

Abstract
As the most mutated gene in cancer, it is no surprise that TP53 has been the center of cancer biology discourse since its discovery in the late 1970s. Although early demonstrations of p53’s role in the modulation of cell proliferation and survival solidified its classification as a tumor suppressor and transcription factor, our conceptualization of p53 is ever-evolving. Here, we present novel evidence of the role of alternative splicing isoforms, truncating/separation-of-function mutations, and hotspot silent mutations in the regulation of p53’s activities.
Keywords: p53, p53 isoforms, p53-psi, Alternative splicing, Loss-of-function, Gain-of-function, Separation-of-function, Truncating mutations, Silent mutations, Hot-spot mutations
INTRODUCTION
p53 has long been revered by cancer biologists as the “Guardian of the Genome” [1]. It first emerged as a character in cancer discourse in the late 1970s and has since become one of the most studied genes with more than 80,000 related publications to date [2]. The functional complexity of p53 is well reflected by the scientific community’s evolving understandings of its role in cancer biology. Initially, p53 was conceptualized as a proto-oncogene; however, further investigations revealed that p53 primarily functions as a tumor suppressor [3–6]. While p53’s activities as a tumor suppressor have since been widely validated, recent findings indicate that certain p53 mutations could actually have pro-tumorigenic activities. These contemporary discoveries once again challenge the scientific community’s classification of this gene [1,3,6–9].
As a tumor suppressor, p53 regulates a number of cellular functions that are necessary for cell growth and survival. Specifically, activated p53 can induce transient growth arrest, DNA repair, autophagy, senescence, apoptosis and necrosis [10–17]. p53 is also shown to regulate physiological processes such as development, reproduction, self-renewal, and metabolism [18–23].
While p53 regulates the majority of the aforementioned processes by modulating the expression of downstream target genes as a transcription factor, p53 regulates several other processes in a transcription-independent manner (Figure 1) [12–15]. Structure-function analysis of p53 demonstrated the presence of five distinct domains that enable its transcriptional activities: i) a transcriptional activation domain (TAD); ii) a proline-rich domain (TAD-II/PRD); iii) a sequence-specific DNA-binding domain (DBD); iv) a tetramerization domain (TET) and; v) nuclear localization sequences (NLS) (Figure 1)[24]. In addition, p53 has also been shown to interact with and modify the function of other proteins, including but not limited to: BCL-2, BCL-XL, Bax, mTOR, AMPK, PTEN, SMAD3, p73 and p63 [10,19,24–31].
Figure 1.

P53 structure and function. A) Structure-function schematic of p53 showing the presence of five distinct domains that enable its transcriptional activities: i) a transcriptional activation domain (TAD); ii) a proline-rich domain (TAD-II/PRD); iii) a sequence-specific DNA-binding domain (DBD); iv) a tetramerization domain (TET) and; v) nuclear localization sequences (NLS).
The breadth of p53’s biological functions and interactions is centered on its crosstalk with multiple signaling networks that ensure the precise regulation of its expression, subcellular localization and stability via multiple covalent modifications (e.g. phosphorylation, acetylation, methylation, ubiquitination) and protein-protein interactions (Figure 1) [24]. In particular, one of the best-characterized primary negative regulators of p53 is the MDM2 gene. MDM2 encodes for an E3 ubiquitin ligase that induces proteasome-based degradation of p53. MDM2 also inhibits the transcriptional activity of p53 by directly binding to its transactivation domain [10,11,32].
In addition to the previously mentioned regulators of p53’s activity, it is now well known that p53’s function may be amended by means of alternative splicing. In fact, TP53 produces multiple protein isoforms with unique biochemical properties (Figure 2) [28,33]. For example, alterative splicing of intron-9 produces the p53-beta and p53-gamma isoforms, while the alternative splicing of intron-2 generates the Δ40p53-alpha, Δ40p53-beta, Δ40p53- gamma isoforms [34]. The expression of some of these isoforms is documented in multiple types of cancer such as breast, AML, melanoma, ovarian and head and neck cancers [33]. Although their specific functional roles are still under investigation, recent data suggest these p53 isoforms participate in tumorigenesis and metastatic dissemination [35].
