

Abstract
Emerging evidence suggests an important role for epigenetic mechanisms in modulating signals during macrophage polarization and inflammation. JMJD3, a JmjC family histone demethylase necessary for M2 polarization is also required for effective induction of multiple M1 genes by lipopolysaccharide (LPS). However, the effects of JMJD3 to inflammation in the context of obesity remains unknown. To address this deficiency, we firstly examined the expression of JMJD3 in macrophage isolated from bone marrow and adipose tissue of diet induced obesity (DIO) mice. The results indicated that JMJD3 was down-regulated in obesity. Adiponectin (APN), a factor secreted by adipose tissue which is down-regulated in obesity, functions to switch macrophage polarization from M1 to M2 (Jenke et al., 2014), thereby attenuating chronic inflammation. Intriguingly, our results indicated that APN contributed to JMJD3 up-regulation, reduced macrophage infiltration in obese adipose tissue, and abolished the up-regulation of JMJD3 in peritoneal macrophages isolated from DIO mice when challenged with Porphyromonas gingivalis LPS (pg.lps). To elucidate the interaction of APN and JMJD3 involved in macrophage transformation in the context of inflammation, we designed the loss and gain-function experiments of APN in vivo with APN−/− mice with experimental periodontitis and in vitro with macrophage isolated from APN −/− mice. For the first time, we found that APN can help to reduce periodontitis-related bone loss, modulate JMJD3 and IRF4 expression and macrophage infiltration. Therefore, it can be inferred that APN may contribute to anti-inflammation macrophage polarization by regulating JMJD3 expression, which provides a basis for macrophage-centered epigenetic therapeutic strategies.
Keywords: macrophage, epigenetic, adiponectin, JMJD3, periodontitis
Introduction
Periodontitis is a chronic inflammatory disease modified by inflammatory host response and characterized by the progressive destruction of the tooth-supporting tissues (Pihlstrom et al., 2005). It has a significant negative impact on a wide range of physical, psychological and social aspects of life in affected individuals (Beikler and Flemmig, 2011; O’Dowd et al., 2010). Based on data from the National Health and Nutrition Examination Survey III, it is estimated that half of the U.S. population aged over 30 years suffers from periodontitis (Albandar, 2011).
In recent years, there has been intense interest in potential associations between periodontal disease and various chronic systemic diseases. Although published studies have identified associations between them, very little is known about the molecular mechanism. The cross-reactivity of local and systemic inflammation remains controversial and the debate continues regarding two questions: which of the local specialized tissues are affected by systemic inflammatory changes, and how local specific inflammatory processes impact the whole body (Amar et al., 2007; Ebersole et al., 2010; Hajishengallis, 2010). Such a dynamic response requires an intricate network of cellular and non-cellular components (Hasturk et al., 2012).
In inflammatory processes, macrophages are key cells, which determine whether the inflammation advances to chronic pathological changes or resolves with no damage. As such, macrophages play an important role in homeostatic processes, which is vital to the host. Emerging evidence suggests an important role for epigenetic mechanisms in modulating signals during macrophage polarization. Gene expression is epigenetically regulated through DNA methylation, acetylation, phosphorylation, ubiquitination, sumoylation, and methylation of histones. Histone methylation state is dynamically regulated by histone methyltransferases and demethylases. JMJD3 was found to catalyze the demethylation of H3K27me2/3 in vitro and activate gene expression. It has been reported that JMJD3 expression was induced in macrophages by Toll-like receptor (TLR) stimuli, and Irf4 was proved to be a JMJD3 target gene, which is crucial for controlling M2 polarization(De Santa et al., 2007; Satoh et al., 2010). However, little has been mentioned about the epigenetic role of macrophage during inflammatory resolution of periodontitis.
Activated macrophages polarize towards various functional phenotypes depending on the cytokines expressed in the microenvironment (Gordon and Martinez, 2010; Lawrence and Natoli, 2011; Mosser and Edwards, 2008; Sica and Mantovani, 2012). The best characterized macrophage activation phenotypes are classical activation (also termed M1) induced by microbial products such as TLR ligands, and alternative activation (M2) induced by the T helper (Th)- cytokines interleukin (IL)-4 and IL-13. M1 macrophages are effective at host defense and pathogen removal, and M2 macrophages are important for tissue repair. Deeper understanding of epigenetic regulation of macrophage phenotype will enable the development of gene-specific therapeutic approaches to enhance host defense while preserving tissue integrity and preventing chronic inflammatory diseases.
