

Abstract
Developing therapies for central nervous system (CNS) diseases is exceedingly difficult due to the blood-brain barrier (BBB). Notably, emerging technologies may provide promising new options for the treatment of CNS disorders. Adeno-associated virus serotype 9 (AAV9) has been shown to transduce cells in the CNS following intravascular administration in rodents, cats, pigs, and non-human primates. These results suggest that AAV9 is capable of crossing the BBB. However, mechanisms that govern AAV9 transendothelial trafficking at the BBB remain unknown. Furthermore, possibilities that AAV9 may transduce brain endothelial cells or affect BBB integrity still require investigation. Using primary human brain microvascular endothelial cells (BMVEC) as a model of the human BBB, we performed transduction and transendothelial trafficking assays comparing AAV9 to AAV2, a serotype that does not cross the BBB or transduce endothelial cells effectively in vivo. Results of our in vitro studies indicate that AAV9 penetrates BMVEC barriers more effectively than AAV2, but has reduced transduction efficiency. In addition, our data suggest that 1) AAV9 penetrates endothelial barriers through an active, cell-mediated process, and 2) AAV9 fails to disrupt indicators of BBB integrity such as transendothelial electrical resistance, tight junction protein expression/localization, and inflammatory activation status. Overall, this report shows how human brain endothelial cells configured in BBB models can be utilized for evaluating transendothelial movement and transduction kinetics of various AAV capsids. Importantly, the use of a human in-vitro BBB model can provide import insight into the possible effects that candidate AVV gene therapy vectors may have on the status of BBB integrity.
Keywords: Blood-brain barrier, adeno-associated virus, gene therapy, neurological disorders
Graphical abstract
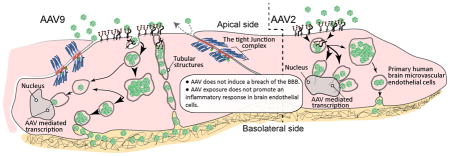
AAV9 vectors penetrate the BBB and are in clinical trials for gene-therapy of CNS disease. Remarkably little is known regarding how AAV9 traverses the BBB, and whether this process affects barrier homeostasis. Here for the first-time, using primary human brain-endothelial cells in a model of the BBB, we provide evidence that AAV9 crosses the BBB by an active-transport mechanism, while not compromising barrier integrity. These results provide insight related to safety of AAV use and a platform for further analysis.
INTRODUCTION
Based on their safety profile in clinical trials and their ability to efficiently transduce target cells, such as neurons and glia, recombinant AAV (rAAV) vectors are the top prospect gene therapy delivery vehicle currently being developed to treat central nervous system (CNS) disorders (Vannucci et al. 2013, Maguire et al. 2014). Transduction of target cells in the CNS using rAAV has previously been achieved through local (i.e. direct parenchymal or intra-cerebroventricular) injections. Unfortunately, due to high tortuosity of the extracellular space, which limits the diffusion of vectors within the CNS, only focal transduction of parenchymal cells is possible (Lo et al. 1999). Global transduction of target cells in the CNS has proven difficult because the most widely studied AAV serotype, AAV2, cannot penetrate the BBB after intravascular injection (Fu et al. 2003). However, recent reports have identified the remarkable ability of AAV serotype 9 (AAV9) to transduce parenchymal brain cells and portions of the BBB endothelium after intravenous injection (Foust et al. 2009, Yang et al. 2014). Due to its apparent efficiency at crossing the BBB, preclinical data obtained from rodents, cats, pigs, and non-human primates indicate that AAV9 may have utility in the treatment of human CNS disorders (Gray et al. 2011, Samaranch et al. 2012, Bevan et al. 2011, Duque et al. 2009). Notably, intravenously delivered AAV9 encoding a therapeutic transgene is currently being tested in clinical trials for the treatment of spinal muscular atrophy (Passini et al. 2014, Wirth et al. 2015, Rashnonejad et al. 2015).
To date, no studies have described the kinetics of AAV9 transport across a human BBB. Furthermore, little is known regarding the mechanisms that regulate AAV9 trafficking across the brain endothelium. For example, AAV9 may cross the BBB by either a transcellular or paracellular route. Understanding how AAV9 interacts with and negotiates blockades typically imposed by the BBB is important for several reasons. First, in order to maintain homeostasis in the CNS, the structural and functional integrity of the BBB must be preserved and not disrupted by AAV9 exposure. Thus, evaluating the effect of AAV9 vectors on key parameters of BBB integrity is important to safeguard patient health. Second, determining the viral peptides that function in BBB penetration might allow for the development of better viral and non-viral therapeutics.
We set out with the goal of employing an in vitro model that would allow us to examine the effects of AAV9 on the human BBB. Using primary human brain microvascular endothelial cell (BMVEC) cultures, we performed transduction and transendothelial trafficking assays to compare the efficiency of AAV9 against AAV2, a vector that does not appreciably cross the BBB or transduce brain endothelial cells in vivo (Varadi et al. 2012, Geoghegan et al. 2014). Our results indicate that AAV9 crosses our human BBB model more efficiently than AAV2; however, AAV2 exhibited more robust transgene expression in BMVEC cultures compared to AAV9. Furthermore, we monitored the effect of AAV9 exposure on multiple indicators of BMVEC barrier integrity including the stability of transendothelial electrical resistance (TEER), permeability, relative expression and cellular localization of tight junction (TJ) proteins, and the surface expression of cellular adhesion molecules, each of which could undermine neuronal function and CNS homeostasis if disrupted (Persidsky et al. 2006b, Abbott et al. 2006). Importantly, our data show no adverse effect on these indicators of BMVEC barrier integrity. Finally, using live-cell two-photon microscopy, we observed and compared the intracellular distribution of AAV9 to AAV2 finding unique distribution patterns between these serotypes in BMVEC cultures. These distinct intracellular patterns may explain the different activities we observed between AAV2 and AAV9 in our transduction and transcytosis assays. Notably, future investigations characterizing the mechanisms that guide these differing patterns may assist in developing innovative gene therapy delivery platforms for either endothelial transduction or transport across the BBB.
MATERIALS AND METHODS
Primary cell culture
Dr. Marlys Witte and Michael Bernas from the University of Arizona (Tucson, AZ) provided the primary human brain microvascular endothelial cell (BMVEC) cultures. These cells were isolated from either temporal lobe or hippocampal tissue resected from human subjects seeking operative treatment for epilepsy. The Temple University Institutional Review Board approved all procedures detailed in this study related to the use of these primary human cultures. As previously described, BMVEC cultures were derived from microvessels outside of the epileptogenic margins of the resected tissue (Bernas et al. 2010). Low passage BMVEC cultures from five different donors (mean age: 25.4 years; range: 18–39 years, both genders) were used to obtain the data presented herein. From each donor, cultures were validated for the presence of endothelial markers and the formation of barrier properties (Bernas et al. 2010). Briefly, the purity of our BMVEC cultures were validated by assessing the presence of astrocyte and pericyte contamination using α-smooth muscle actin (α-SMA) and glial fibrillary acidic protein (GFAP) labeling, along with positive expression of endothelial markers such as glucose tranporter-1 (GLUT-1), platelet endothelial cell adhesion molecule-1 (PECAM-1), zonula occludens-1 (ZO-1), occludin (Occ), and claudin-5 (CLD-5) using western blot, real time PCR, fluorescence activated cell sorting, and immunohistochemical techniques. Furthermore, from each donor, our BMVEC cultures have been shown to generate absolute TEER values greater than 1000 ohms (Bernas et al. 2010). Of note, quantification of α-SMA labeling has revealed approximately 2–5% pericytes in our BMVEC cultures, while GFAP labeling is negative. For expansion, BMVECs were cultured in pre-medium: DMEM/F12 media containing 10% fetal bovine serum, heparin (1 mg/ml), penicillin (100U/mL), streptomycin (10μg/ml), amphotericin B (2.5μg/ml), and endothelial cell growth supplement. After expansion, BMVEC cultures were maintained in heparin-free post-medium: DMEM/F12 media containing 10% fetal bovine serum, penicillin (100U/mL), streptomycin (10μg/ml), and amphotericin B (2.5μg/ml). Cultures were incubated at 37°C with 5% CO2 and 95% humidity. Before initiating any in vitro experiments, cells were rinsed with 1X-PBS and maintained in post-medium for at least 24 hours to remove any heparin from the culture.
