIRT7 was knocked down. To confirm these results, we overexpressed SIRT7 in MDA-MB-231 cells and pulled down endogenous FKBP51 to detect the interactions between FKBP51 and Akt/PHLPP. Overexpressing SIRT7 greatly increased the binding of FKBP51 with Akt and PHLPP, resulting in decreased phosphorylation of Akt Ser473 (Fig. 4B). Collectively, these findings demonstrated that SIRT7 regulated Akt activity through its deacetylase activity and modulating the interaction between FKBP51 and Akt/PHLPP. We then tested whether SIRT7 regulation of Akt activity was FKBP51-dependent. Using MEF cells derived from FKBP51 WT and KO embryos, we found that overexpression of SIRT7 inhibited Akt activation in fkbp5+/+ cells but not in fkbp5−/− cells (Fig. 4C). Consistently, overexpressing of SIRT7 in the WT MEFs, but not FKBP51 deficient MEFs, rendered cells more sensitive to taxane treatment (Fig. 4D). These results suggest that SIRT7 regulates Akt activity and response to chemotherapy through the regulation of FKBP51 scaffolding function.
Figure 4. SIRT7 Regulates FKBP51 Binding to Akt/PHLPP.

(A) MDA-MB-231 cells were transfected with the indicated siRNAs and endogenous binding of FKBP51 with Akt/PHLPP was detected by IP with anti-FKBP51 antibody and blotting with anti-PHLPP and anti-Akt antibodies.
(B) HEK293T cells were transfected with empty vector (EV) or HA-SIRT7 plasmids, and the interactions between FKBP51 and Akt/PHLPP were analyzed.
(C) Empty vector or HA-SIRT7 plasmids were transfected into either fkbp5+/+ or fkbp5−/− MEF cells. Cell lysates were subjected to Western Blot analyses with the indicated antibodies; or (D) cells were treated with the indicated drugs and cell survival was determined by MTS assay as described in Methods. Data are shown as mean values for three independent experiments and error bars represent standard error of the mean (n=3).
SIRT7 Regulates FKBP51 Acetylation
We found that SIRT7 interacted with FKBP51 and regulated its scaffolding function. Because SIRT7 is a protein deacetylase that regulates a wide range of proteins (Barber et al., 2012; Lee et al., 2014; Tsai et al., 2012), we further hypothesized that SIRT7 might directly regulate FKBP51 acetylation. To test this, we treated the cells with a pan-sirtuin family inhibitor, nicotinamide and found that treatment with NAM led to increased FKBP51 acetylation (Fig. 5A). To confirm whether SIRT7 might deacetylate FKBP51, we knocked down SIRT7, followed by IP using antibodies against either FKBP51 or acetylated lysine to determine the levels of acetylated FKBP51. Depletion of SIRT7 led to an increase in acetylated FKBP51 (Fig. 5B), but not Akt or PHLPP (Fig. S4A). Furthermore, we observed that overexpression of SIRT7 decreased acetylation of endogenous FKBP51 (Fig. 5C). Collectively, these data suggest that SIRT7 interacts with FKBP51 and regulates FKBP51 deacetylation.
Figure 5. SIRT7 collaborates with CBP in regulating FKBP51 acetylation.

(A) HEK293T cells transfected with empty vector or Flag-FKBP51 were treated with vehicle or nicotinamide (NAM, 10 mM) overnight. FKBP51 acetylation was determined by IP with Flag beads followed by Western Blot.
(B) MDA-MB-231 breast cancer cells were transfected with negative siRNA or SIRT7 siRNA, and endogenous FKBP51 was examined by IP with anti-FKBP51 antibody (left panel) or acetylated lysine beads (right panel) and immunoblot using anti-FKBP51 or anti-acetylation antibodies. See also Figure S4.
(C) MDA-MB-231 cells transfected with empty vector or Flag-SIRT7 were subjected to IP with anti-FKBP51antibody to detect endogenous FKBP51 acetylation.
(D) HEK293T cells transfected with Flag-tagged FKBP51 were co-transfected with empty vector or plasmids encoding full-length HA-P300, DMOF, CBP and TIP60. FKBP51 acetylation was then examined. * represents the targeted protein that expressed.
(E) Endogenous interaction of FKBP51 and CBP was detected by IP followed by immunoblotting using the anti-CBP and anti-FKBP51 antibodies.
(F) In vitro deacetylation assay demonstrated that CBP acetylated FKBP51 and SIRT7 deacetylated FKBP51.
(G) Immunopurified Wild Type (WT) or catalytic-dead HY mutant Flag-SIRT7 were used to perform the in vitro deacetylation assay. Acetylation of lysine residues was detected with anti-pan-Ac-K antibody.