APN, a factor secreted by adipose tissue, is down-regulated in obesity. It functions to switch macrophage polarization from M1 to M2 and then attenuates chronic inflammation (Jenke et al., 2014). APN has been reported to counteract the effects of P. gingivalis on oral epithelial cells. Low levels of APN expression in obese individuals can increase the risk for periodontal destruction (Kraus et al., 2012). The epigenetic mechanism underlying how APN affects the macrophage state might enable the development of potential associations between periodontitis and obesity.
The study exhibits that APN can ameliorate the periodontal bone loss and macrophage infiltration in DIO mice with periodontitis. According to the counteractions of APN and pg.lps, we hypothesize that APN might contribute to anti-inflammation by involving in JMJD3-IRF4 axis, which is required for M2 polarization in macrophages. The study may shed light on macrophage-centered epigenetic therapeutic strategies for periodontitis in the context of obesity.
Materials and Methods
Cell culture
Bone marrow-derived macrophages (BMDM) were obtained by differentiating bone marrow from 12-week-old C57BL/6J mice (wild-type or APN−/− mice) in the presence of recombinant mouse Colony Stimulating Factor-1 (10 ug/ml, CSF-1) for 7 days. On day 7, BMDM were harvested and plated in complete media containing CSF-1 for treatment on day 8.
Thioglycollate-elicited peritoneal macrophages (TEPM) were generated by injecting 1 ml of 3% thioglycollate broth into the peritoneal cavity of 12-week-old C57BL/6J mice (wild-type & dietinduced obesity mice, and APN−/− mice), followed by peritoneal lavage with PBS 5 days later. All animal studies were reviewed and approved by the appropriate Tufts University animal ethics committee.
The RAW264.7 cell line was obtained from ATCC. BMDM and TEPM were cultured in RPMI-1640 media supplemented with 10% FBS, 20 U/mL penicillin, 20 U/mL streptomycin and 2 mM L-glutamine. RAW264.7 cells were cultured as for BMDM and TEPM. All cells were cultured at 37 °C and 5% CO2.
Cell treatment with pg.lps and/or APN
RAW264.7 cells were stimulated with ultrapure LPS from P. gingivalis (Cayla-InvivoGen, Toulouse, France) with different concentrations (10 ng/ml, 50 ng/ml, 100 ng/ml) at variant time points(1 h, 2 h, 4 h,). According to the initial results, pg.lps with the concentration of 10 ng/ml for 2 and 4 hours was applied in the further experiment in vitro. When the cells (RAW264.7 cells, BMDM, TEPM) were stimulated by pg.lps and recombinant mouse APN (1 mg/ml; R&D Systems, Wiesbaden, Germany), the concentrations of the APN applied were based on the results from other studies to ensure that data are comparable (Kamio et al., 2009; Kraus et al., 2012).
Quantification of mRNA by real-time PCR
Total RNAs were extracted from samples from cultured cells and tissues with Trizol reagent (Invitrogen) to study the expressions of JMJD3, IRF4 proinflammatory cytokines (TNF-a, IL-1β), and markers of macrophage phenotype (F4/80, Arginase 1, CCL3, CCL4). Levels of mRNA encoding various gene products were determined by quantitative real-time PCR (qRT-PCR). qRT-PCR assays were performed with VeriQuest Fast SYBR Green qPCR Master Mix (Affymetrix) using a Bio-Rad iQ5 thermal cycler. Primers used for amplification are listed in sTable 1.
Immunofluorescence
Cells were passaged on coverslips and cultured for 24 hours, fixed in 4% paraformaldehyde (Sigma-Aldrich) and treated with PBS containing 0.1% Triton X-100 (Sigma-Aldrich) for 15 min. Next, slides were blocked with 10% BSA and incubated with a rabbit anti-JMJD3 antibody (Abcam) in 1:300 dilutions. The slides were then cultured with Alexa Fluor 488 Dye (2 ug/ml, Life Technologies) at 37°C for 1 hour. ProLong® Gold Antifade Reagent with DAPI (Life Technologies) was used to stain the nucleus and mount for fluorescence microscopic analysis.
Western blot analysis
Cell proteins were separated by SDS–PAGE as previously described (Tu et al., 2011) and transferred to Immobilon (PVDF) transfer membranes. Blots were blocked in 5% non-fat dry milk in Tween-Tris buffered saline (TTBS) and incubated overnight at 4 °C with primary antibodies NF-κB and pNF-κB (1:500), respectively, in blocking buffer. Blots were then treated with secondary antibody (horseradish peroxidase-linked goat-anti-rabbit IgG,Santa Cruz Biotechnology, Inc.) in TBS buffer (1:2,000) for 1 hour at room temperature and developed using SuperSignal® West Dura Extended Duration Substrate (Thermo Fisher Scientific Inc., Waltham, MA). Bound antibody was visualized by chemiluminescence exposure of X-ray film.