Figure 2.
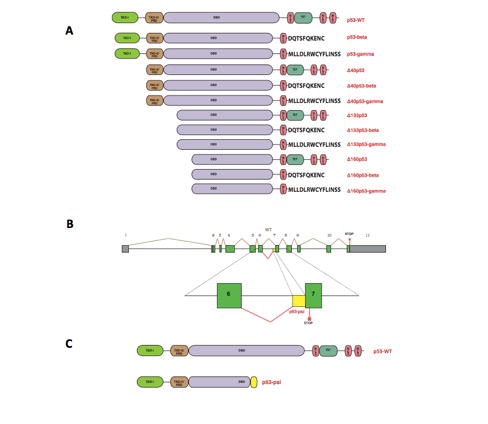
TP53 alternative splicing isoforms. A) Diagram of twelve p53 protein isoforms. B) Schematic of human p53 gene structure and of the alternative splicing mechanism leading to the generation of the p53-psi isoform. C) Comparison of p53-WT and p53-psi protein structures.
Recently, Senturk et al., identified a thirteenth p53 splicing isoform. This novel isoform is generated by the use of a cryptic acceptor site in intron-6, which leads to a frame shift in the coding region that introduces a stop codon in exon-7 (Figure 2) [36]. As a result, this alternative splicing generates a truncated gene product, named p53-psi, that is primarily expressed in tissues exposed to chronic stress conditions and in tumors with high metastatic potential [36]. Further molecular analysis of p53-psi showed that this isoform lacks the majority of the DNA binding domain, nuclear localization sequences, and tetramerization domain that are present in the wild-type protein. Thus, p53- psi is devoid of transcriptional activities and canonical tumor suppressor functions [36]. However, p53-psi is still able to localize to the mitochondria where it can interact with Cyclophilin D (CypD), a major regulator of the mitochondria permeability transition pore (MPTP) [36] (Figure 3). Consequently, p53- psi increases the mitochondria permeability and the levels of mitochondria reactive oxygen species [36]. Interestingly, Senturk et al., also demonstrated that the p53-psi/CypD interaction is necessary for the reprograming of cells to acquire mesenchymal features (Figure 3) [36].
Figure 3.
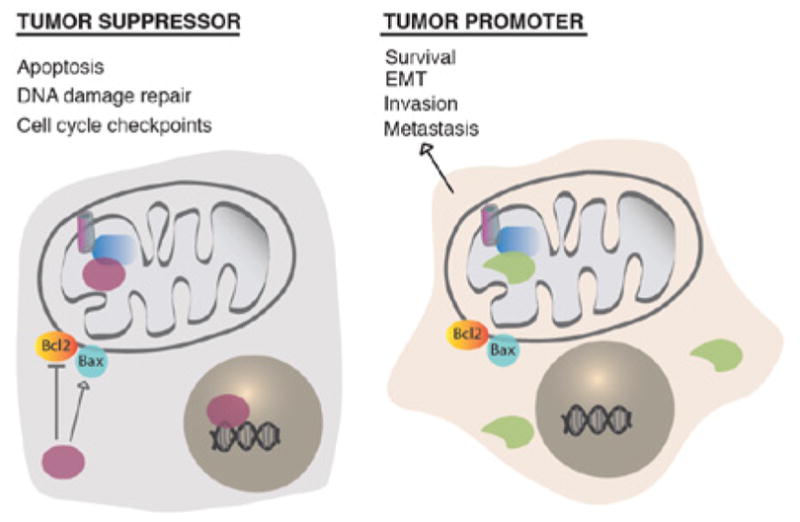
Comparison of p53-WT p53-psi activities. A) Diagram depicting p53-WT’s role in regulating apoptosis, DNA damage repair and cell-cycle checkpoints. P53-WT’s activities are attributed to its possession of a functional DNA-binding domain, nuclear localization sequences, and a tetramerization domain. B) Diagram depicting p53-psi’s ability to modulate cell survival, epithelial-to-mesenchymal transition (EMT), invasion and metastasis. Unlike p53-WT, p53-psi is generated by the use of an alternative 3’ splice site in intron 6 and lacks the functional domains necessary to promote canonical tumor suppressor functions.