Since acetylation is catalyzed and controlled by opposing actions of both acetyltransferases and deacetylases, we set out to identify which acetyltransferases are responsible for FKBP51 acetylation. In order to do this, we co-transfected Flag-FKBP51 with a set of acetyltransferases in HEK293T cells, including p300 (EP300) and CBP (CREB-binding protein), both of which belong to the p300/CBP family, TIP60 (TAT-interactive protein) and hMOF (human males absent on the first), both of which belong to the MYST family. These acetyltransferases were chosen because they are members belonging to three major categories of HATs and are involved in acetylation of many substrates (Yang, 2004). Flag-FKBP51 was immunoprecipitated with an anti-Flag antibody, followed by immunoblotting with an anti-acetylated-lysine antibody. We found that FKBP51 acetylation was increased by CBP and to a lesser extent, p300, but not by hMOF or TIP60 (Fig. 5D). Furthermore, we also detected a specific interaction between endogenous FKBP51 and CBP (Fig. 5E).
In addition, direct SIRT7 acetylation and deacetylation of FKBP51 was evaluated using in vitro acetylation and deacetylation assays. As shown in Fig. 5F and Fig. S4B, CBP robustly acetylated FKBP51, while p300 displayed moderate acetylation activity. Furthermore, CBP-induced acetylation of FKBP51 was reverted by SIRT7 (Fig. 5F). To further confirm SIRT7 catalytic activity is essential for the deacetylation, we used the SIRT7HY mutant. SIRT7 WT, and not SIRT7 HY, deacetylated FKBP51 in an NAD-dependent manner (Fig. 5G). Moreover, the SIRT7 mediated deacetylation of FKBP51 was significantly inhibited by NAM, a pan-sirtuin family inhibitor.
Identification of Lysine Residues on FKBP51 Regulated by SIRT7
Since we showed that SIRT7 modulated the acetylation of FKBP51, we next evaluated the potential acetylation sites regulated by SIRT7. FKBP51 lysine residues were analyzed by mass spectrometry in both control and SIRT7-depleted cells. Acetylation signals increased for six lysine residues (28, 155, 248, 414, 415, and 441) of FKBP51 when SIRT7 was depleted (Fig. 6A). To confirm which lysine residues were regulated by SIRT7, we performed site directed mutagenesis for each of these six candidate lysine residues, changing from lysine (K) to arginine (R) to mimic the nonacetylated form. Depletion of SIRT7 increased the acetylation of WT FKBP51; however, in cells expressing each single FKBP51 mutants, following SIRT7 depletion, increased FKBP51 acetylation levels were partially compromised with K28R and K155R mutants (Fig. 6B), suggesting these two sites might be the major acetylation sites regulated by SIRT7. We then mutated these two lysine residues to arginine simultaneously (2KR) and expressed the mutants in cells. Knockdown of SIRT7 increased the acetylation of WT FKBP51, however this effect is abolished in the 2KR mutant, indicating that these two lysine residues are the main sites regulated by SIRT7 (Fig. 6C). Furthermore, overexpression of CBP increased the acetylation of FKBP51 WT, but not the 2KR mutant (Fig. 6D). These results demonstrated that K28 and K155 were the major acetylation sites regulated by SIRT7 and CBP.
Figure 6. Identification of FKBP51 Acetyl Sites Regulated by SIRT7.

(A) A list of acetylation sites in FKBP51 protein analyzed by mass spectrometry.
(B) HEK293T cells stably expressing control siRNA or SIRT7 siRNA were transfected with the indicated constructs. After 72 hours, FKBP51 acetylation was examined.
(C) HEK293T cells were transfected with the indicated constructs and siRNAs. FKBP51 acetylation was then examined by pulling down using anti-Flag antibody and acetylation was determined by Western Blot.
(D) HEK293T cells were transfected with the indicated constructs. FKBP51 acetylation was examined as in (B). Arrows indicates endogenous FKBP51 bands or Flag-FKBP51 bands.
(E) fkbp5+/+ or fkbp5−/− MEF cells were transfected with empty vector and the indicated SIRT7 constructs. Cell lysates were subjected to Western Blot.
(F) fkbp5+/+ and fkbp5−/− MEF cells were transfected with the indicated constructs and were treated with increased dosage of paclitaxel for 72 hours. Cell survival was determined by MTS assays. Data are shown as mean values for three independent experiments and error bars represent standard error of the mean (n=3).