Culture and preparation of Porphyromonas gingivalis
Porphyromonas gingivalis (P. gingivalis, ATCC®) was cultured and maintained in supplemented tryptic soy broth (ATCC®) in an anaerobic chamber with 85% N2, 10% H2, and 5% CO2 at 37°C. 6-0 silk sutures were presoaked in the broth containing P. gingivalis (108/ml) prior to periodontitis induction.
Establishment of mouse experimental periodontitis model
To elucidate whether APN may counteract critical actions of P. gingivalis on periodontal tissues, experimental periodontitis was established in APN−/− mice (Jax #008195) and WT mice (Jax #000664). APN−/− mice were maintained as reported previously (Tu et al., 2011). 12-week-old male mice were used in this study. APN−/− mice were divided into 3 groups (n = 5): periodontitis only, periodontitis+ APN infusion, and control without periodontitis and APN infusion. WT mice (n = 5) were used as negative control. Experimental periodontitis was induced as previously described (Zhang et al., 2014). For APN infusion, an Alzet micro-osmotic pump (model 1004, Durect Corporation) containing 1 mg/ml of APN was inserted subcutaneously in the back of each mouse following periodontitis induction. The pumps delivered 2.5 μg of recombinant APN per day. Mice were euthanized 10 days after periodontitis induction.
Alveolar bone loss analysis
After euthanasia, the mouse palatal bone samples were dissected and defleshed after 15 minutes in boiling water, immersed overnight in 3% hydrogen peroxide, and stained with 1% methylene blue. The buccal and palatal faces of the molars were photographed at 30× magnification using a dissecting microscope with the occlusal faces of the molars positioned perpendicular to the base. The distance from the cementoenamel junction to the alveolar crest was measured at six sites of secondary molar: mesio-buccal, mid-buccal, disto-buccal, disto-palatal, mid-palatal and mesio-palatal using Image-Pro Plus software. All bone measurements were repeated twice per site(Amar et al., 2007; Zhang et al., 2014).
Histology analysis
Palatal bone samples were fixed in 4% paraformaldehyde and decalcified in 10% EDTA. Tissue sections were stained with hematoxylin and eosin (H&E), and interdental areas between the first and second molars were examined. The total number of inflammatory cells was manually counted from 4 separate fields captured at 400× magnification on H&E-stained sections. Data were reported as the numbers of polymorphonuclear and mononuclear leukocytes per square millimeter.
Results
JMJD3 was transiently up-regulated in macrophage while challenged with pg.lps
To explore the role of demethylases in macrophages cell fate while challenged with pg.lps, expression of histone demethylases was systemically profiled in RAW264.7 cells stimulated with different concentrations of pg.lps at variant time points by real-time PCR. JMJD3 was identified to be up-regulated in RAW264.7 cells while stimulated with pg.lps (10 ng/ml, 50 ng/ml, 100 ng/ml) for 2h and 4h (Fig. 1A), and pro-inflammatory cytokines, such as TNF-a and IL-1β were up-regulated earlier than JMJD3 (Fig. 1B&C).
Fig. 1. LPS increased JMJD3 expression via regulating NF-κB.
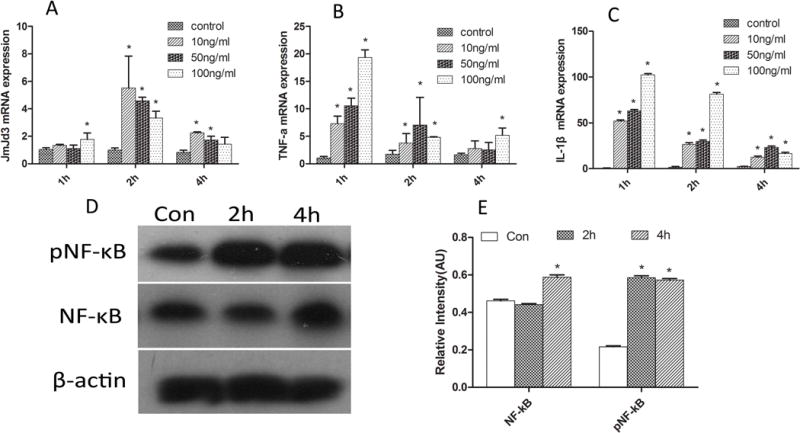
A: JMJD3 was transiently up-regulated in macrophage while challenged with pg.lps; B,C: The expression of inflammatory cytokines such as TNF-a and IL-1β were up-regulated; D&E: western blots results exhibited that pNF-κB and NF-κB increased significantly in RAW264.7 cells while stimulated with pg.lps (10ng/ml) for 2h and 4h. * P<0.05 vs control group
The results of western blots exhibited that pNF-κB and NF-κB increased significantly in RAW264.7 cells while stimulated with pg.lps (10 ng/ml) for 2 and 4 hours (Fig. 1D&E). The results of immunocytochemistry confirmed the up-regulation of JMJD3 in RAW264.7 cells (Fig. 2A–F). The up-regulation of JMJD3 was confirmed in primary BMDM and TEPM macrophage isolated from WT while stimulated with pg.lps (data was not shown).
