

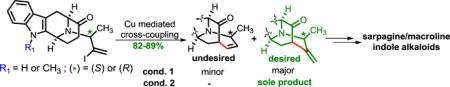
Abstract
An enolate driven copper-mediated cross-coupling process enabled a cheaper and greener access toward the key pentacyclic intermediates required for the enantiospecific total synthesis of a number of C-19 methyl substituted sarpagine/macroline indole alkaloids. Replacement of palladium (60–68%) with copper iodide (82–89%) resulted in much higher yields. The formation of an unusual 7-membered cross-coupling product was completely inhibited by using TEMPO as a radical scavenger. Further functionalization led to the first enantiospecific total synthesis of macrocarpine D and E.
Graphical Abstract
The medicinal plants of the Alstonia (Apocynaceae) genus have been used in traditional medicine in many countries of the world from antiquity.1 Their traditional uses include treatment of ulcers, dysentery, malaria, anthelmintics, diabetes, rheumatism, snake bites, etc.1 The presence of indole alkaloids that are the secondary metabolites of these plants is the most probable source of their medicinal activity.2 According to a review by Cordell et al. among the 60 plant-derived alkaloids of medicinal significance, 39 were directly related to their traditional uses.3 Macroline/sarpagine type indole alkaloids are one of the major classes of alkaloids isolated from these species to date by Le-Quesne, Elderfield, Schmid, Kam, and others.4–7 Macrocarpine A–C (1–3) were isolated from the bark extract of Alstonia macrophylla in 2004.8 Several other alkaloids of the same series, macrocarpine D (4) and macrocarpine E–H (5–8), were isolated in 2014 from the stem-bark and leaf extracts of A. macrophylla and A. angustifolia respectively by Kam et al.9,10 All of these macroline type indole alkaloids, macrocarpine A–H (1–8) share the common feature of a β-methyl substituent at the C-19 position. This is a distinct difference from earlier Alstonia alkaloids isolated by LeQuesne and Schmid.5,7 To date, around 30 alkaloids of the sarpagine/macroline/ajmaline family, which bear a diastereomeric methyl function at C-19 have been isolated.4,11 Macroline related alkaloids N(4)-methyl-N(4),21-secotalpinine (9)8,12, N(4)-methyltalpinine (10)12, and 19-epitalcarpine (11)10 as well as sarpagine related alkaloids macrosalhine chloride (12)13, and deoxyperaksine (13)14 also possess the C-19 methyl substitution. Among these, 11 and 13 have a diastereomeric α-methyl group at C-19. The synthesis of these alkaloids (1–13, Figure 1) has not been reported yet. Moreover, N(4)-methyltalpinine (10) and N(4)-methyl-N(4),21-secotalpinine (9) have been reported recently to have potent anticancer (NF-ƙB inhibitor, ED50 1.2 μM) activity and profound leishmanicidal12 activity, respectively. The unique structural features and potential medicinal properties prompted attempts at the first total synthesis of this class of alkaloids via a general strategy for the entire series. This is the basis of the approach toward chemical economy described here.
Figure 1.
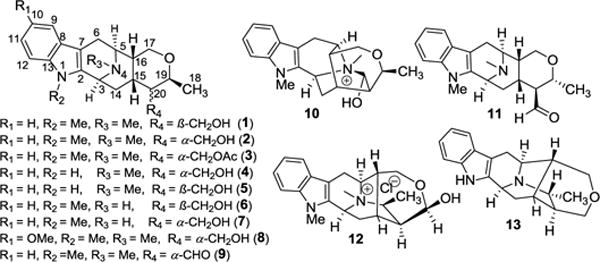
Examples of some C-19 methyl substituted sarpagine/macroline indole alkaloids
Copper-mediated carbon-carbon bond formation is more than a century old.15 Although palladium-catalyzed cross-coupling reactions have been the dominant method in the field of total synthesis of complex natural products, copper has proven itself to be an essential alternative, as indicated by the increase in copper-mediated cross-coupling processes over the last decade.16 As a result, it was decided to investigate a copper catalyzed, or mediated coupling process to offer a less expensive and less toxic alternative to catalytic palladium, while avoiding phosphine based ligands for easier purification. Moreover, potential improvements in yields, as well as workup and purification would be important.17
Wang et al.18 developed an enolate driven palladium catalyzed α-vinylation of a ketone in 2000 (Scheme 1, entry 1). This process has been employed in the total synthesis of several sarpagine/macroline/ajmaline indole alkaloids.19–21 Although this palladium catalyzed process was effective in accessing the key intermediate 15 from vinyl iodide 14, it provided only 60–68% yield in the case of vinyl iodides 16 or 18 (Scheme 1, entry 2), wherein there was a diastereomeric methyl function along with a terminal olefin in place of the ethylidene function in 14 (internal olefin, Wang, 2000). These features in 16 and 18 make them structurally and chemically different than 14. Since the diastereomeric methyl function was essential for the synthesis of C-19 methyl substituted alkaloids, further improvement was required for better access to the key intermediates 17, 19, 21, and 23. More importantly, replacement of palladium with the cheaper and less toxic copper, would greatly facilitate use of this enolate-mediated process by others and formed much of the driving force in this research.