To test the functional impact of these two lysine residues on Akt activation, we further reconstituted the WT, the 2KR or 2KQ (lysine-to-glutamine substitutions, mimicking the constitutively acetylated form) mutants into the fkbp5 KO MEFs (fkbp5−/−) and determined Akt Ser473 phosphorylation. As shown in Fig. 6E, reconstitution of WT FKBP51 or 2KR FKBP51, but not the FKBP51 2KQ reversed the increase of Akt p473 level in fkbp5−/− MEF cells. Additionally, reconstitution of FKBP51 WT or FKBP51 2KR, but not the FKBP51 2KQ, significantly increased the complex formation of FKBP51-Akt-PHLPP. These results suggest that FKBP51 lysine residues K28 and K155 are deacetylated by SIRT7, which allows for the interaction of the FKBP51-Akt-PHLPP complex and subsequent regulation of Akt activity.
We also evaluated whether FKBP51 acetylation was important for cell survival in response to genotoxic agents, such as paclitaxel, in fkbp5−/− MEF cells reconstituted with WT, 2KR, or 2KQ mutant FKBP51. As shown in Fig. 6F, reconstituting cells with FKBP51 WT or 2KR mutants, but not the 2KQ mutant, reversed paclitaxel resistance caused by FKBP51 deficiency. These results indicate that the regulation of FKBP51 acetylation at residues K28 and K155 by SIRT7 is important for cell sensitivity to genotoxic stresses, a downstream marker of Akt activation. Taken together, our results suggest that SIRT7 regulates Akt signaling through the regulation FKBP51 acetylation at lysine 28 and 155 sites.
2DG Sensitizes Cancer Cells to Chemotherapy
We have shown that under energy stress, such as glucose deprivation or 2DG treatment, the binding of FKBP51 and Akt/PHLPP was significantly increased and led to suppressed Akt activity (Fig. 1). Consistent with the importance of FKBP51 deacetylation in complex with Akt/PHLPP, under energy stress conditions, a strong interaction between FKBP51 and SIRT7 was observed (Fig. 7A). We next examined whether this regulation is dependent on SIRT7. When we knocked down SIRT7, the increased interactions between FKBP51 and Akt/PHLPP were abolished (Fig. 7B). Hence, glucose deprivation or 2DG treatment induced increase of FKBP51 and Akt/PHLPP binding is dependent on intact SIRT7.
Figure 7. 2DG Increases SIRT7-FKBP51 Interaction and Sensitize Cells to Chemotherapy.

(A) HEK293T cells stably expressing Flag-FKBP51 were serum starved for 3 hours followed by full medium with 10 mM 2DG or glucose free medium for 6 hours. Flag-FKBP51 was IP with anti-Flag beads and elute was subjected to Western Blot with indicated antibodies.
(B) HEK293T cells stably expressing Flag-FKBP51 were transfected with control siRNA or siRNA targeting SIRT7. The treatments were similar as those in (A).
(C) sirt7+/+ and sirt7−/− MEF cells were treated with either the indicated drugs alone or combined with 2 mM 2DG for 72 hours. Cell survival was determined by MTS assays. Data are shown as mean values for three independent experiments. See also Figure S5.
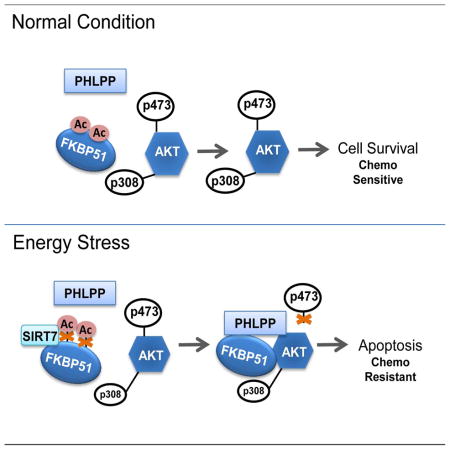
(D) Proposed model for SIRT7 in regulation of FKBP51 acetylation and Akt activation.
Since suppressed Akt activity is expected to lead to increased sensitivity to chemotherapy, we predicted that combination of 2DG and chemotherapies might have a synergetic effect on killing cancer cells. To test this hypothesis, MDA-MB-231 and sirt7+/+ or sirt7−/− MEFs were treated with low dose 2DG combined with different chemotherapeutic agents. As shown in Fig. 7C, the addition of low dose 2DG significantly sensitized sirt7+/+ MEFs but not sirt7−/− MEFs to gemcitabine, paclitaxel and epirubicin. Similar observations were found in MDA-MB-231 cells (Fig. S5). These results indicate a potential therapeutic strategy to overcome resistance to standard chemotherapy by adding 2DG in the combination therapy.