Adeno-associated viral (AAV) vectors
AAV vectors were produced in 293T cells as previously described (Maguire et al. 2012). Briefly, a triple transfection of AAV and helper plasmids was performed using the calcium phosphate method. AAV vectors were extracted from cell lysates and purified by iodixanol density gradient ultracentrifugation. Next, iodixanol was removed and vector concentrated using Amicon Ultra 100 kDa molecular weight cutoff (MWCO) centrifugal devices (Millipore, Billerica, MA) and phosphate buffered saline (PBS). The final preparation was filtered through a 0.22 um Millex-GV Filter Unit (Millipore). Vectors were stored at −80°C until use. The following AAV vector constructs were used: (1) single stranded (ss) AAV construct encoding firefly luciferase (Fluc) driven by the CBA promoter (Gyorgy et al. 2014); (2) self-complementary (sc) AAV construct encoding eGFP driven by the CBA promoter kindly provided by Dr. Miguel Sena-Esteves (University of Massachusetts Medical School, Worcester, MA); (3) a ss-AAV9 construct encoding IRES-GFP driven by the CBA promoter. To titer AAV preparations, a quantitative TaqMan PCR that detects AAV vector genomes (polyA region of the transgene cassette) was performed as previously described (Maguire et al. 2012). AAV vectors encoding Fluc or eGFP using these plasmids were subsequently packaged into either AAV2 or AAV9 capsids using plasmids encoding these structural proteins. For confocal microscopy analysis of AAV capsid trafficking on BMVEC monolayer cultures, AAV2 or AAV9 capsids were labeled using the DyLight™ 488 Amine-Reactive Dye (ThermoFisher Scientific, Waltham, MA). Briefly, approximately 5 × 1011 genome copies (gc) of AAV vector were diluted into labeling buffer (0.1M sodium phosphate, 0.15M NaCl, pH 7.2) containing 50 μg of dye. After mixing thoroughly, the solution was incubated 1 hour at room temperature in the dark. Next, free dye was removed by dialyzing into PBS overnight at 4°C using 20,000 molecular weight cutoff Slide-A-Lyzer™ MINI dialysis devices (ThermoFisher Scientific). AAV capsid labeling was confirmed by placing an aliquot of each sample diluted 1:100 into PBS in a black 96 well plate and measuring fluorescence in a FlexStation 3 Multi-Mode Microplate Reader (excitation wavelength, 500 nm; emission wavelength 518 nm; cutoff filter, 515 nm). Fluorescence values of the 1:100 diluted labeled capsids samples were determined to be 10-fold over background (PBS only) levels. We observed fluorescence values of labeled AAV2 and AAV9 capsids to be within 10% of one another, indicating similar levels of labeling efficiency. Aliquots of labeled vector were prepared and stored at −80°C until use.
Immunofluorescence and microscopy
BMVEC cultures were prepared in 6-well plates on sterile 25 mm, circular glass microscopy coverslips coated with rat-tail collagen type I. Cells were seeded at a density of 2×105 and expanded in pre-medium for 2 weeks. BMVEC cultures were then rinsed with 1X-PBS and maintained in post-medium for 24 hours to remove heparin from the culture. Next, either sc-AAV2-eGFP or sc-AAV9-eGFP (both at 1×104 genome copies (gc)/cell) were suspended in post-medium and incubated with BMVECs for 4 hours. After incubation, the media was changed to remove unbound virus. BMVEC cultures were then further maintained in virus-free post-medium for 5 days to allow for eGFP expression before imaging.
A separate set of experiments was performed in under-confluent BMVEC cultures. Again, cells were seeded at a density of 2×105 and expanded in pre-medium but for only 3 days. Under-confluent cultures were then rinsed with 1X-PBS and maintained in post-medium for another 24 hours to remove heparin from the culture. BMVECs were then exposed to either sc-AAV2-eGFP or sc-AAV9-eGFP as described above. For both confluent and under-confluent conditions, control coverslips were incubated in virus-free post-medium. At the end of all experiments, cultures were rinsed with 1X-PBS, fixed with 3% ultra-pure formaldehyde, and mounted on glass microscopy slides using prolong anti-fade reagent without DAPI.
Cultures were prepared as previously described for immunocytochemistry. BMVECs (2×104) were incubated with AAV9-Fluc (2.5×105 gc/cell) for 24 hours, then rinsed, treated with pre-extraction buffer (0.2% Triton X-100), fixed using 3% ultra-pure paraformaldehyde, and permeablized with 0.05% Triton X-100. Cells were incubated overnight at 4°C with primary antibodies against Occ (1:50; Santa Cruz clone: N-19). Next, cells were washed and incubated with secondary antibodies, Alexa-488 (1:100), for 1 hour at room temperature. Finally, cells were mounted and counterstained with DAPI. Occludin signal intensity was evaluated by measurement of pixel intensity across intercellular borders. Images were captured at 10X magnification from monolayers with or without AAV exposure with camera settings kept uniform (i.e., exposure time) throughout all imaging sets. Using the NIH ImageJ (1.48v) imaging software, 5 plot profile histograms (based on the intensity/pixel) from top to bottom were generated for each image. Intensity profiles for each image were added and then averaged to the image set from each experimental condition (no virus vs virus) and graphed.
For the multi-photon experiments, BMVECs were plated at a density of 2×104 on collagen-coated MatTek glass-bottomed cultureware dishes outfitted with a 14 mm microwell (Ashland, MA). Cultures were expanded and maintained for one week as described above to remove heparin. BMVEC cultures were incubated with 2.5×106 gc/cell DyLight™-488 conjugated ss-AAV2-CBA-Fluc or ss-AAV9-CBA-Fluc for either 4 or 24 hours. At the indicated time points, BMVEC cultures were washed three times with 1X-PBS to remove unbound virus. Cells were then incubated with CellTracker-CMTPX (Invitrogen), a viability tracker dye, for 30 minutes. After CMTPX staining, cells were washed with 1X-PBS, the microwell containing BMVECs was filled with DMEM/F12 without phenol red (Life Technologies), and cultures were secured under a glass microscopy coverslip for live cell imaging.
Imaging was performed using epi-fluorescence microscopy using a CoolSNAP EZ CCD camera (Photometrics) fitted to a Nikon 80i Eclipse (Nikon). Bright field images were acquired with a DS-Fi2 Nikon camera also configured to the above microscope. Two-photon imaging was performed with a Leica TCS SP5 II MP multi-photon microscope (Leica Microsystems) configured with a tunable femto-second pulsed Mai Tai Ti:Sapphire laser (Spectra Physics) featuring a resonant scanner and non-descanned detector or NDD external detectors. A 20x water immersion objective (NA 0.95) was used for observation of the specimen and images were acquired using Leica’s LAS imaging software. Images were processed with either NIS Elements (Nikon) or the Imaris (Bitplane) imaging analysis software.
Luciferase assay
A clear bottomed, black polystyrene 96-well plate (Corning Life Sciences) was used to assess luciferase activity in BMVEC cultures incubated with AAV vectors expressing firefly luciferase. First, wells were coated with rat-tail collagen type I. Cells were then seeded at a density of 2×104 cells/well and expanded in pre-medium for 3 days. After expansion, BMVEC cultures were washed with 1X-PBS and then maintained in post-medium for another 24 hours to remove heparin from the culture. Next, vectors (AAV2-Fluc or AAV9-Fluc) were suspended in post-medium and applied to BMVEC cultures at concentrations of either 1×104 gc/cell or 1×105 gc/cell. Cultures were then incubated with the AAV vectors at 37°C with 5% CO2 for 24, 48, 72, or 120 hrs. A Luciferase Assay System (Promega) was used to evaluate AAV-mediated expression of firefly luciferase (Fluc). After incubation with AAV-Fluc vectors, cells were rinsed with 1X-PBS, lysed, and exposed to luciferase substrate, D-luciferin. An Infinite M200 PRO microplate reader with i-control™ software (Tecan) was then used to detect luciferase activity (in relative light units) in the BMVEC cultures. Luciferase activity was normalized to read-outs from 0-hour controls treated with either AAV2-Fluc or AAV9-Fluc. All treatments were performed in quadruplicate. Within the groups for each serotype, luciferase activity was analyzed as a function of the 24-hour read-outs. Between serotypes, luciferase activity was analyzed as a function of the read-outs for each time point.