Scheme 1.
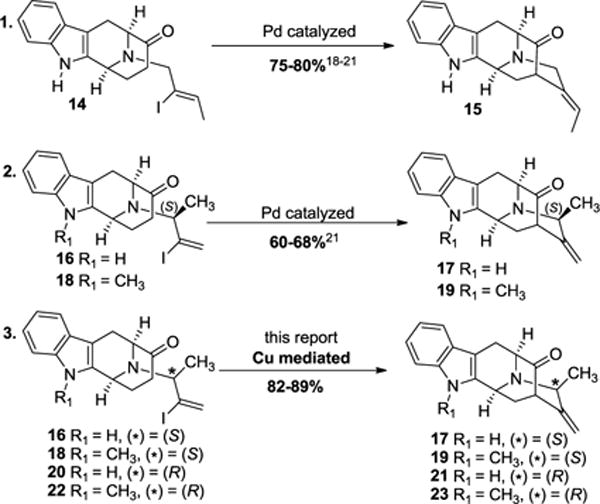
Regiospecific access to the pentacyclic core system via palladium and copper-catalyzed cross-coupling process
The copper-catalyzed conditions22 that were used for the α-vinylation of 14 gave lower yields along with an unusual, undesired product in the case of 22. Numerous attempts were made to optimize the desired yield of the olefin 23, eliminate the unusual side product 23′ and understand the mechanism of multiple competing reactions. The implementation, improvement and extension of the scope of this process to access the C-19 methyl substituted sarpagine/macroline/ajmaline alkaloids with either an (R) or (S) C-19 methyl substituent in the Na-H as well as Na-CH3 series (Scheme 1, entry 3) form the basis of this communication.
All of the vinyl iodide intermediates (16, 18, 20 and, 22) were prepared according to the previously reported procedures21 (Scheme 2) beginning from the tetracyclic ketones 24/25 which had been prepared in the standard two pot process on 300 gram scale.23 As depicted in Scheme 2, the Nb alkylation via SN2 substitution of the chiral tosylates (26/27) in CH3CN with K2CO3 and subsequent deprotection of the TIPS protecting group with wet TBAF in THF furnished the Nb-alkylated terminal alkynes (28–31) in excellent yields. Haloboration21 of the terminal alkynes with I-B(Cy)2 in DCM followed by protodeboronation with HOAc, resulted in the vinyl iodides (16, 18, 20, and, 22) in 74–79% yield with complete regioselectivity.
Scheme 2.
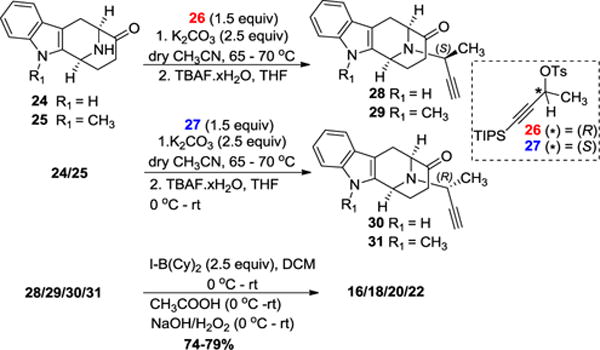
Completely Regioselective access to the vinyl iodides 16, 18, 20, and 22
Initial experiments with vinyl iodide (22) and CuI under the reported conditions22 resulted in the desired product (23) in lower yields (~42%) along with an unusual/unexpected cyclization product (23′), a seven-membered ring with an internal alkene (Scheme 3).