Fig. 2. Immunocytochemistry results confirmed the up-regulation of JMJD3 in RAW264.7 cells while challenged with LPS.
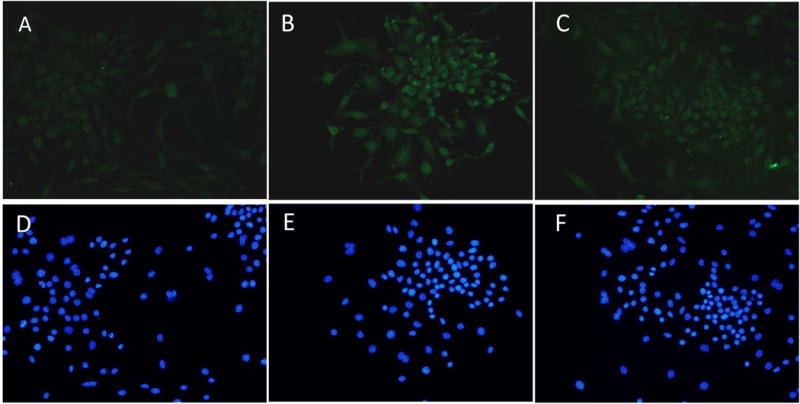
A&D: control group; B&E: RAW264.7 cells were stimulated with pg.lps (10ng/ml) for 2h; C&F: RAW264.7 cells were stimulated with pg.lps (10ng/ml) for 4h
Regulation of JMJD3 contributed to the weakened innate immunity in the context of DIO
TEPM were isolated separately from DIO mice and challenged with pg.lps (10 ng/ml). RT-PCR results exhibited that JMJD3 was significantly up-regulated in macrophages of DIO while challenged with pg.lps (10 ng/ml) (Fig. 3A), although proinflammatory cytokines (TNF-a, IL-1β) were unable to be up-regulated simultaneously (Fig. 3B&C). To further explore the modification of JMJD3 in the context of obesity, a JMJD3 expression was detected by RT-PCR in bone marrow, gingival tissues, and adipose tissues of DIO mice. The results exhibited that JMJD3 expression was significantly decreased in the context of obesity (Fig. 3D).
Fig. 3. JMJD3 contributed to the weakened innate immunity in the context of DIO.
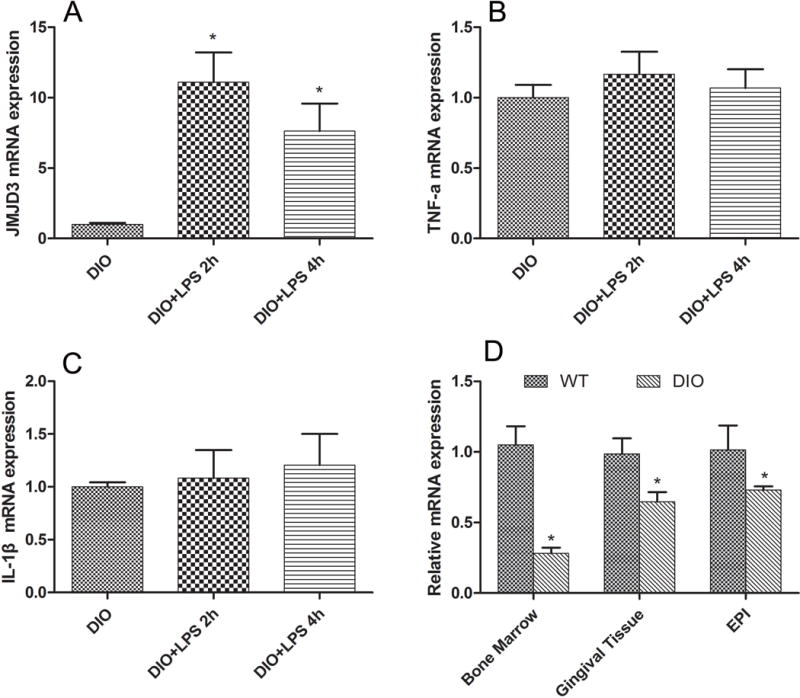
A: The gene expression of JMJD3 was significantly up-regulated in macrophages of DIO while challenged with pg.lps (10ng/ml) * P<0.05 vs DIO group; B&C: The gene expression of proinflammatory cytokines (TNF-a, IL-1β) cannot be up-regulated in DIO macrophage while challenged with LPS; D: The JMJD3 gene expression was significantly decreased in gingival tissue, bone marrow and adipose tissues in the context of obesity. EPI: adipose tissue, * P<0.05 vs WT group.