BBB model and transendothelial assay
Cell culture inserts featuring transparent polyethylene terephthalate (PET) track-etched membranes (0.3 μm pores) were coated with rat-tail collagen type I and rinsed with 1X-PBS. BMVEC were then seeded onto cell culture inserts at a density of 4×104 cells/insert. BMVEC cultures were expanded in pre-medium for 3 days, washed, and then maintained in post-medium (changes every 3 days) until a confluent monolayer with a stable transendothelial electrical resistance was established, as previously described (Persidsky et al. 2006a, Ramirez et al. 2008). After assessing the integrity of the monolayer, AAV2-Fluc or AAV9-Fluc vectors were applied to the upper chamber at 2×105 gc/cell. Cultures were then incubated with the virus at 37°C with 5% CO2 for 2, 3, 6, 8, and 24 hours. At the indicated times, media was collected separately from both the upper and lower chambers, then analyzed by qPCR to quantify the number of AAV genomes present in each compartment. Media was diluted 1:10 and analyzed using the same Taqman qPCR assay as described above for the titration of purified AAV vectors. In additional experiments, BMVEC cultures were incubated with either AAV2-Fluc or AAV9-Fluc vectors at 4°C to reduce metabolic activity related to cell-mediated transport. A third set of experiments was performed as proof of concept, whereby 2×104 primary human astrocytes (ScienCell™ Research Laboratories) were seeded at the bottom of poly-L-lysine-coated wells and incubated with BMVEC cultures grown on cell culture inserts. These cultures were treated with AAV9-Fluc vectors applied to the upper chamber at 2×105 gc/cell and incubated with the virus for 24, 72, and 120 hours. At the indicated time points, astrocyte cultures were washed, lysed and evaluated for AAV9-mediated expression of luciferase using a luciferase assay as described in Materials and Methods. Luciferase activity was normalized to read-outs from 0-hour controls treated with AAV9-Fluc. All treatments were performed in quadruplicate, and luciferase activity was analyzed as a function of the 24-hour read-outs.
Permeability assay
As described above, BMVECs were grown in 24-well format cell culture inserts with collagen-coated PET track-etched membranes with 0.1 μm pores. Cultures were incubated with 2×105 gc/cell AAV9-IRES-GFP vectors for 2, 4, 6, 24, and 48 hours. At the indicated time points, BMVEC monolayers were rinsed with 1X-PBS, and phenol red minus DMEM/F12 was dispensed into wells below cell culture inserts. Tetramethylrhodamine (TMR)-labeled dextrans of two different molecular weights (3,000 and 40,000 daltons) were reconstituted in phenol minus DMEM/F12 and dispensed into the upper chamber of separate BMVEC cultures. Cell monolayers were incubated with the different weight, labeled dextrans for 20 minutes at room temperature. Following incubation, cell culture inserts were removed and media remaining in the well (bottom chamber) was analyzed for the presence of dextran-TMR using a Synergy 2 Multi-Mode microplate reader with Gen5 2.0 software (BioTek Instruments, Inc.). All treatments were performed in quadruplicate, and data were analyzed as a function of fluorescence recorded from control BMVEC monolayers exposed to AAV9 vectors at 0 hours.
Transendothelial electrical resistance
An Electrical Cell-substrate Impedance Sensor (ECIS) Zθ system produced by Applied BioPhysics (Troy, NY) was used to measure BMVEC transendothelial electrical resistance (TEER). First, cells were plated at a density of 2×104 BMVEC/well on 96W20idf PET cultureware arrays (Applied Biophysics) coated with rat-tail collagen type I. Cultures were expanded in pre-medium for 3 days, rinsed with 1X-PBS, and then maintained in post-medium for another 4 days to remove heparin. Throughout the expansion and maintenance phase, BMVEC monolayer formation was continually assessed until stable TEER baseline values were established. One-week after plating, BMVEC cultures were treated with sc-AAV2-eGFP or sc-AAV9-eGFP vectors at three different concentrations: 1×104, 1×105, and 1×106 gc/cell. Cultures were then incubated with the AAV vectors for an additional 48 hours to assess the effect of viral exposure on stable BMVEC TEER recordings. TEER measurements were continuously obtained at 1000 Hz alternating current (AC) signal settings in 30-minute intervals. TEER recordings were obtained from a single BMVEC donor with each treatment performed in triplicate for all time points. Control cultures that were never exposed to virus were used to compare TEER stability after AAV exposure. EDTA was used as a positive assay control for the reduction of baseline TEER values.
Western blotting
BMVECs were expanded to confluence and maintained as previously described in collagen-coated 6-well cell culture plates. Cultures were then exposed to AAV9 at 2.5×105 gc/cell for 24 hours. Next, cells were lysed and a BCA assay (Thermo Scientific) was used to determine protein content. Equal protein was then loaded onto a gel for each condition. Samples were mixed with 4x Laemmli buffer and boiled for 5 minutes, then loaded onto a 4–20% Mini-Protean TGX gel (Biorad). Gels were transferred to nitrocellulose membranes using the Trans-blot Turbo™ transfer system (Biorad) following the manufacturer’s protocol. Membranes were blocked with 5% milk in PBST (0.5%). All primary antibodies (ZO-1, BD Biosciences clone: 1/ZO-1; Occ, Abcam clone: EPR8208; CLD-5, Abcam clone: EPR7583, and β-actin, Sigma clone: AC-15) and secondary antibodies were dissolved in 5% milk-PBST (0.5%). After incubation with antibodies, blots were imaged with a G:Box Chemi HR16 (Syngene, Frederick, MD) gel documentation system. Control cultures never exposed to virus were used for comparison.
Flow cytometry
BMVECs were expanded and maintained as previously described, then incubated with AAV9 at 2.5×105 gc/cell for either 8 or 24 hours. At the indicated time points, cells were washed using 1X PBS without calcium and magnesium, trypsinized, and pelleted by centrifugation at 1000 rpm for 5 minutes. Next, cells were suspended in fixation buffer (ebioscience) and incubated for 10 minutes. Following fixation, cells were washed with flow cytometry buffer (ebioscience) and re-pelleted. Cells were re-suspended in 100 μL of flow cytometry buffer with pre-conjugated antibodies for ICAM-1 (PE; ebioscience clone: HA58) and VCAM-1 (APC; BD Biosciences clone 51-10C9) for 1 hour. Cells were then washed, pelleted, and re-suspended in flow cytometry buffer for analysis using a FACS BD Canto II flow cytometer (BD Biosciences). Acquisition parameters and gating were controlled by BD FACSDiva software (BD Biosciences). Data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA). All treatments were performed in triplicate. Control cultures never exposed to virus were prepared to determine baseline expression of ICAM-1 and VCAM-1. Cultures treated with recombinant IL-1β (100 ng/mL) for 18 hours were used as positive controls for ICAM-1 and VCAM-1 up-regulation.
Statistical analysis
The values shown in all figures and those mentioned throughout the text represent the average ± SEM of experiments that were performed at least 3 times (or as indicated). Statistical significance (P < 0.05) was determined by performing unpaired two-tailed Student’s t-test or via multiple group comparisons (where warranted) by ANOVA with Dunnett’s post hoc test utilizing Prism v6.04 software (GraphPad Software, Inc.).
RESULTS
Comparative analysis of AAV9 and AAV2 transgene expression in primary human BMVEC cultures
We first compared transgene expression of AAV2 and AAV9 vectors encoding eGFP under the ubiquitously utilized chicken beta actin (CBA) promoter in primary human brain microvascular endothelial cell (BMVEC) cultures. To ensure the integrity of our cultures, BMVECs from all donors were validated for both the presence of endothelial markers and the formation of barrier properties (see methods section). Furthermore, whenever AAV9 and AAV2 were compared herein, BMVECs from the same donor were utilized for both serotypes. To begin analysis of transgene expression, BMVECs from two separate donors (one male, one female) were seeded and expanded into confluent monolayers for 2 weeks, then exposed to either AAV2 or AAV9 particles expressing the eGFP reporter. Cells were incubated with the virus for a period of 4 hours, after which the media was changed to remove unbound particles. BMVEC cultures were further incubated for 5 days to allow for eGFP expression (Figure 1A, are representative images from a single BMVEC donor).
Figure 1. AAV2 outperforms AAV9 at transgene expression in primary human BMVEC cultures.

(A) Confluent BMVEC monolayers from two donors were incubated with sc-AAV2-eGFP or sc-AAV9-eGFP vectors for 4 hours at 1×104 gc/cell. Cells were then washed to remove unbound virus and incubated for 5 days to allow for eGFP expression. Fluorescent images were acquired using equal capture parameters between serotypes. Representative images acquired from one of the BMVEC donors evaluated are presented. (i–iv) Bright field and GFP images obtained from confluent BMVEC monolayers incubated with either AAV2 or AAV9; 20X objective magnification. (v–vi) GFP filter images from under-confluent BMVEC cultures incubated with either AAV2 or AAV9 with bright field inserts; 40X objective magnification. Arrows indicate expression of eGFP in BMVEC cultures. (B) Additional experiments with a firefly luciferase reporter. BMVEC cultures from a third donor were incubated with either AAV2 or AAV9 expressing firefly luciferase (Fluc) at two different concentrations (1×104 gc/cell and 1×105 gc/cell) for 24, 48, 72, and 120 hours. At the indicated time points, viral-mediated transgene expression was assessed using a luciferase assay system. Data were normalized to the 0-hour treatments for each group and are presented as mean luciferase activity in relative light units (RLU) for treatments performed in quadruplicate + SEM. Statistical significance was analyzed as a function of the 24-hour read-outs for each group. Asterisks denote p < 0.05.