DISCUSSION
Resistance to genotoxic chemotherapeutics remains a major challenge in cancer chemotherapy. Dysregulation of the PI3K/Akt pathway has been associated with resistance to many different classes of chemotherapies (Brown and Toker, 2015). We and other groups have identified the FKBP51-Akt-PHLPP axis that plays an important role in the regulation of Akt activation and, subsequently, the response to various clinically important chemotherapies in cancer cells (Mulholland et al., 2011; Pei et al., 2009). Recently, SIRT7, the least understood member of the Sirtuin family, has gained attention in the field with respect to the pathogenesis of different diseases (Li and Bhatia, 2013). However, little is known about SIRT7 function in the context of treatment resistance. Our present work has demonstrated that SIRT7 regulates cancer cell response to chemotherapy by regulating the FKBP51-Akt-PHLPP axis. Specifically, in breast cancer cells, SIRT7 deacetylation activity opposes the CBP-dependent acetylation of FKBP51. Constitutive or transient SIRT7 insufficiency in breast cancer cells leads to a significant elevation of FKBP51 acetylation and, therefore, inhibits the binding of PHLPP to Akt, a process facilitated by the scaffolding protein, FKBP51. This also leads to increased activation of the Akt pathway and cell resistance to chemotherapy. Consistently, these abnormalities are reversed by overexpression of WT SITR7, but not catalytically-dead mutant SIRT7 in cells. Energy stress (via glucose deprivation) or pharmacologic interventions (treated with 2DG) activates SIRT7 and further enhances the binding of PHLPP to Akt (Fig. 7A), promoting cell death under genotoxic stress. Overall, our results have not only provided mechanisms by which SIRT7 regulates the activation of the Akt pathway but also revealed a potential therapeutic strategy to help sensitize patients to chemotherapy by adding 2DG to therapeutic options in the treatment of breast cancer.
Previous reports on the subcellular localization of SIRT7 seem controversial. Most studies have demonstrated that SIRT7 is predominantly localized in the nucleolus (Ford et al., 2006; Michishita et al., 2005). However, we reveal that SIRT7 is present in both nuclear fractions and cytoplasmic fractions in both breast cancer cells and MEF cells (Fig. S1A). This finding is consistent with recent studies (Kiran et al., 2013; Zhang et al., 2016). Although different interactome data have revealed that SIRT7 interacts with dozens of proteins that are exclusively localized in the cytoplasm (Lee et al., 2014; Tsai et al., 2012), the molecular function of SIRT7 in the cytoplasm remains unknown (Zhang et al., 2016), and our studies have revealed one potential function of SIRT7 in the cytoplasm.
Activation of the phosphoinositide 3-kinase (PI3K)/Akt pathway represents one of the most frequent genetic or epigenetic alterations that occur in breast cancer (Thorpe et al., 2015). Acetylation is one mechanism involved in the regulation of Akt activity either directly or indirectly. Most previous studies are focused on the role of SIRT1 in the regulation of this pathway (Sundaresan et al., 2011; Zhang, 2007). Deacetylation of two lysine residues in the Akt PH domain by SIRT1 is essential for the binding of Akt to PIP3 for its membrane localization and activation (Sundaresan et al., 2011). SIRT1 is also reported to deacetylate insulin receptor substrate 2 (IRS2), which in turn, increases its activity and activates downstream Akt (Zhang, 2007). Additionally, studies have shown that SIRT1 can also regulate Akt activity by deacetylation of p53 (Tang et al., 2008). Besides SIRT1, studies have also shown the involvement of other SIRT family members in the regulation of Akt activity. SIRT3 is found to directly control hyperactivation of Akt by regulating mitochondrial ROS production and ROS- mediated Ras-PI3K-Akt activation (Pillai et al., 2014). SIRT6 is also reported to negatively regulate Akt signaling at the level of chromatin (Sundaresan et al., 2012). Our data demonstrate the involvement of SIRT7 in Akt regulation (Fig. 2 and 3). Mechanistically, SIRT7 deacetylates FKBP51 at the K28 and K155 residues (Fig. 6A–E). Deacetylated FKBP51 promotes the binding of the phosphatase, PHLPP, to Akt and resultes in dephosphorylation of p-Akt S473, thereby, inactivating Akt signaling (Fig. 4A & 4B, Fig. 6E). Therefore, our study has revealed a critical role of the SIRT7-FKBP51 interaction in Akt activation and cellular response to genotoxic stress. It has also added a regulatory mechanism of the Akt pathway by SIRT7 in addition to the reported function of other SIRT family members.