Considering the critically important roles p53 plays in the modulation of cell growth and survival in response to a variety of cellular cues, it is not surprising that p53 is the most mutated gene in cancer [37]. In fact, a mutated version of TP53 is detected on average in 50% of tumors [38]. The types of mutations commonly affecting TP53 are loss of function (LOF), gain of function (GOF) and, as will be introduced in the present review, separation of function (SOF) mutations [39,40]. Most LOF mutations in TP53 adhere to the two-hit hypothesis proposed by Alfred G. Knudson in 1971 [41]. In fact, the most common cause of TP53 loss of function is an inactivating missense mutation in one allele and simultaneous deletions in regions of the 17 pchromosome encompassing the TP53 locus [42,43].
It has become apparent that certain TP53 GOF mutations confer p53 with capacities that are different than those resulting only from the loss of tumor-suppressing functions. Interestingly, these GOF mutations occur at higher than expected frequencies and, consequently, are usually referred to as “hot-spot” mutations [44,45]. Knock-in mouse models of some of these p53 GOF hot-spot mutations have been shown to produce carcinomas, adenomas and osteosarcomas [44–47]. These models’ outcomes differ from those of TP53 null models, which primarily develop lymphoma. The mechanism behind these processes is highly complex and primarily centered on changes that occur in the transcriptional activities of p53 [44–47].
Functional differences between GOF and LOF mutations in the TP53 gene are perhaps best exemplified by Li-Fraumeni Syndrome (LFS). LFS is a relatively rare and hereditary disorder that renders those affected susceptible to the development of various cancers [46–48]. LFS patients harbor different germline mutations in the TP53 gene (Figure 4). While LFS patients harboring LOF TP53 mutations tend to develop tumors later in life, carriers of the GOFTP53 mutation have an increased probability of developing cancers as children and adolescents [49].
Figure 4.
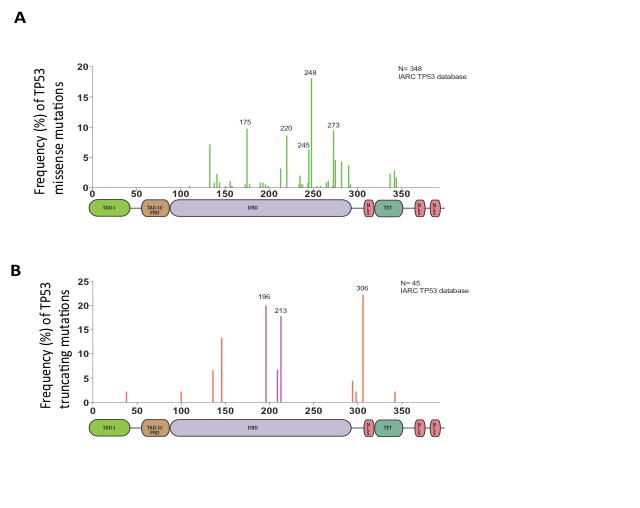
Distribution of TP53 missense and nonsense mutations in patients with Li-Fraumeni Syndrome (LFS).A) Histogram featuring p53 missense mutations present in the germline of LFS patients. B) Histogram featuring truncating mutations present in the germline of LFS patients. Together, these plots shows that the majority of TP53 in patients with LFS mutations occur in the core domain. Labeled residues are mutation hot-spots. Exon- 6/exon-7 truncating mutations are indicated in purple.