As can be seen in Figure 1A-iii, epi-fluorescence microscopy imaging revealed robust eGFP expression in BMVECs transduced by self-complimentary AAV2 (sc-AAV2) vectors. In contrast, BMVECs exposed to sc-AAV9 (Figure 1A-iv) exhibited marginal to no evidence of eGFP reporter expression, indicating that unlike AAV2, AAV9 does not efficiently permit transgene expression in brain endothelial cultures under these conditions. Interestingly, when the same experiment described above was repeated with under-confluent BMVEC cultures (same donors), the number of cells expressing eGFP following AAV2 exposure was greatly enhanced at the monolayer edge (Figure 1A-v), along with some evidence of AAV9-mediated transgene expression (Figure 1A-vi) near the edge. These results suggest that cellular transduction by both AAV serotypes may be more efficient in BMVECs that have a pro-angiogenic status.
In order to test the possibility that AAV9 transduction and reporter expression may be occurring in BMVEC cultures to a degree that is simply well below the detection threshold for fluorescent microscopy, experiments were performed with AAV2 and AAV9 vectors encoding the gene for firefly luciferase (Fluc), an extremely sensitive reporter. Figure 1B demonstrates a time course analysis of enzymatic/luciferase activity (in relative light units) in BMVEC cultures (derived from a third donor; female) incubated with either AAV2 or AAV9 for 0, 24, 48, 72 or 120 hours. Within the AAV2 group (1×104 gc/cell), expression-mediated luciferase activity increased 7.3 fold (48 hours), 8.5 fold (72 hours), and 24.7 fold (120 hours), as a function of the 24-hour read-outs. Luciferase activity measurements for cells exposed to AAV9 (1×104 gc/cell) showed the following fold differences within the group: no change (48 hours), 6.6 fold (72 hours), and 12.6 fold (120 hours), again as a function of the 24-hour read-outs. In additional experiments with AAV9, we tested to see if BMVEC transduction efficiency was increased when monolayers were established on cell culture inserts. Results of these studies showed no significant change in luciferase activity fold differences at 72 hours (6.6 fold) and 120 hours (10.7 fold) when compared to BMVEC monolayers grown at the bottom of cell culture wells (Supplemental Figure 2). Notably, marked differences were observed across the groups (i.e. AAV9 versus AAV2). For example, within the lower concentration (1×104 gc/cell) at 48, 72 and 120 hours, BMVEC cultures incubated with AAV2-Fluc showed greater luciferase activity compared to cultures incubated with AAV9-Fluc, in the order of: 1501.7, 266.1 and 405.4 fold, respectively. Together, our luciferase assay and fluorescent microscopy analyses clearly show that in as many as three donors, AAV9 is much less efficient in the transduction of primary human BMVEC cultures when compared to AAV2. This is in contrast to AAV9’s performance in vivo, in which robust transduction of brain vasculature occurs after systemic delivery (Foust et al. 2009, Nature Biotech), while AAV2 transduction is minimal (Chen et al. 2009, Geoghegan et al. 2014). Supporting these earlier findings, we also observed transduction of brain endothelial cells after systemic injection of adult mice with AAV9-GFP (Supplemental Figure 1).
AAV9 transendothelial trafficking dynamics in an in vitro model of the human BBB
Using our in vitro model of the human BBB, we optimized an assay to study the properties of AAV9 trafficking across brain endothelial cells. Primary human BMVECs from two different donors (both female) were seeded on cell culture inserts featuring transparent polyethylene terephthalate (PET) membranes with pores, 0.3 microns in diameter. The two-compartment configuration allows for an upper and lower chamber to be separated by polarized BMVECs. Similar to endothelial polarity in vivo, BMVECs on cell culture inserts define an apical or “luminal” surface (exposed to the upper chamber) from a basolateral aspect of the cell (exposed to the lower chamber), which is attached to a collagen-coated membrane (acting as basal lamina), thus completing the construct validity of our in vitro model of the human BBB endothelium.
Vectors were diluted in media and added to the upper chamber of cell culture inserts, exposing the “luminal” surface of BMVECs in our BBB model to either AAV2 or AAV9. At the indicated time points, media was collected from the lower chambers and analyzed by qPCR for the presence of vector genomes, enabling the monitoring of AAV particles that traffic across the BMVEC monolayer (Figure 2A). Two hours after the addition of viral vectors, we observed a trend with AAV9 outperforming AAV2 in the number of particles trafficking into the bottom chamber. AAV9 achieved statistical significance compared to AAV2 at 3, 8, and 24 hours (Figure 2A). Therefore, similar to results produced in animal models, these analyses show that AAV9 has the ability to cross an in vitro model of the human BBB composed of primary human brain endothelial cells more efficiently than AAV2. Moreover, we performed proof-of-concept experiments with primary human astrocytes grown in the lower chamber of our in vitro BBB model in order to confirm that AAV9 vectors are able to transduce parenchymal brain cells after crossing human BMVEC monolayers. Primary human astrocytes treated with AAV9-Fluc vectors directly (no cell culture inserts containing BMVECs) were used as a positive control and show strong luciferase transgene activity, indicating efficient AAV9-mediated transduction of primary human astrocyte cultures (Figure 2B). Similarly, Figure 2B depicts luciferase activity measures recorded from primary human astrocytes 24, 72, and 120 hours after the addition of AAV9-Fluc vectors to the upper chamber of the BBB model, confirming that AAV9 vectors are intact and able to transduce parenchymal brain cells after trafficking across human BMVEC monolayers.
Figure 2. Trafficking of AAV9 across an in vitro BBB model composed of primary human BMVECs is more efficient when compared to AAV2.

(A) BMVEC cultures from two different donors exhibiting monolayer formation indicative of barrier properties were prepared on collagen-coated cell culture inserts, creating a two-compartment design to model the BBB. Cultures were incubated with either AAV2 or AAV9 vectors (2×105 gc/cell) for 2, 3, 6, 8, or 24 hours. At the indicated time points, media was collected from the bottom chamber (below the cell culture insert) and analyzed for the presence of vector DNA by qPCR. All treatments were performed in quadruplicate for both donors. Data from a single donor are presented as the mean of total AAV genomes retrieved from the bottom chamber ± SEM. Asterisks denote p < 0.05. (B) Proof-of-concept experiments were performed to show that AAV9 vectors are able to transduce parenchymal brain cells after trafficking across human BMVECs. Primary human astrocytes were seeded in the lower compartment of our BBB model, and AAV9-Fluc vectors (2×105 gc/cell) were applied to the upper compartment and incubated for 24, 72, and 120 hours. Primary astrocyte cultures incubated directly with AAV9-Fluc (no BMVECs) for 120 hours served as a positive control. At the indicated time points, AAV9-mediated transgene expression in primary human astrocytes was evaluated using a luciferase assay. Data were normalized to 0-hour controls and are presented as mean luciferase activity in relative light units (RLU) for treatments performed in quadruplicate + SEM. Statistical significance was analyzed as a function of the 24-hour read-outs. Asterisks denote p < 0.05. (C) Additional experiments were performed as described above in (A); however, BMVEC cultures from one of the donors were incubated with AAV vectors at either 37°C (physiological temperature) or 4°C, to reduce active, cell-mediated transport of AAV particles. After 3 hours, media was collected from the bottom chamber and analyzed for the presence of vector DNA by qPCR. All treatments were performed in quadruplicate. Data are presented as the mean of total AAV genomes retrieved from the bottom chamber + SEM. Asterisks denote p < 0.05.
Given the results of our transendothelial trafficking assays, additional experiments were performed to examine the various possibilities that may explain the greater movement of AAV9 vectors into the lower compartment of our in vitro BBB model. For instance, AAV9 could simply negotiate the barrier more efficiently by moving via a paracellular route (i.e. around or between endothelial cells). BMVECs were maintained under reduced metabolic conditions by placing the cultures at 4°C to slow cell-mediated transport mechanisms across the BBB model. Figure 2C demonstrates the results of transcytosis assays that were performed with AAV2 and AAV9 incubated with BMVECs from a single donor (female) at normal, physiological conditions (37°C) or at lower temperatures (4°C), to reduce cell-mediated transportation. These results show the amount of virus retrieved from the lower chamber after 3 hours of incubation. As can be seen in Figure 2C, paracellular movement of AAV2 is significantly affected by the decrease in temperature, showing a slight increase in the amount of AAV2 detected in the lower chamber at 4°C compared to 37°C. Conversely, AAV9 was drastically affected by the temperature change in the opposite direction compared to AAV2, exhibiting a steep reduction in the number of viral particles trafficking into the lower chamber from 37°C to 4°C. Further analysis of this data revealed a reduction of 89.7 % in the number of AAV9 particles recovered from the bottom chamber at 4°C. Together, these results suggest that AAV9 greatly depends on active, energetic cellular systems to transport the virus across BMVECs, while endothelial crossing by AAV2 appears to be independent of active cell-mediated transport and is most likely a function of paracellular diffusion.