APN ameliorated the periodontal bone loss in vivo
The experimental periodontitis was successfully established in the APN−/− mice, and the staining results of methylene blue exhibited that the alveolar bone loss in APN−/− mice with periodontitis was much severe than that in WT mice with periodontitis (0.290483 ± 0.025713 mm vs 0.13237 ± 0.0512 mm). More interestingly, APN infusion significantly ameliorated alveolar bone loss (0.2225 ± 0.002894 mm) (Fig. 4). There was no alveolar bone loss in WT and APN−/− mice without periodontitis.
Fig. 4. Adiponectin ameliorated the periodontal bone loss in vivo.
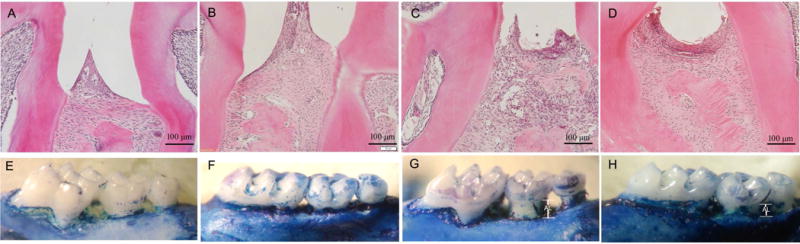
A~D: H&E staining of thepalatal bone; E~H: Palatal bone samples stained with 1% methylene blue. The palatal bone samples exhibited no difference between control group and APN−/− group, APN−/− mice with periodontitis exhibited much severe bone loss and system APN treatment significantly decreased alveolar bone loss associated with experimental periodontitis in APN−/− mice. A, E: control group; B,F: APN−/−; C,G: APN−/− with periodontitis; G,H: APN−/− with periodontitis and APN treatment.
Involvement of APN with the JMJD3-IRF4 axis in the context of periodontitis
To investigate whether APN regulates periodontitis via JMJD3-IRF4 in vivo, RT-PCR was performed to detect the expression of JMJD3 and IRF4. The results exhibited that the expression of JMJD3 and IRF4 was lower in gingival tissues from APN−/− mice with periodontitis than that in WT mice, and systemic APN treatment reversed the down-regulation of JMJD3 and IRF4 expressions (Fig. 5 A&B).
Fig. 5. Adiponectin contributed to anti-inflammation by regulating jmjd3-irf4 axis.
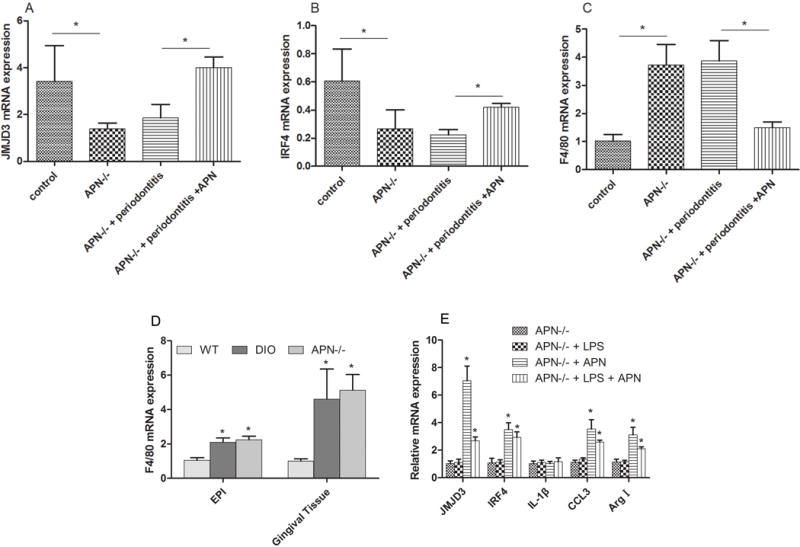
A: The expression of JMJD3 was decreased in gingival tissues from APN−/− mice and APN−/− mice with periodontitis, and systemic APN treatment rescued the downregulation effect of JMJD3 expression.