The Akt pathway plays an anti-apoptotic role in response to genotoxic stress and contributes to cancer cell tolerance of genotoxic therapeutics. Consistent with this, SIRT7 also appears to be anti-apoptotic. In breast cancer cells or mouse embryonic fibroblast cells, deletion of SIRT7 renders the cells resistant to genotoxic agents including paclitaxel, gemcitabine and epirubicin (Fig. 3A–C). This anti-apoptotic effect is further confirmed by reconstitution of SIRT7 KO cells with WT SIRT7 (Fig. 3D). SIRT7 was reported to be involved in the regulation of DNA damage response. γH2AX is an important marker for DNA damage response. In previous publication (Li et al., 2016), the authors observed different DNA damage response in different cell lines. In MCF7 cells, depletion of SIRT7 significantly enhanced γH2AX level. However, in U2OS cells, down regulation of SIRT7 suppressed γH2AX. In our study, no significant difference in γH2AX was observed between sirt7+/+ and sirt7−/− MEFs with or without Gemcitabine treatments (Fig. S2C), indicating that DNA damage might not be the major pathway regulated by SIRT7 that contributing to the phenotype (chemo-response) here. However, obviously, we could not totally exclude the possibility that there might be other mechanisms involved in SIRT7 mediated chemo-sensitivity or resistance.
Cancer cells undergo metabolic reprograming involving high rates of glycolysis even in the presence of oxygen, known as the Warburg effect (Ward and Thompson, 2012). This process facilitates cancer cell survival, proliferation and resistance to therapies. Because of the dependence of tumors on glycolysis, the glycolytic inhibitor 2-deoxy-D-glucose (2DG), which inhibits glucose metabolism (Ben Sahra et al., 2010) and transport (Dwarakanath and Jain, 2009), was initially considered as a potential anticancer agent. It has been tested as an adjuvant treatment in combination with radiation (Dwarakanath and Jain, 2009; Heminger et al., 2006; Lin et al., 2003) or chemotherapy (Dwarakanath and Jain, 2009) and has shown enhanced clinical efficacy. The mechanism of 2DG involved in the cytotoxicity effect is mainly mediated through an effect on oxidative stress, a process that is regulated by many redundant pathways in tumor cells. Our research has provided additional insight into the mechanisms of 2DG treatment and its effect on the sensitization of cancer cells to chemotherapy, a process that may partially be through SIRT7 regulation of the FKBP51-Akt-PHLPP axis.
In summary, our study identifies a direct role of SIRT7 in regulating Akt activation. We found that SIRT7 could regulate FKBP51 acetylation and affect complex formation of FKBP51-Akt-PHLPP. Under energy stress or 2DG treatment, enhanced interaction of SIRT7 with FKBP51 further enhances FKBP51-Akt-PHLPP complex formation and sensitizes cancer cells to cytotoxic agents (Fig. 7D). This observation provides evidence supporting the potential application of 2DG as an adjuvant agent to enhance chemo-sensitivity in the treatment of solid tumors.
EXPERIMENTAL PROCEDURES
Cell Culture
HEK293T cells and Su86 cells were cultured in RPMI 1640, supplemented with 10% fetal bovine serum (FBS). Human breast cancer cell lines, MDA-MB-231 and MCF7 (HTB-22™) were purchased from the American Type Culture Collection (ATCC) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS. Chemical compounds were administered in high-glucose DMEM or glucose-free DMEM supplemented with 10% FBS for 24 hours, unless otherwise indicated. sirt7+/+ and sirt7−/− mouse embryonic fibroblast (MEFs) were kindly provided by Dr. Danica Chen (University of California, Berkeley) and were cultured in DMEM supplemented with 15% FBS as described (Shin et al., 2013).
Chemicals and Plasmids
Generation of the S/Flag/SBP-tagged FKBP51 construct was described previously (Pei et al., 2009). SIRT7 was cloned into the pIRES-EGFP vector containing a Flag tag or to a pCMV plasmid with an HA tag. All mutants were generated by site-directed mutagenesis (Stratagene) and sequences were verified by Sanger sequencing. The expression constructs for acetyltransferases, p300, CBP, PCAF and Tip60; deacetyltransferases, SIRT1-7 have been previously described (Liu et al., 2014).
RNA Interference
For siRNA transfection, cells were transfected with the indicated siRNA using lipofectamine RNAiMAX (Life Technologies Invitrogen) following the manufacturer’s instructions. Small interfering RNA (siRNA) oligonucleotides targeting human sirt7 were purchased from Dharmacon (Thermo Scientific).