Recent studies from our laboratory have demonstrated that several TP53 truncating mutations occur at the exon-6/exon- 7 boundary. These splicing mutations, deletions, and nonsense mutations produce mutants that induce cell proliferation and the acquisition of metastatic features in cancer cells [50]. Notably, these p53-truncated proteins have molecular features that are nearly indistinguishable from those of the p53-psi isoform (Figure 5) [50]. Like p53-psi, these exon-6 truncating mutants retain the capacity to partially localize to the mitochondria, to interact with CypD and to regulate the MPTP [50].
Figure 5.
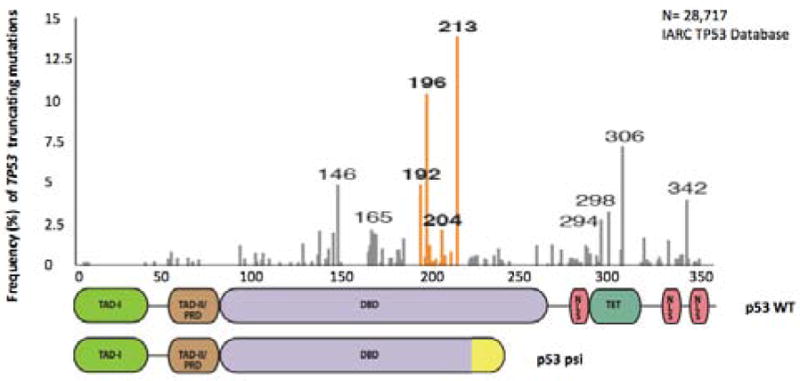
Tumor Distribution of TP53 truncating mutations. A) Histogram depicting the frequency of somatic TP53 truncating mutations in tumors. This plot demonstrates that exon-6/exon-7 truncating mutations are particularly abundant and occur at higher than expected frequencies. Exon-6/exon-7 truncating mutation hot-spots are indicated in orange.
Given the similarity of the molecular and phenotypic features of exon-6 truncating mutants and p53-psi, these truncating mutations are perhaps best characterized as separation-of-function (SOF) [50]. In fact, SOF mutations produce stable proteins that are marked by the loss of certain biochemical properties, while leaving other activities of the wild-type allele undisrupted. Notable examples of SOF mutations are certain RAG2 mutants. RAG2 is a nuclease that cleaves and rejoins different segments of the V(D)J locus during V(D)J recombination. Indeed, it has been observed that several RAG2 mutants resulting from SOF mutations retain the capacity to perform cleavage despite being severely defective in the joining step in V(D)J recombination [51].
Analysis of multiple cancer genome datasets indicated that, much like TP53 GOF mutations, SOF mutations occur at higher than expected frequencies at particular hot-spot locations. Although these p53 SOF mutations are not druggable, CypD inhibitors are readily accessible and have been developed by pharmaceutical companies for other indications. Hence, these observations consummately imply that these SOF mutations could constitute a good precision medicine target. Notably, the frequency of SOF mutations in tumors for which the current therapeutic opportunities are very limited, such as pancreatic and ovarian cancers, occur at frequencies comparable to other actionable precision medicine targets like the EGFR oncogenic-mutation in non-small cell lung carcinomas [52].
Interestingly, as has also been suggested by the reports of Cartegni et al. [53], our analysis of the IARC TP53 database indicated that certain TP53 silent mutations also occur at higher than expected frequencies at specific hot-spots (Figure 6) [54]. In principle, this observation suggests that the presence of these mutations could confer an advantage of growth to tumors. Though these silent mutations do not impact the amino acid composition of p53, the resulting nucleotide changes could instead lead to alterations in splicing patterns and/or impinge upon p53 expression and activity. The effects of these TP53 silent mutations may mirror certain mutations in the MAPT gene that have been documented in cases of frontotemporal dementia with Parkinsonism [55].
Figure 6.