B: The mRNA expression of IRF4 in gingival tissues was decreased in APN−/− mice and APN−/− mice with periodontitis compared with control group, and APN treatment rescued the downregulation effect of IRF4 expression.
C: the expression of F4/80 was up-regulated in gingival tissues from APN−/− mice and APN−/− mice with periodontitis, and systemic APN treatment decreased the expression of F4/80 expression in gingival tissues.
D: RT-PCR results exhibited thatloss function of adiponectin increased the expression of F4/80 in the adipose tissue and gingival tissues; EPI:adipose tissue;
E: RT-PCR results exhibited that while challenged with pg.lps (10ng/ml), APN−/− BMDM cannot have adequate up-regulation of JMJD3, IRF4, CCL3, arginase I and pro-inflammatory cytokines (IL-1β). Exogenous adiponectin rescued the expression of JMJD3, IRF4, CCL3 and arginase 1, and had no effect on the expression of IL-1β. * P<0.05
To determine the possible involvement of macrophage in the periodontitis, F4/80 expression was detected by RT-PCR. The results exhibited that compared with WT group, the expression of F4/80 showed 3.7-fold and 3.86-fold increase in APN−/− mice and APN−/− mice with periodontitis, respectively (Fig 5C). APN infusion decreased the expression of F4/80 by 61% compared with APN−/− mice with periodontitis.
APN contributed to the infiltration of macrophage by involving in JMJD3-IRF4 axis
To investigate whether APN contributes to the infiltration of macrophage, RT-PCR was performed to detect the expression of F4/80, which is a marker of macrophage. The results exhibited that compared with WT mice, the expression of F4/80 in adipose tissue from DIO mice and APN−/− mice showed 2.1-fold and 2.3-fold increase respectively. In gingival tissues, the expression of F4/80 exhibited even higher increase, with 4.6-fold and 5.1-fold respectively (Fig. 5D).
To further elucidate the interaction of APN and JMJD3 involved in macrophage transformation in the context of inflammation, loss and gain-function experiments of APN were designed in vitro with BMDM isolated from the APN−/− mice. RT-PCR results exhibited that while challenged with pg.lps (10 ng/ml), APN deficiency blocked the up-regulation of JMJD3, IRF4, CCL3, arginase I and pro-inflammatory cytokines (IL-1) in BMDM isolated from APN−/− mice as shown in WT BMDM. However, APN infusion induced a 7.1-fold increase of JMJD3 expression, 3.5-fold increase of IRF4 expression, 3.5-fold increase of CCL3 and 3.1-fold increase of arginase I in these BMDMs (Fig. 5E).
Discussion
Cumulated evidences indicated that the phenotype of the macrophage is central to determine the fate of the resolving or chronic lesion. However, little is known about the epigenetic molecular events in this complex system. In the present study, the possible epigenetic regulation was initially explored in the context of periodontitis and obesity. Intriguingly, we found that APN could ameliorate the periodontal bone loss by regulating the JMJD3-IRF4 axis, which was necessary for M2 macrophage transformation.
It has been reported that JMJD3 was essential to the effective induction of multiple LPS-inducible genes (De Santa et al., 2007; Lee et al., 2014). In the present study, we found that LPS up-regulated the expression of NF-κB and promoted the phosphorylation of NF-κB. Besides, LPS also induced the up-regulation of JMJD3 expression. As previous research reported that JMJD3 was quickly induced by NF-κB in primary mouse macrophages when stimulated with inflammatory (Das et al., 2012; De Santa et al., 2007), these results indicated that LPS might increase JMJD3 expression via regulating NF-κB activity.
The expressions of TNF-α and IL-6 was reduced in DIO animals exposed to oral infection, which indicated that DIO animals developed a blunted inflammatory response. Previous research also demonstrated that when exposed to P. gingivalis, peritoneal macrophages from DIO mice exhibited reduced levels of proinflammatory cytokines, which means that DIO in mice induced changes in host immune responses to bacterial challenge (Amar et al., 2007). Our results demonstrated that when macrophages from DIO mice were treated with LPS, the expression of JMJD3 was up-regulated, however, the expressions of IL-1 and TNF-α were not affected. As IL-1 and TNF-α played important roles in M1 polarization (He et al., 2015), which was effective at host defense and pathogen removal, these results indicated that M1 polarization was blunted during the process of inflammation in the context of DIO (Mills, 2012). However, the promotion of JMJD3 by LPS was not affected in DIO mice, which demonstrated that JMJD3 might not be involved in the process of M1 polarization. These results were consistent with the previous research by De Santa (De Santa et al., 2007), which proved that although JMJD3 is a TLR-inducible gene in macrophages, JMJD3 is dispensable for M1 macrophages.