Immunoprecipitation and Immunoblotting Assays
Methods used for performing immunoprecipitation and immunoblotting were described previously (Qin et al., 2015). Specifically, cells were lysed with NETN buffer (20mM Tris-HCl, pH 8.0, 100mM NaCl, 1mM EDTA, 0.5% Nonidet P-40) containing 50mM β-glycerophosphate, 10mM NaF, and protease inhibitor cocktail on ice for 30 mins. Whole cell lysates were obtained by centrifugation. The cell lysates were then incubated with 2 μg of antibody and protein A or protein G Sepharose beads (Amersham Biosciences) at 4°C for 2–3 hours or overnight. The immunocomplexes were washed three times with ice cold NETN buffer. Proteins were then eluted in SDS loading buffer and detected by Western Blot analysis.
In vitro Acetylation and Deacetylation Assays
The in vitro acetylation and deacetylation assays were performed as previously described (Ryu et al., 2014). Specifically, 1 μg of human recombinant GST-FKBP51 protein (LS-G28483, LifeSpan BioSciences) was incubated with 500 ng of recombinant CPB (catalytic domain) protein (Millipore) or p300 protein (Active Motif) in acetylation buffer (50 mM Tris-HCL [pH 8.0]), 100 mM NaCl, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM dithiothreitol [DTT], 1 μg/ml bepstatin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM sodium butyrate, and 150 μM acetyl-CoA) for 1 hr at 30 °C.
For deacetylation assay, 1 μg of acetylated FKBP51 was incubated with 500 ng of SIRT7 protein (TP305658, Origene), or immuno-purified WT or HY mutant Flag-SIRT7, in the deacetylation buffer (50 mM Tris-HCL [pH 9.0]), 4 mM MgCl2, 0.2 mM DTT, 1 μg/ml bepstatin, 1 μg/ml leupeptin, 1 μg/ml pepstatin and 1mM NAD+) for 30 min. The reactions were terminated by boiling in SDS sample buffer. Samples were subjected to SDS-PAGE and analyzed by Western Blot.
Flag tagged wild-type SIRT7 or catalytic-inactive SIRT7 H187Y mutant (HY mutant) constructs were transfected in HEK293T cells using lipofectamine 2000. Flag-SIRT7 was purified from HEK293T cells with M2 anti-Fag beads (Sigma) in cell lysis buffer supplemented with 0.1% NP-40, protease inhibitors and HDAC inhibitors as previously described (Chen et al., 2016). The proteins were then eluted with FLAG peptide (Sigma).
Acetyl Lysine Residues Detected by Mass Spectrometry
HEK293T cells stably expressing S/Flag/SBP-tagged FKBP51 were transfected with control or SIRT7 siRNA. After 72 hours, cells were lysed by NETN buffer. Cell lysates were incubated with streptavidin sepharose beads (Amersham Biosciences) at 4°C for 4 hours. After washing with NETN buffer three times, S-tagged FKBP51 was eluted and protein samples were subjected to SDS–PAGE followed by staining with Coomassie blue. S-tagged FKBP51 protein bands were cut out from the gel and digested with trypsin in-gel as previously described (Liu et al., 2014). Tryptic peptides were extracted from the gel, separated on a C18 column and were analyzed by LTQ-Orbitrap Velos (Thermo, Germany) with a 75-min gradient. The survey scan range was set to m/z 375 to 1600. Proteins with possible modification by acetylation, trimethylation, and phosphorylation were identified with the Proteome Discovery 1.3 using the MASCOT search engine against the human NCBI RefSeq protein databases. The false discovery rate (FDR) of protein identification was set to 5% for the initial analysis. Area under the curve (AUC) was used to calculate the quantities of a specific peptide. Acetylated peptides within 1% FDR were further manually verified and the acetylation sites were selected for functional evaluation.
MTS Assay
2-Deoxy-D-glucose (2DG) and paclitaxel were purchased from Sigma-Aldrich (St. Louis, MO, USA); and gemcitabine was provided by Eli Lilly (Indianapolis, IN). Cytotoxicity was determined with the MTS assay. After incubation with drugs for 72 hr, 20 μl MTS was added to each well and the plates were measured in a Safire2 microplate reader (Tecan AG, Switzerland).
Statistical Analysis and Data Visualizations
Statistical analysis was performed by Student t-test for two group comparisons and by ANOVA for multiple groups. Statistical significance is represented in figures by: *, p<0.05; **, p<0.01.
Supplementary Material
Acknowledgments
We thank Dr. Danica Chen for providing wild-type or SIRT7 deficient MEFs. This work was supported by National Institutes of Health grants U19 GM61388 (The Pharmacogenomics Research Network, PGRN), RO1 CA196648 (L. Wang), CA203561 (Z. Lou) and CA130996 (Z. Lou). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Conflict of Interest: None.
Supplemental Information includes five figures, Supplemental Experimental Procedures and Supplemental References. The information can be found with this article online.