Effects of AAV9 on BMVEC barrier integrity
As a front-runner gene therapy delivery vehicle for the treatment of CNS disorders in humans, it is important to determine whether AAV9 exposure may compromise BBB integrity by destabilizing cellular adhesion throughout the endothelium. Furthermore, in continuing to address the different possibilities that may explain the greater movement of AAV9 across our in vitro BBB model, experiments were prepared to assess an alternative mechanism of paracellular trafficking. The results from Figure 2A show a steady increase in AAV9 accumulation in the lower chamber over time, raising the possibility that the virus may induce a breach in barrier integrity, allowing viral particles to pass into the lower chamber through a compromised paracellular route. To this end, studies were performed to evaluate the status of endothelial barrier integrity by acquiring measurements of transendothelial electrical resistance (TEER) in BMVEC cultures. Once BMVECs form a monolayer, mature TJ complexes develop, which bestow upon the endothelial monolayer characteristics typical of the BBB, such as TEER. This measure of electrical resistance provides an analytical method for directly evaluating experimental conditions that may induce barrier “tightness”/higher resistance or “leakiness”/lower resistance. TEER measurements were acquired as described in Materials and Methods. BMVEC cultures from a single donor (female) were allowed to reach confluence and steady state TEER for one-week (last 10 hours are shown) prior to the addition of increasing concentrations of either AAV2 or AAV9 (arrow, Figure 3A). As can be seen, both AAV2 and AAV9 had no significant effect on BMVEC barrier integrity when compared to control cultures never exposed to virus (blue line). Furthermore, the presence of AAV particles (at concentrations ranging from 1×104 to 1×106 gc/cell) in BMVEC cultures did not appear to affect BMVEC barrier integrity either acutely or in a chronic manner, as TEER measurements do not fluctuate significantly during the time course tested. As expected, EDTA treatment caused an immediate drop in TEER, as EDTA chelates calcium needed to maintain TJ complex assembly and cellular adhesion thus inducing barrier “leakiness”.
Figure 3. AAV9 does not affect in vitro indicators of BMVEC barrier integrity.

(A) BMVEC cultures from a single donor were prepared on collagen-coated electrode arrays designed for use with an Electric Cell-substrate Impedance System. Transendothelial electrical resistance (TEER) readings were acquired continuously in 30-minute intervals for one week. After BMVEC cultures established stable TEER readings, cells were exposed to either AAV2 or AAV9 at three different concentrations (1×104 gc/cell, 1×105 gc/cell, and 1×106 gc/cell) for 48 hours. All treatments were performed in triplicate. Cultures never exposed to virus were used as stable baseline controls. EDTA was used as a control for the reduction of baseline TEER values. Data are presented as the normalized change in resistance from baseline TEER + SEM over a 45-hour period. Right-hand y-axis shows absolute TEER values (Ohms • cm2). Arrow indicates addition of AAV vectors to BMVEC cultures with stable TEER readings (last 10 hours shown). Statistical analysis revealed no significant changes in TEER between AAV treated cultures and controls. (B) BMVECs exhibiting monolayer formation indicative of barrier properties were prepared on collagen-coated cell culture inserts and incubated with AAV9 vectors (2×105 gc/cell) for 0, 2, 4, 6, 24 and 48 hours. At the indicated times, permeability measures were determined using two tetramethylrhodamine (TMR)-labeled dextrans (3 kDa and 40 kDa) applied to the upper compartment for 20 minutes. Media from the lower compartment was then analyzed for the presence of dextran-TMR tracers. Data are presented as mean fluorescence (RU) + SEM for treatments performed in quadruplicate. Statistical analyses revealed no significant difference between cultures incubated with AAV9 and 0-hour controls. NS denotes p > 0.05. (C-E) Additional BMVEC cultures were incubated with AAV9 (2.5×105 gc/cell) for 24 hours in order to determine the cellular localization and relative expression of key tight junction (TJ) proteins that help to stabilize BBB integrity. BMVEC cultures not exposed to AAV were used as controls. (C) Immunocytochemistry reveals cellular localization of occludin (Occ) at intercellular borders consistent with TJ formation both in control cultures and in the presence of AAV9. (D) The histogram represent inserts values from intensity/pixel from intensity line profile drawn across the images. (E) On the basis of the intensity line profiles from sets of N=5, the graph of the average ± SEM shows no statistical significance observed between no virus and AAV9 exposed cultures. (F) Western blots depict similar expression levels of ZO-1, Occ, and claudin-5 (CLD-5) in BMVEC control cultures and those incubated with AAV9. β-actin is presented as a loading control for equal protein.
A second set of experiments was performed to test whether BMVEC barrier integrity is compromised by the presence of AAV; and furthermore, to establish that AAV9 does not cross our in vitro model of the human BBB more efficiently due to increased paracellular trafficking. BMVECs were seeded on cell culture inserts featuring PET membranes with pores, 0.1 micron in diameter. Cultures were prepared as previously described and exposed to AAV9 vectors for 2, 4, 6, 24, and 48 hours. At the indicated time points, permeability measures were obtained using two different molecular weight tetramethylrhodamine (TMR)-labeled dextrans (3,000 and 40,000 daltons) as described in Material and Methods. BMVEC monolayers exposed to AAV9 vectors at 0 hours were used as controls. As anticipated, the 3 kDa dextran penetrated our endothelial barriers to a slightly greater extent than the 40 kDa dextran. Importantly, the detection of fluorescent signal from each tracer appeared constant in the presence of AAV9 when compared to controls (Figure 3B). Furthermore, statistical analyses revealed no significant difference in fluorescence between control cultures or time zero when compared to those exposed to AAV9 (Figure 3B), thus corroborating the TEER data.
Additional studies were performed to determine whether AAV9 affects the cellular localization and relative expression of ZO-1, claudin-5 (CLD-5), and occludin (Occ), three principle components of TJ complexes. Figure 3C indicates that the cellular distribution or pattern of Occ at intercellular borders is unaffected by AAV9 exposure (2.5×105 gc/cell for 24 hours). Quantitation via intensity line profiles of images taken form the immunolabeling of Occ revealed no significant difference from controls in the presence of AAV9 (Figure 3D and E). Likewise, western blot analyses of whole cell lysates collected from BMVEC cultures incubated with AAV9 for 24 hours (2.5×105 gc/cell) show no change in the relative expression of ZO-1 (195 kDa), CLD-5 (20 kDa), and Occ (65 kDa) when compared to controls (Figure 3F). Together, results of from the TEER, permeability, immunostaining, and western blot supports the notion that transendothelial trafficking of AAV9 unlikely occurs by an increase in paracellular flux or TJ remodeling. Moreover, these results suggest that exposure to AAV9 does not compromise BMVEC barrier integrity in multiple human donors.
Activation status of BMVECs after AAV9 exposure
Viral infection and exposure to viral proteins may initiate an inflammatory response that can have deleterious effects on the BBB endothelium. Notably, few studies have investigated the effect of AAV exposure on endothelial activation, or more specifically, the pro-inflammatory activation status of human BMVECs. Although a variety of tests can be performed to assess pro-inflammatory status, up-regulation of cellular adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) is a clear and universal sign of endothelial activation. Notably, these adhesion molecules facilitate leukocyte-endothelial interactions at the BBB. Endothelial cells respond to warning signals such as viral DNA by quickly enhancing surface expression of ICAM-1 and VCAM-1 in order to recruit immune cells to sites where pathogens or damage have been detected. To test the possibility that exposure to AAV9 could activate a pro-inflammatory response in endothelial cells of the human BBB, surface expression of ICAM-1 and VCAM-1 were analyzed in primary human BMVEC cultures. As before, BMVECs from a single donor (male) were incubated with AAV9 for either 8 or 24 hours. Figure 4A-D show representative histograms acquired from cytometric analysis of cells immunostained with antibodies against ICAM-1 (Figure 4A, B) and VCAM-1 (Figure 4C, D). Detection of ICAM-1 and VCAM-1 in cultures exposed to AAV9 nearly overlap with untreated BMVEC controls, indicating that primary human brain endothelial cells are not activated by incubation with AAV9 for either of the two time points tested. In comparison, BMVEC cultures exposed to IL-1β for 18 hours were used as positive controls for the induction of endothelial adhesion molecules. Figure 4A–D show a distinct shift in the detection of both adhesion molecules in BMVECs after the addition of IL-1β. Bar graphs demonstrating the average mean fluorescence Intensity (MFI) of ICAM-1 and VCAM-1 from multiple experiments can be seen in Figure 4E and Figure 4F, respectively. Furthermore, statistical analyses reveal no significant difference in the MFI of either ICAM-1 or VCAM-1 in BMVEC cultures exposed to AAV9 compared to untreated controls. Therefore, results of these experiments support the hypothesis that AAV9 exposure does not promote a pro-inflammatory response in brain endothelial cells.