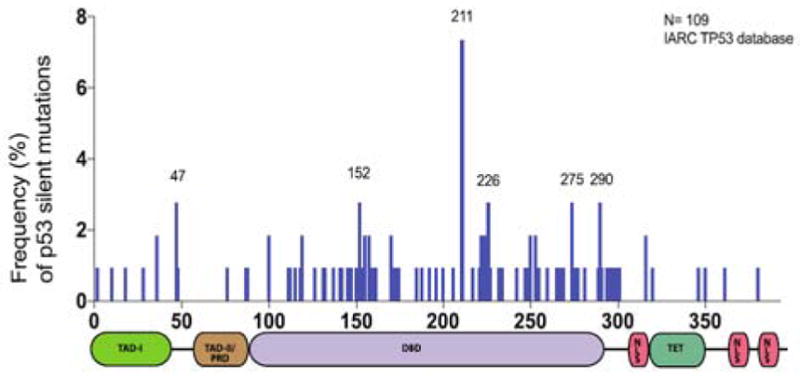
Distribution of TP53 silent mutations. A) Histogram depicting the frequency of somatic TP53 silent mutations in tumors. This plot demonstrates that silent mutations occur at higher than expected frequencies at certain locations on the TP53 gene. Labeled residues are mutation hot-spots.
More than 40 years have elapsed since the initial discovery of p53; nonetheless, progress continues to be made in the characterization of the most studied gene in cancer biology. As was presented, a growing body of evidence indicates the importance of novel alternative splicing isoforms, truncating/separation-of-function mutations, and possibly hot-spot silent mutations in the modulation of p53’s activity. These findings challenge pre-existing conceptions of p53 as a mere tumor suppressor protein and transcription factor. Importantly, these results also provide a basis for the development of novel therapeutic options for multiple tumor types that could ultimately bring us closer to a cure for cancer.
ABBREVIATIONS
- CypD
Cyclophilin D
- GOF
Gain-of-Function
- LOF
Loss-of- Function
- SOF
Separation-of-Function
- MPTP
Mitochondria Permeability Transition Pore
- LFS
Li-Fraumeni Syndrome
References
- 1.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 2.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40- transformed cells. Nature. 1979;278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 3.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 4.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 5.Lane DP, Benchimol S. p53: oncogene or anti-oncogene? Genes & Development. 1990;4:1–8. doi: 10.1101/gad.4.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–1093. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 7.Dippold WG, Jay G, Deleo AB, Khoury G, Old LJ. p53 transformation-related protein: Detection by monoclonal antibody in mouse and human cells. Proc Natl Acad Sci USA. 1981;78:1695–1699. doi: 10.1073/pnas.78.3.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci USA. 1989;86:8763–8767. doi: 10.1073/pnas.86.22.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mercer WE, Shields MT, Amin M, Sauve GJ, Appella E, Romano JW, et al. Negative growth regulation in a glioblastoma tumor cell line that conditionally expresses human wild-type p53. Proc Natl Acad Sci USA. 1990;87:6166–6170. doi: 10.1073/pnas.87.16.6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Junttila MR, Evan GI. p53--a Jack of all trades but master of none. Nat Rev Cancer. 2009;9:821–829. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- 12.Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, et al. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- 13.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 14.Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol Cell Biol. 1992;12:2866–2871. doi: 10.1128/mcb.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farmer G, Bargonetti J, Zhu H, Friedman P, Prywes R, Prives C. Wild-type p53 activates transcription in vitro. Nature. 1992;358:83–86. doi: 10.1038/358083a0. [DOI] [PubMed] [Google Scholar]
- 16.Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352:345–347. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- 17.Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci U S A. 1992;89:4495–4499. doi: 10.1073/pnas.89.10.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995;5:931–936. doi: 10.1016/s0960-9822(95)00183-7. [DOI] [PubMed] [Google Scholar]
- 19.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Molecular cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 20.Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature. 2007;450:721–724. doi: 10.1038/nature05993. [DOI] [PubMed] [Google Scholar]
- 21.Meletis K, Wirta V, Hede SM, Nistér M, Lundeberg J, Frisén J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- 22.Muller PA, Vousden KH, Norman JC. p53 and its mutants in tumor cell migration and invasion. J Cell Biol. 2011;192:209–218. doi: 10.1083/jcb.201009059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donehower LA, Harvey M, Slagle BL, McArthuri MJ, Montgomery CA., Jr Mice deficient for p53 are developmentally normal but susceptible to spontaneous. Nature. 1992;356:19. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 24.Harms KL, Chen X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006;13:890–897. doi: 10.1038/sj.cdd.4401904. [DOI] [PubMed] [Google Scholar]