AUTHOR CONTRIBUTIONS
J.Y. and B.Q. planned and performed experiments, analyzed the data and wrote the manuscript. F.W. performed experiments. S.Q., S.N. and S.S. provided methodological and technical assistance as well as assistance in analyzing the data. J.Z. analyzed the data and contributed to manuscript preparation. H.P. performed the Mass Spectrometry experiments. Z.L. provided specific expertise and knowledge in the area of SIRTs and L.W. was responsible for the concept generation and entire study design. Z.L. and L.W. are responsible for all the data described in the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Current Biology. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Andjelković M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proceedings of the National Academy of Sciences. 1996;93:5699–5704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf N, Zino S, MacIntyre A, Kingsmore D, Payne AP, George WD, Shiels PG. Altered sirtuin expression is associated with node-positive breast cancer. Brit J Cancer. 2006;95:1056–1061. doi: 10.1038/sj.bjc.6603384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber MF, Michishita-Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL, Chen K, et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012;487:114–118. doi: 10.1038/nature11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012;11:443–461. doi: 10.1038/nrd3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010;70:2465–2475. doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–931. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Brown KK, Toker A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015;7:13. doi: 10.12703/P7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Blank MF, Iyer A, Huang B, Wang L, Grummt I, Voit R. SIRT7-dependent deacetylation of the U3-55k protein controls pre-rRNA processing. Nat Commun. 2016;7:10734. doi: 10.1038/ncomms10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nigris F, Cerutti J, Morelli C, Califano D, Chiariotti L, Viglietto G, Santelli G, Fusco A. Isolation of a SIR-like gene, SIR-T8, that is overexpressed in thyroid carcinoma cell lines and tissues. Br J Cancer. 2002;87:1479. doi: 10.1038/sj.bjc.6600636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwarakanath B, Jain V. Targeting glucose metabolism with 2-deoxy-D-glucose for improving cancer therapy. Future Oncol. 2009;5:581–585. doi: 10.2217/fon.09.44. [DOI] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E, Voit R, Liszt G, Magin C, GrumMt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Gene Dev. 2006;20:1075–1080. doi: 10.1101/gad.1399706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Guo S, Rena G, Cichy S, He X, Cohen P, Unterman T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999;274:17184–17192. doi: 10.1074/jbc.274.24.17184. [DOI] [PubMed] [Google Scholar]
- Hajduch E, Litherland GJ, Hundal HS. Protein kinase B (PKB/Akt)--a key regulator of glucose transport? FEBS Lett. 2001;492:199–203. doi: 10.1016/s0014-5793(01)02242-6. [DOI] [PubMed] [Google Scholar]
- Heminger K, Jain V, Kadakia M, Dwarakanath B, Berberich SJ. Altered gene expression induced by ionizing radiation and glycolytic inhibitor 2-deoxy-glucose in a human glioma cell line - Implications for radiosensitization. Cancer Biol Ther. 2006;5:815–823. doi: 10.4161/cbt.5.7.2812. [DOI] [PubMed] [Google Scholar]
- Hou JM, Wang LW. FKBP5 as a Selection Biomarker for Gemcitabine and Akt Inhibitors in Treatment of Pancreatic Cancer. PLoS One. 2012;7 doi: 10.1371/journal.pone.0036252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Noh JH, Jung KH, Eun JW, Bae HJ, Kim MG, Chang YG, Shen Q, Park WS, Lee JY, et al. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors MiR-125a-5p and MiR-125b. Hepatology. 2013;57:1055–1067. doi: 10.1002/hep.26101. [DOI] [PubMed] [Google Scholar]
- Kiran S, Chatterjee N, Singh S, Kaul SC, Wadhwa R, Ramakrishna G. Intracellular distribution of human SIRT7 and mapping of the nuclear/nucleolar localization signal. Febs J. 2013;280:3451–3466. doi: 10.1111/febs.12346. [DOI] [PubMed] [Google Scholar]
- Lai CC, Lin PM, Lin SF, Hsu CH, Lin HC, Hu ML, Hsu CM, Yang MY. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumour Biol. 2013;34:1847–1854. doi: 10.1007/s13277-013-0726-y. [DOI] [PubMed] [Google Scholar]
- Lee N, Kim DK, Kim ES, Park SJ, Kwon JH, Shin J, Park SM, Moon YH, Wang HJ, Gho YS, et al. Comparative interactomes of SIRT6 and SIRT7: Implication of functional links to aging. Proteomics. 2014;14:1610–1622. doi: 10.1002/pmic.201400001. [DOI] [PubMed] [Google Scholar]