Figure 4. BMVECs maintain baseline activation status after AAV9 exposure.

BMVEC cultures from a single donor were incubated with AAV9 (2.5×105 gc/cell) for either 8 or 24 hours. At the indicated time points, BMVECs were harvested to measure the surface expression of cellular adhesion molecules (ICAM-1 and VCAM-1) by flow cytometry. All treatments were run in triplicate. Cultures never exposed to virus were used as controls for the baseline expression of ICAM-1 and VCAM-1. BMVECs treated with recombinant IL-1β for 18 hours were used as positive controls. (A–D) Representative histograms depicting the shift in ICAM-1 and VCAM-1 expression in BMVEC cultures treated with AAV9 compared to controls after 8 or 24 hours as indicated. (E–F) Data are presented as the average mean fluorescence intensity (MFI) + SEM of ICAM-1 and VCAM-1 in BMVEC cultures exposed to AAV9 for either 8 or 24 hours compared to controls. NS denotes p > 0.05. Hashtags denote p < 0.05.
AAV localization and the effect of endothelial trafficking on transduction and transcytosis
To better understand how AAV9 traffics across the BBB, live cell imaging with multiphoton microscopy was employed with BMVEC monolayers exposed to either AAV2 or AAV9 for a comparative analysis. BMVEC cultures from a single donor (female) were prepared as previously described and exposed to AAV particles conjugated to a fluorescent dye. Cultures were incubated with the fluorescently labeled AAV particles for either 4 or 24 hours. Unbound virus was removed prior to imaging as described in the Materials and Methods. Also, prior to imaging, the BMVEC cytoplasm was labeled with a viability tracker dye for observation under the red channel, while labeled AAV particles were visualized under the green channel. Figure 5A, C, I, and K all show volumetric images from a field of view from BMVEC cultures exposed to either AAV2 (A and C) or AAV9 (I and K). The white circle and arrow were inserted to identify areas under close observation in Figure 5B, D, J and L. In these figures, the volumetric rendered Z-stacks are subdivided into XY, YZ and XZ planes. X and Y are the top coordinates, and the Z plane segments through both the X and Y planes (color coded). Once segmented, the volume can be flipped and visualized on its side, generating YZ and XZ planes.
Figure 5. Cellular localization of AAV9 and AAV2 in primary human BMVEC cultures.

BMVECs from a single donor were seeded in MatTek dishes outfitted with a 14 mm glass-bottomed microwell. Next, cultures were expanded and maintained for one week as previously described to remove heparin. BMVEC cultures were then incubated with DyLight™-488 (green signal) conjugated AAV2 or AAV9 particles (2.5×106 gc/cell) for either 4 or 24 hours. Following incubation, cells were washed to remove unbound virus, stained with a viability tracker dye, CMTPX (red signal), and imaged using multiphoton live cell microscopy. (A, C, I, K) Representative fields of view acquired from BMVEC cultures exposed to fluorescently labeled AAV2 or AAV9 for 4 or 24 hours as indicated. White circles with arrows identify areas under close observation. (B, D, J, L) High-resolution images of fluorescently labeled AAV capsid clusters visualized in XZ and YZ planes. White arrowheads point to intracellular accumulations of either AAV2 or AAV9 as indicated. Green circles with arrows identify fluorescently labeled AAV capsid clusters at the basolateral aspect of the XZ plane (below BMVEC monolayers). (E, G, M, O) Extracellular, DyLight™-488 conjugated AAV capsid clusters at the basolateral side of BMVEC cultures incubated with either AAV2 or AAV9 for 4 or 24 hours as indicated. Using NIH Image J software, AAV capsid clusters were quantified by converting the fluorescent images to binary and performing particle counting based on area and circularity. (F, H, N, P) Inverted images derived from binary conversion of the fluorescent AAV capsid signal used for particle counting in NIH Image J software.
As can be seen in Figure 5B and J, conjugated AAV2 and AAV9 particles both localize to the apical surface of BMVECs at 4 hours (particles identified by arrows in the YZ and XZ planes). Notably, in this example, and others that were investigated (data not shown), labeled virus was not found at areas where two cells meet (i.e. intercellular junctions). Interestingly, upon closer inspection, funnel-like structures appeared to form at the contact points where labeled AAV particle clusters interact with the apical surface of BMVECs. Another observation, particularly for AAV2 (Figure 5B, XZ), is the presence of oval shaped cellular inclusions that are likely vesicular accumulations of AAV2 undergoing endocytosis. Furthermore, diffuse nuclear localization of the AAV capsid could be observed at 24 hours in additional BMVEC cultures exposed to fluorescently labeled AAV2 (Movie 1). Alternatively, internalized AAV9 capsids were clearly visible inside of the cytoplasmic compartment of BMVECs (arrows at XZ and YZ). As depicted in the XZ aspect of Figure 5L, vesicular accumulations of AAV9 appear smaller and more tubular than the AAV2 conditions shown in Figure 5. In some instances, these tubular structures appear to span the entire cell from apical to basolateral surfaces. Interestingly, in contrast to AAV2, there was no accumulation of fluorescently labeled AAV9 capsid clusters either surrounding or within BMVEC nuclei (Movie 2).
Outside of the cell, AAV capsid clusters are readily apparent at high magnification. Figure 5E, G, M and O all show the basolateral aspect of BMVEC cultures (XY of the XZ plane) taken from images under close observation (Figure 5B, J, D, and L; follow the green arrow). These images represent what is immediately below the BMVEC monolayer. Image analysis performed by particle counting corroborates the results of our transcytosis assays, which revealed an increase in the number of AAV genomes in the bottom chamber over time, and more importantly, greater penetration of the endothelial monolayer by AAV9 compared to AAV2. Figure 5F and H show a low level of AAV2 in the basolateral aspect of BMVEC cultures at 4 hours, which increases 6 fold by 24 hours. Likewise, analysis of AAV9 revealed an 11-fold increase in the number of particles present at the basolateral surface of BMVEC cultures from 4 to 24 hours. More importantly, quantification of the number of labeled viral capsid clusters passing through the BMVEC culture to the basolateral surface indicates a substantial increase in the amount of AAV9 compared to AAV2 at both time points: 9 fold at 4 hours, and 16 fold at 24 hours. Moreover, image analyses at 24 hours clearly show a large portion of fluorescently labeled AAV2 capsid clusters remaining inside of BMVECs; whereas, almost all of the AAV9 capsid cluster signal can clearly be seen in the extracellular plane below the endothelial monolayer (Supplementary Movie 1 and 2).
The results of our multiphoton imaging strongly suggest that AAV9 does not use a paracellular but rather a transcellular route for negotiating passage across primary human BMVEC cultures. Furthermore, differences between the intracellular, tubular-shaped transport vesicles observed between AAV2 and AAV9 may explain 1) how AAV2 may be more efficient in the transduction of BMVECs in vitro by endosomal trafficking to the nucleus, while 2) AAV9 is more efficient in moving across endothelial cells and entering the basolateral compartment.
DISCUSSION
In summary, we have validated and characterized an in vitro model of the human BBB as a means to study AAV transduction and transcytosis. With regards to the transduction of primary human BMVECs, our data have shown that AAV2 outcompetes AAV9. On the contrary, the results of our transendothelial trafficking assays suggest that AAV9 achieves greater penetration across BMVEC monolayers when compared to AAV2, supporting observations in vivo (Foust et al. 2009, Gray et al. 2011, Samaranch et al. 2012, Bevan et al. 2011, Duque et al. 2009, Yang et al. 2014). Furthermore, our data are the first to indicate that AAV9 passage across human BMVECs is an energetic process. Most reports have focused on the ability of AAV9 to transduce parenchymal brain cells in animal models after intravascular delivery and rarely address the mechanism regulating AAV9 passage into the CNS. In addition, this report uniquely addresses the importance of BBB integrity specifically in primary human BMVEC cultures. The safety profile of AAV9 toward BBB integrity appears very favorable, as our data indicate that human BMVECs maintain TEER, permeability measures, relative expression and cellular localization of TJ proteins, and baseline expression of adhesion molecules. Notably, these data also rule against paracellular trafficking of AAV9 across a compromised BBB, and instead support a hypothesis of transcellular extravasation of systemically injected AAV9 across an intact barrier. Finally, using multiphoton microscopy, we have observed three-dimensional localization of AAV2 and AAV9 in primary human BMVECs. These images reveal an almost complete basolateral distribution of AAV9 below BMVEC monolayers, while AAV2 maintains greater intracellular distribution with some perinuclear/nuclear accumulation.