More interestingly, we found that JMJD3 expression decreased in the bone marrow, gingival tissue and adipose tissue in DIO mice compared with WT mice. This provided the information that although JMJD3 is dispensable for M1 polarization, JMJD3 might still involve in regulation of obesity. Another macrophage polarization is M2 polarization, which has been proved involve in tissue remodeling and repair (Sugg et al., 2014). However, little has been known about the role of M2 polarization in obesity and the regulation of JMJD3 in this process.
APN is a hormone secreted by adipocytes and it regulates energy homeostasis and lipid metabolism (Yamauchi et al., 2002). The protein expression was decreased in diabetes and insulin resistance (Fisman and Tenenbaum, 2014). However, very little is known about epigenetic events involved in APN under different conditions of host such as obesity. Whether APN regulated M2 polarization via JMJD3 is still not clear. In order to detect the effects of APN on JMJD3-IRF4 axis, we created experimental periodontitis in APN−/− mice. Our results exhibited that in the gingival tissues of APN−/− mice and DIO mice, the expression of JMJD3 and IRF4 were down-regulated. As JMJD3-IRF4 axis has been proved regulating M2 macrophage polarization, these results indicated that APN might epigenetically regulate M2 polarization via JMJD3-IRF4 axis.
As we know, M2 polarization played an important role in tissue repair. These results might explain the reason why tissue repair was delayed in diabetes. In order to further verify the effects of APN on tissue repair, we created periodontitis model in APN−/− mice. APN deficiency increased the bone resorption of periodontitis and exogenous APN reflesh inhibited bone resorption and promoted bone repair in periodontitis. These results were consistent with the previous research done by Lan Zhang et al (Zhang et al., 2014). They demonstrated that APN reduced bone loss in experimental periodontitis; besides, APN reduced the number of osteoclasts and ameliorated the infiltration of inflammatory cells in APN−/− mice with periodontitis. Taken together, the results suggested that APN epigenetically regulated inflammatory response via JMJD3 and inhibited bone resorption in periodontitis.
The current research found that APN−/− promoted the migration of macrophage toward adipose and gingival tissues. We also found that APN−/− decreased JMJD3 expression in vivo. Next, we further explored the epigenetic regulation of APN on M2 polarization in vitro. It was reported that JMJD3-IRF4 axis regulates M2 macrophage polarization and host responses (Satoh et al., 2010), APN promotes macrophage polarization to an anti-inflammatory phenotype. The loss and gain-of function experiments exhibited that JMJD3 and IRF4 were down-regulated in APN−/− macrophage, and exogenous APN can rescue the abnormal expression of JMJD3 and IRF4. We postulated that APN might regulate M2 polarization via JMJD3-IRF4 axis. Next, we detected the expression of CCL3, and Agr1, which are the core factors of M2 macrophage polarization, in the macrophages in APN−/− mice. We found that in loss of-function experiment APN decreased the expressions of Agr1 and CCL3, and exogenous addition of APN could rescue the expression of these factors. These results indicated that APN regulated the process of M2 macrophage polarization. More interestingly, our results demonstrated that the expression tendency of Agr1, CCL3 and CCL4 is corresponding with JMJD3 and IRF4. The results above proved that JMJD3 involved in the process of M2 polarization. Therefore, our research demonstrated that APN regulated M2 polarization and JMJD3-IRF4 axis involved in this process. However, the detailed mechanism about APN and JMJD3-IRF4 axis on M2 polarization needs further research.
In conclusion, decreased JMJD3 contributed to the weakened immunity response for the periodontitis in the context of obesity by regulating macrophage polarization, and APN ameliorated the periodontal bone loss by up-regulating the expression of JMJD3-IRF4 axis.
Supplementary Material
Acknowledgments
This project was supported by National Institutes of Health (NIH) grants R01DE16710 and R01DE21464, and the International Association of Dental Research (IADR) and the Academy of Osseointegration (AO) Innovation in Implant Science Award (to J.C).