- Li L, Bhatia R. The controversial role of Sirtuins in tumorigenesis - SIRT7 joins the debate. Cell Res. 2013;23:10–12. doi: 10.1038/cr.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shi L, Yang SD, Yan RR, Zhang D, Yang JG, He L, Li WJ, Yi X, Sun LY, et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun. 2016;7 doi: 10.1038/ncomms12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Zhang F, Bradbury CM, Kaushal A, Li L, Spitz DR, Aft RL, Gius D. 2-Deoxy-D-glucose-induced cytotoxicity and radiosensitization in tumor cells is mediated via disruptions in thiol metabolism. Cancer Res. 2003;63:3413–3417. [PubMed] [Google Scholar]
- Liu T, Lin YH, Leng W, Jung SY, Zhang H, Deng M, Evans D, Li Y, Luo K, Qin B, et al. A divergent role of the SIRT1-TopBP1 axis in regulating metabolic checkpoint and DNA damage checkpoint. Mol Cell. 2014;56:681–695. doi: 10.1016/j.molcel.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosashvilli D, Kahl P, Mertens C, Holzapfel S, Rogenhofer S, Hauser S, Buttner R, Von Ruecker A, Muller SC, Ellinger J. Global histone acetylation levels: prognostic relevance in patients with renal cell carcinoma. Cancer Sci. 2010;101:2664–2669. doi: 10.1111/j.1349-7006.2010.01717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. 2014;114:368–378. doi: 10.1161/CIRCRESAHA.113.300536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin B, Minter-Dykhouse K, Yu J, Zhang J, Liu T, Zhang H, Lee S, Kim J, Wang L, Lou Z. DBC1 functions as a tumor suppressor by regulating p53 stability. Cell Rep. 2015;10:1324–1334. doi: 10.1016/j.celrep.2015.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D, Jo YS, Lo Sasso G, Stein S, Zhang H, Perino A, Lee JU, Zeviani M, Romand R, Hottiger MO, et al. A SIRT7-dependent acetylation switch of GABPbeta1 controls mitochondrial function. Cell Metab. 2014;20:856–869. doi: 10.1016/j.cmet.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, Wang Q, Chia D, Goodglick L, Kurdistani SK. Global levels of histone modifications predict prognosis in different cancers. Am J Pathol. 2009;174:1619–1628. doi: 10.2353/ajpath.2009.080874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, Qiu X, Nabavi N, Mohrin M, Wojnoonski K, et al. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep. 2013;5:654–665. doi: 10.1016/j.celrep.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Gene Dev. 1997;11:241–254. doi: 10.1101/gad.11.2.241. [DOI] [PubMed] [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P. Protein kinase B kinases that mediate phosphatidylinositol 3, 4, 5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, Raghuraman H, Cunningham JM, Gupta M, Gupta MP. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal. 2011;4:ra46. doi: 10.1126/scisignal.2001465. [DOI] [PubMed] [Google Scholar]
- Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med. 2012;18:1643–1650. doi: 10.1038/nm.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zhao WH, Chen Y, Zhao YM, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Greco TM, Boonmee A, Miteva Y, Cristea IM. Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Molecular & cellular proteomics : MCP. 2012;11:60–76. doi: 10.1074/mcp.A111.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Greco TM, Cristea IM. Sirtuin 7 plays a role in ribosome biogenesis and protein synthesis. Mol Cell Proteomics. 2014;13:73–83. doi: 10.1074/mcp.M113.031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakhrusheva O, Braeuer D, Liu Z, Braun T, Bober E. Sirt7-Dependent Inhibition of Cell Growth and Proliferation Might Be Instrumental to Mediate Tissue Integrity during Aging. J Physiol Pharmacol. 2008a;59:201–212. [PubMed] [Google Scholar]
- Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008b;102:703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- Vazquez BN, Thackray JK, Simonet NG, Kane-Goldsmith N, Martinez-Redondo P, Nguyen T, Bunting S, Vaquero A, Tischfield JA, Serrano L. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. Embo J. 2016 doi: 10.15252/embj.201593499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004;32:959–976. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem. 2007;282:34356–34364. doi: 10.1074/jbc.M706644200. [DOI] [PubMed] [Google Scholar]
- Zhang PY, Li G, Deng ZJ, Liu LY, Chen L, Tang JZ, Wang YQ, Cao ST, Fang YX, Wen F, et al. Dicer interacts with SIRT7 and regulates H3K18 deacetylation in response to DNA damaging agents. Nucleic Acids Res. 2016;44:3629–3642. doi: 10.1093/nar/gkv1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gan L, Pan H, Guo S, He X, Olson ST, Mesecar A, Adam S, Unterman TG. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J Biol Chem. 2002;277:45276–45284. doi: 10.1074/jbc.M208063200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.