The difference between AAV2 gene transfer and that of AAV9 is most likely related to how the wild type capsid of these serotypes interacts with BMVEC surface receptors to promote cellular transduction. Studies have shown that during viral transduction, AAV2 binds to heparin sulfate proteoglycans (HSPGs), is packaged inside of endosomal-like structures that traffic to the nucleus along microtubule bundles, followed by migration into the nucleus through nuclear pore complexes (Xiao & Samulski 2012, Xiao et al. 2012, Nicolson & Samulski 2014). Collectively, our data with AAV2 reflect these processes as we observed greater intracellular accumulation with nuclear/perinuclear distribution of fluorescently labeled AAV2 capsids, followed by more efficient transduction compared to AAV9, and fewer AAV2 genomes in the bottom chamber during our transendothelial trafficking assays.
Interestingly, using confocal microscopy, recent studies have shown that HSPGs are primarily expressed at the basolateral aspect of endothelial cells (Stoler-Barak et al. 2014). Therefore, the observation that more BMVECs were transduced by AAV2 at the monolayer edge (Figure 1A-v) may be a function of increased HSPG availability or the absence of mature TJ complexes. Conversely, within confluent regions, AAV2 transduction may be limited by lower HSPG expression at the apical surface of the BMVEC monolayer or the presence of fully formed TJ complexes refracting viral transduction. Although AAV2 transduction requires cell-mediated activity, Figure 2B indicates that AAV2 transcytosis may occur due to passive diffusion. Again, the unique distribution pattern of HSPGs on the endothelial surface may explain the significant increase in AAV2 passage across BMVECs incubated at 4°C compared to 37°C. Apical HSPGs are enriched at paracellular endothelial junctions where TJ complexes are expressed (Stoler-Barak et al. 2014). At normal physiological temperatures, BMVEC TJ complexes retain functional integrity of the endothelial monolayer, potentially limiting the penetration of AAV2 particles that localize near paracellular junctions by binding to HSPGs expressed in this region. However, under reduced metabolic conditions at 4°C, BMVEC TJ complexes may be destabilized, allowing AAV2 particles to pass between endothelial cells. Importantly, our results reveal the opposite effect for AAV9, whereby the transcytosis of virus is significantly elevated at 37°C compared to 4°C (Figure 2B). This finding suggests that unlike AAV2, cell-mediated activity likely accounts for the movement of AAV9 across our in vitro model of the human BBB.
Unlike AAV2, the AAV9 capsid does not appear to use HSPGs for cellular transduction. Current reports have provided evidence that AAV9 possesses a galactose binding domain required for cellular transduction (Bell et al. 2012, Shen et al. 2011, Bell et al. 2011). Although the galactose binding domain of AAV9 has been identified as a functional site for cellular binding/transduction, perhaps galactose binding also facilitates transendothelial trafficking at the BBB (Bell et al. 2011). Considering the results of our transduction and transcytosis assays along with our multiphoton imaging of fluorescently labeled AAV, it is likely that AAV9 accesses a pathway that allows vectors to pass through endothelial cells without leading to nuclear translocation. This conclusion is supported by a previous study that characterized the transport of multiple AAV serotypes across a variety of endothelial cells, finding that transduction efficiencies could be enhanced by blocking AAV transcytosis with tannic acid (Di Pasquale & Chiorini 2006). Results of this study suggest that some AAV serotypes, such as AAV4 and bovine AAV, may exhibit poor transduction efficiency due to an enhanced capacity for cellular transcytosis (Di Pasquale & Chiorini 2006). Furthermore, by inhibiting transcytosis without altering capsid structure, and in turn enhancing the transduction efficiency of select serotypes, these data indicate AAV transduction and transcytosis may be regulated by binding to the same residue (i.e. HSPGs or galactose, depending on serotype), while subtleties in intracellular trafficking ultimately determine the distribution and subsequent effect of AAV vectors.
Notably, one of the first reports to demonstrate that AAV9 requires galactose binding for viral transduction also showed that ricin agglutinin (RCA), a β-galactose binding protein, co-localizes with CD31, a marker for endothelial cells, in tissue sections obtain from the mouse brain (Bell et al. 2011, Raub & Audus 1990). These results suggest that AAV9 may utilize galactose residues expressed on BMVECs to penetrate the CNS. This possibility is not unfounded as studies in microbiology have shown that Balamuthia mandrillaris, a protozoan that crosses the BBB leading to life-threatening infections (namely Balamuthia amoebic encephalitis), expresses a galactose binding protein that interacts with primary human BMVECs and facilitates entry into the brain (Matin et al. 2007). Furthermore, prior studies have shown that the surface of bovine BMVECs is dominated by galactosylated glycoconjugates that are rapidly internalized upon RCA binding through a process known as adsorptive endocytosis (Raub & Audus 1990). Notably, these investigations have also shown that endosomes produced during this process are recycled back to the endothelial surface within minutes, where RCA is released into the extracellular media (Raub & Audus 1990). Thus, AAV9 may traffic across the BBB and be released into the CNS through a similar process. Furthermore, just as we report with AAV9, studies have shown that both the endocytosis and efflux of RCA are energy-dependent processes (Raub & Audus 1990). Intriguingly, adsorptive endocytosis is a mechanism that blood-borne human immunodeficiency virus may use to cross the BBB and infect susceptible cell types in the CNS (Banks et al. 1998, Banks et al. 2001). Therefore, the possibility that AAV9 uses similar mechanisms such as galactose binding and adsorptive endocytosis to cross the BBB endothelium and access parenchymal cell types in the CNS deserves further investigation.
We foresee many applications for our primary human BBB model relevant to current gene therapy research. First, identifying receptors specifically utilized by AAV9 or AAV capsid mutants with enhanced transduction efficiency may help to target apical BMVEC surface receptors that access desirable pathways, such as those leading to transcytosis versus those leading to cellular transduction. Second, we have previously found that CNS transduction with systemically administered AAV9 is significantly higher in female mice than male mice, subsequently observed in in rats (Maguire et al. 2013, Jackson et al. 2015). Therefore, it will be interesting to compare the ability of AAV9 to cross human BMVECs derived from male versus female patients. Furthermore, BMVECs from male and female donors may be tested in the presence or absence of sex hormones, as estrogen is a known stimulator of transcytosis (Burgess & Stanley 1997, Diebel et al. 2011). Such a study would have direct relevance to potential dose efficacy in putative female versus male patients receiving intravenous AAV9 gene therapy. Third, our in vitro BBB model should be useful for screening novel AAV’s and AAV capsid libraries for serotypes with more efficient transendothelial trafficking. This approach may help to identify novel proteins and cellular pathways involved in BBB transcytosis.
In conclusion, we have shown that AAV2 mediates gene transfer to primary human brain endothelial cells more efficiently than AAV9, while AAV9 engages in more permissive transendothelial trafficking across BMVEC monolayers compared to AAV2. Our data also indicate that AAV9 trafficking across an in vitro BBB model does not occur due to passive paracellular diffusion, but instead occurs because of active cell-mediated transport mechanisms. Furthermore, we found that AAV9 does not appreciably alter barrier integrity or the activation status of BMVECs that form the human BBB. Future work aimed at uncovering the different mechanisms that regulate AAV9 transcytosis from AAV2 transduction in human BMVEC cultures may provide valuable information for gene therapy researchers developing neurotherapeutics for the treatment of CNS disorders.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health/National Institute on Drug Abuse (NIH/NIDA) T32 DA007237 (SFM and AMA), NIH/National Institute of Neurological Disorders and Stroke (NINDS) R01 NS086570-01 (SHR), The Shriners Hospitals for Children 85110-PHI-14 (SHR), and the American Brain Tumor Association Discovery Grant (CAM). We would like to thank the laboratory of Dr. Yuri Persidsky for the use of key equipment and technical expertise of lead scientist Nancy Lee Reichenbach.