- 25.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 26.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, et al. The regulation of AMPK beta, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulat. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 28.Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13:962–72. doi: 10.1038/sj.cdd.4401914. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Prives C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene. 2007;26:2220–2225. doi: 10.1038/sj.onc.1210311. [DOI] [PubMed] [Google Scholar]
- 30.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 31.Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell. 2003;113:301–314. doi: 10.1016/s0092-8674(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 32.Shi D, Gu W. Dual roles of MDM2 in the regulation of p53 ubiquitination dependent and ubiquitination independent mechanisms of MDM2 repression of p53 activity. Genes Cancer. 2012;3:240–248. doi: 10.1177/1947601912455199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khoury MP, Bourdon JC. p53 isoforms an intracellular microprocessor? Genes Cancer. 2011;2:453–465. doi: 10.1177/1947601911408893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–2137. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth I, Campbell H, Rubio C, Vennin C, Wilson M, et al. The Δ133p53 isoform and its mouse analogue Δ122p53 promote invasion and metastasis involving pro-inflammatory molecules interleukin-6 and CCL2. Oncogene. 2016;35:4981–4989. doi: 10.1038/onc.2016.45. [DOI] [PubMed] [Google Scholar]
- 36.Senturk S, Yao Z, Camiolo M, Stiles B, Rathod T, Walsh AM, et al. p53Ψ is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc Natl Acad Sci USA. 2014;111:3287–3296. doi: 10.1073/pnas.1321640111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogelstein B, Sur S, Prives C. p53: the most frequently altered gene in human cancers. Nature Education. 2010;3:6. [Google Scholar]
- 38.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 39.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- 41.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Chen C, Xu Z, Scuoppo C, et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016;531:471–475. doi: 10.1038/nature17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Menon AG, Anderson KM, Riccardi VM, Chung RY, Whaley JM, Yandell DW, et al. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinghausen neurofibromatosis. Proc Natl Acad Sci USA. 1990;87:5435–5439. doi: 10.1073/pnas.87.14.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, et al. Gain of function mutations in p53. Nat Genet. 1993;4:42–46. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 45.Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 46.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 47.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 48.Malkin D. Li-Fraumeni syndrome. Genes Cancer. 2011;2:475–484. doi: 10.1177/1947601911413466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol. 2015;33:2345–2352. doi: 10.1200/JCO.2014.59.5728. [DOI] [PubMed] [Google Scholar]
- 50.Shirole NH, Pal D, Kastenhuber ER, Senturk S, et al. TP53 exon-6 truncating mutations produce separation of function isoforms with pro-tumorigenic functions. Elife. 2016:5. doi: 10.7554/eLife.17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qiu JX, Kale SB, Yarnell Schultz H, Roth DB. Separation-of-function mutants reveal critical roles for RAG2 in both the cleavage and joining steps of V(D)J recombination. Mol Cell. 2001;7:77–87. doi: 10.1016/s1097-2765(01)00156-3. [DOI] [PubMed] [Google Scholar]
- 52.Korpanty GJ, Graham DM, Vincent MD, Leighl NB. Biomarkers That Currently Affect Clinical Practice in Lung Cancer: EGFR, ALK, MET, ROS-, and KRAS. Front Oncol. 2014;4:204. doi: 10.3389/fonc.2014.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nature Reviews Genetics. 2002;3:285–98. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 54.Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, et al. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat. 2016;37:865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 55.Boeve BF, Hutton M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN) Arch Neurol. 2008;65:460–464. doi: 10.1001/archneur.65.4.460. [DOI] [PMC free article] [PubMed] [Google Scholar]