Footnotes
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Albandar JM. Underestimation of periodontitis in NHANES surveys. Journal of periodontology. 2011;82:337–341. doi: 10.1902/jop.2011.100638. [DOI] [PubMed] [Google Scholar]
- Amar S, Zhou Q, Shaik-Dasthagirisaheb Y, Leeman S. Diet-induced obesity in mice causes changes in immune responses and bone loss manifested by bacterial challenge. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:20466–20471. doi: 10.1073/pnas.0710335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beikler T, Flemmig TF. Oral biofilm-associated diseases: trends and implications for quality of life, systemic health and expenditures. Periodontology 2000. 2011;55:87–103. doi: 10.1111/j.1600-0757.2010.00360.x. [DOI] [PubMed] [Google Scholar]
- Das ND, Jung KH, Choi MR, Yoon HS, Kim SH, Chai YG. Gene networking and inflammatory pathway analysis in a JMJD3 knockdown human monocytic cell line. Cell biochemistry and function. 2012;30:224–232. doi: 10.1002/cbf.1839. [DOI] [PubMed] [Google Scholar]
- De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Ebersole JL, Steffen MJ, Holt SC, Kesavalu L, Chu L, Cappelli D. Systemic inflammatory responses in progressing periodontitis during pregnancy in a baboon model. Clinical and experimental immunology. 2010;162:550–559. doi: 10.1111/j.1365-2249.2010.04202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisman EZ, Tenenbaum A. Adiponectin: a manifold therapeutic target for metabolic syndrome, diabetes, and coronary disease? Cardiovascular diabetology. 2014;13:103. doi: 10.1186/1475-2840-13-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G. Complement and periodontitis. Biochemical pharmacology. 2010;80:1992–2001. doi: 10.1016/j.bcp.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A, Van Dyke TE. Oral inflammatory diseases and systemic inflammation: role of the macrophage. Frontiers in immunology. 2012;3:118. doi: 10.3389/fimmu.2012.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, Kou X, Luo Q, Yang R, Liu D, Wang X, et al. Enhanced m1/m2 macrophage ratio promotes orthodontic root resorption. J Dent Res. 2015;94:129–139. doi: 10.1177/0022034514553817. [DOI] [PubMed] [Google Scholar]
- Jenke A, Holzhauser L, Lobel M, Savvatis K, Wilk S, Weithauser A, et al. Adiponectin promotes coxsackievirus B3 myocarditis by suppression of acute anti-viral immune responses. Basic research in cardiology. 2014;109:408. doi: 10.1007/s00395-014-0408-y. [DOI] [PubMed] [Google Scholar]
- Kamio N, Akifusa S, Yamaguchi N, Nonaka K, Yamashita Y. Anti-inflammatory activity of a globular adiponectin function on RAW 264 cells stimulated by lipopolysaccharide from Aggregatibacter actinomycetemcomitans. FEMS immunology and medical microbiology. 2009;56:241–247. doi: 10.1111/j.1574-695X.2009.00573.x. [DOI] [PubMed] [Google Scholar]
- Kraus D, Winter J, Jepsen S, Jager A, Meyer R, Deschner J. Interactions of adiponectin and lipopolysaccharide from Porphyromonas gingivalis on human oral epithelial cells. PloS one. 2012;7:e30716. doi: 10.1371/journal.pone.0030716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nature reviews Immunology. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- Lee HT, Kim SK, Kim SH, Kim K, Lim CH, Park J, et al. Transcription-related element gene expression pattern differs between microglia and macrophages during inflammation. Inflammation research: official journal of the European Histamine Research Society [et al] 2014;63:389–397. doi: 10.1007/s00011-014-0711-y. [DOI] [PubMed] [Google Scholar]
- Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Critical reviews in immunology. 2012;32:463–488. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature reviews Immunology. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dowd LK, Durham J, McCracken GI, Preshaw PM. Patients’ experiences of the impact of periodontal disease. Journal of clinical periodontology. 2010;37:334–339. doi: 10.1111/j.1600-051X.2010.01545.x. [DOI] [PubMed] [Google Scholar]
- Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nature immunology. 2010;11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. The Journal of clinical investigation. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugg KB, Lubardic J, Gumucio JP, Mendias CL. Changes in macrophage phenotype and induction of epithelial-to-mesenchymal transition genes following acute Achilles tenotomy and repair. Journal of orthopaedic research: official publication of the Orthopaedic Research Societ. 2014;32:944–951. doi: 10.1002/jor.22624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Q, Zhang J, Dong LQ, Saunders E, Luo E, Tang J, et al. Adiponectin inhibits osteoclastogenesis and bone resorption via APPL1-mediated suppression of Akt1. The Journal of biological chemistry. 2011;286:12542–12553. doi: 10.1074/jbc.M110.152405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nature medicine. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- Zhang L, Meng S, Tu Q, Yu L, Tang Y, Dard MM, et al. Adiponectin ameliorates experimental periodontitis in diet-induced obesity mice. PloS one. 2014;9:e97824. doi: 10.1371/journal.pone.0097824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.