Abbreviations
- CNS
Central nervous system
- BBB
blood-brain barrier
- AAV
adeno-associated virus
- BMVEC
brain microvascular endothelial cells
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature reviews. Neuroscience. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Banks WA, Akerstrom V, Kastin AJ. Adsorptive endocytosis mediates the passage of HIV-1 across the blood-brain barrier: evidence for a post-internalization coreceptor. Journal of cell science. 1998;111(Pt 4):533–540. doi: 10.1242/jcs.111.4.533. [DOI] [PubMed] [Google Scholar]
- Banks WA, Freed EO, Wolf KM, Robinson SM, Franko M, Kumar VB. Transport of human immunodeficiency virus type 1 pseudoviruses across the blood-brain barrier: role of envelope proteins and adsorptive endocytosis. Journal of virology. 2001;75:4681–4691. doi: 10.1128/JVI.75.10.4681-4691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CL, Gurda BL, Van Vliet K, Agbandje-McKenna M, Wilson JM. Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. Journal of virology. 2012;86:7326–7333. doi: 10.1128/JVI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CL, Vandenberghe LH, Bell P, Limberis MP, Gao GP, Van Vliet K, Agbandje-McKenna M, Wilson JM. The AAV9 receptor and its modification to improve in vivo lung gene transfer in mice. The Journal of clinical investigation. 2011;121:2427–2435. doi: 10.1172/JCI57367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernas MJ, Cardoso FL, Daley SK, et al. Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nature protocols. 2010;5:1265–1272. doi: 10.1038/nprot.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan AK, Duque S, Foust KD, et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Molecular therapy : the journal of the American Society of Gene Therapy. 2011;19:1971–1980. doi: 10.1038/mt.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess JW, Stanley KK. Estrogen-stimulated transcytosis of desialylated ligands and alpha2 macroglobulin in rat liver. Biochimica et biophysica acta. 1997;1359:48–58. doi: 10.1016/s0167-4889(97)00087-6. [DOI] [PubMed] [Google Scholar]
- Chen YH, Chang M, Davidson BL. Molecular signatures of disease brain endothelia provide new sites for CNS-directed enzyme therapy. Nature medicine. 2009;15:1215–1218. doi: 10.1038/nm.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pasquale G, Chiorini JA. AAV transcytosis through barrier epithelia and endothelium. Molecular therapy : the journal of the American Society of Gene Therapy. 2006;13:506–516. doi: 10.1016/j.ymthe.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Diebel ME, Diebel LN, Liberati DM. Gender dimorphism in the gut: mucosal protection by estrogen stimulation of IgA transcytosis. The Journal of trauma. 2011;71:474–479. doi: 10.1097/TA.0b013e318228239d. [DOI] [PubMed] [Google Scholar]
- Duque S, Joussemet B, Riviere C, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Molecular therapy : the journal of the American Society of Gene Therapy. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nature biotechnology. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Muenzer J, Samulski RJ, Breese G, Sifford J, Zeng X, McCarty DM. Self-complementary adeno-associated virus serotype 2 vector: global distribution and broad dispersion of AAV-mediated transgene expression in mouse brain. Molecular therapy : the journal of the American Society of Gene Therapy. 2003;8:911–917. doi: 10.1016/j.ymthe.2003.08.021. [DOI] [PubMed] [Google Scholar]
- Geoghegan JC, Keiser NW, Okulist A, Martins I, Wilson MS, Davidson BL. Chondroitin Sulfate is the Primary Receptor for a Peptide-Modified AAV That Targets Brain Vascular Endothelium In Vivo. Molecular therapy. Nucleic acids. 2014;3:e202. doi: 10.1038/mtna.2014.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Molecular therapy : the journal of the American Society of Gene Therapy. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorgy B, Fitzpatrick Z, Crommentuijn MH, Mu D, Maguire CA. Naturally enveloped AAV vectors for shielding neutralizing antibodies and robust gene delivery in vivo. Biomaterials. 2014;35:7598–7609. doi: 10.1016/j.biomaterials.2014.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KL, Dayton RD, Klein RL. AAV9 supports wide-scale transduction of the CNS and TDP-43 disease modeling in adult rats. Molecular therapy. Methods & clinical development. 2015;2:15036. doi: 10.1038/mtm.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WD, Qu G, Sferra TJ, Clark R, Chen R, Johnson PR. Adeno-associated virus-mediated gene transfer to the brain: duration and modulation of expression. Hum Gene Ther. 1999;10:201–213. doi: 10.1089/10430349950018995. [DOI] [PubMed] [Google Scholar]
- Maguire CA, Balaj L, Sivaraman S, et al. Microvesicle-associated AAV vector as a novel gene delivery system. Molecular therapy : the journal of the American Society of Gene Therapy. 2012;20:960–971. doi: 10.1038/mt.2011.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire CA, Crommentuijn MH, Mu D, Hudry E, Serrano-Pozo A, Hyman BT, Tannous BA. Mouse gender influences brain transduction by intravascularly administered AAV9. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:1470–1471. doi: 10.1038/mt.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire CA, Ramirez SH, Merkel SF, Sena-Esteves M, Breakefield XO. Gene therapy for the nervous system: challenges and new strategies. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2014;11:817–839. doi: 10.1007/s13311-014-0299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matin A, Siddiqui R, Jung SY, Kim KS, Stins M, Khan NA. Balamuthia mandrillaris interactions with human brain microvascular endothelial cells in vitro. Journal of medical microbiology. 2007;56:1110–1115. doi: 10.1099/jmm.0.47134-0. [DOI] [PubMed] [Google Scholar]
- Nicolson SC, Samulski RJ. Recombinant adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. Journal of virology. 2014;88:4132–4144. doi: 10.1128/JVI.02660-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Bu J, Richards AM, et al. Translational fidelity of intrathecal delivery of self-complementary AAV9-survival motor neuron 1 for spinal muscular atrophy. Human gene therapy. 2014;25:619–630. doi: 10.1089/hum.2014.011. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE) Blood. 2006a;107:4770–4780. doi: 10.1182/blood-2005-11-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2006b;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- Ramirez SH, Heilman D, Morsey B, Potula R, Haorah J, Persidsky Y. Activation of peroxisome proliferator-activated receptor gamma (PPARgamma) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV-1 infected monocytes. Journal of immunology. 2008;180:1854–1865. doi: 10.4049/jimmunol.180.3.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashnonejad A, Chermahini GA, Li S, Ozkinay F, Gao G. Large-Scale Production of Adeno-Associated Viral Vector Serotype-9 Carrying the Human Survival Motor Neuron Gene. Molecular biotechnology. 2015 doi: 10.1007/s12033-015-9899-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raub TJ, Audus KL. Adsorptive endocytosis and membrane recycling by cultured primary bovine brain microvessel endothelial cell monolayers. Journal of cell science. 1990;97(Pt 1):127–138. doi: 10.1242/jcs.97.1.127. [DOI] [PubMed] [Google Scholar]
- Samaranch L, Salegio EA, San Sebastian W, et al. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Human gene therapy. 2012;23:382–389. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Bryant KD, Brown SM, Randell SH, Asokan A. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. The Journal of biological chemistry. 2011;286:13532–13540. doi: 10.1074/jbc.M110.210922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler-Barak L, Barzilai S, Zauberman A, Alon R. Transendothelial migration of effector T cells across inflamed endothelial barriers does not require heparan sulfate proteoglycans. International immunology. 2014;26:315–324. doi: 10.1093/intimm/dxt076. [DOI] [PubMed] [Google Scholar]
- Vannucci L, Lai M, Chiuppesi F, Ceccherini-Nelli L, Pistello M. Viral vectors: a look back and ahead on gene transfer technology. The new microbiologica. 2013;36:1–22. [PubMed] [Google Scholar]
- Varadi K, Michelfelder S, Korff T, Hecker M, Trepel M, Katus HA, Kleinschmidt JA, Muller OJ. Novel random peptide libraries displayed on AAV serotype 9 for selection of endothelial cell-directed gene transfer vectors. Gene therapy. 2012;19:800–809. doi: 10.1038/gt.2011.143. [DOI] [PubMed] [Google Scholar]
- Wirth B, Barkats M, Martinat C, Sendtner M, Gillingwater TH. Moving towards treatments for spinal muscular atrophy: hopes and limits. Expert opinion on emerging drugs. 2015;20:353–356. doi: 10.1517/14728214.2015.1041375. [DOI] [PubMed] [Google Scholar]
- Xiao PJ, Li C, Neumann A, Samulski RJ. Quantitative 3D tracing of gene-delivery viral vectors in human cells and animal tissues. Molecular therapy : the journal of the American Society of Gene Therapy. 2012;20:317–328. doi: 10.1038/mt.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao PJ, Samulski RJ. Cytoplasmic trafficking, endosomal escape, and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network. Journal of virology. 2012;86:10462–10473. doi: 10.1128/JVI.00935-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Li S, Wang H, et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Molecular therapy : the journal of the American Society of Gene Therapy. 2014;22:1299–1309. doi: 10.1038/mt.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.