Scheme 3.
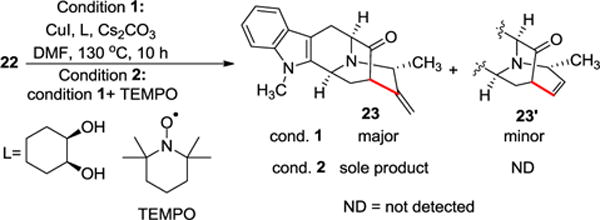
Enolate driven copper mediated cross-coupling of the vinyl iodide (22)
The structures of both the desired (23) and unexpected seven-membered ring (23′) cross-coupling products have been confirmed by MS, 1D and 2D NMR, and X-ray crystallographic analysis (see Supporting Information (SI) for details). In the absence of CuI, the same conditions yielded the 7-membered product to a greater extent. Increasing the equivalents of CuI and ligand to 1.0 equivalent and the base to 4.0 equivalents (entry 3 of Table 1) resulted in a higher overall yield of the desired material (67%), while the unexpected product was still present (~21%).
Table 1.
Optimization of reaction conditions for the Cu-mediated cross-coupling reaction of 22
| entry | CuI or cat. | L | Cs2CO3 | scv. | 23:23′ (by NMR)a | overall yield (%)b |
|---|---|---|---|---|---|---|
| 1 | 0.5 | 0.5 | 2.0 | – | 89:11 | 47 |
| 2 | – | 0.5 | 2.0 | – | 63:37 | 45 |
| 3 | 1.0 | 1.0 | 4.0 | – | 79:21 | 67 |
| 4 | – | – | 2.0 | – | 64:36 | 35 |
| 5 | 0.5 | 0.5 | 2.0 | 2.5 | 97:3 | 45 |
| 6 | 0.5 | 0.5 | 2.0 | 3.0 | 100:0 | 66 |
| 7 | 1.0 | 1.0 | 4.0 | 3.0 | 100:0 | 89 |
| 8 | 0.1 | 0.1 | 4.0 | 3.0 | 100:0 | 25 |
| 9 | 0.5 | 0.5 | 4.0 | 3.0 | 100:0 | 40 |
| 10 | 1.0 | – | 4.0 | 3.0 | 100:0 | 11 |
| 11 | 1.0 | 1.0 | – | 3.0 | – | NDc |
| 12 | – | 1.0 | 4.0 | 3.0 | – | ND |
| 13 | cat.-2 (1.0) | 1.0 | 4.0 | 3.0 | 100:0 | 51 |
| 14 | cat.-3 (1.0) | 1.0 | 4.0 | 3.0 | 100:0 | 24 |
| 15 | cat.-4 (1.0) | 1.0 | 4.0 | 3.0 | 100:0 | 28 |
Unit for all reagent amounts is equivalent (equiv); scv. = TEMPO scavenger
Ratio determined by 1H NMR spectroscopy
Overall isolated yield after flash chromatography on neutral alumina
Starting material was recovered; ND = not detected; L= cis-1-2-cyclo-hexanediol; cat.-2 = Cu(CH3CN)4ClO4; cat.-3 = Cu(CH3CN)4PF6; cat.-4 = Cu(CH3CN)4OTf (see SI for detailed experimental procedures)
It was not surprising that this series of vinyl iodides (16, 18, 20 and, 22) would have different reactivities than vinyl iodides reported earlier.18 Differences in the substrates included the presence of a methylidene (a terminal alkene) instead of the ethylidene in 14 (an internal alkene) as well as the presence of the chiral methyl function at C-19 instead of an achiral methylene. In order to rationalize this unprecedented seven-membered ring cyclization, it was felt that a radical mechanism may have been involved in its formation. To test this hypothesis, it was decided to use 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO), the well-known radical scavenger24, to inhibit any radical step or species that may have diverted the mechanism. In support of this hypothesis, an experiment with 2.5 equivalents of TEMPO (entry 5 of Table 1) resulted in almost complete inhibition of the formation of the undesired 7-membered internal alkene (~3% by NMR spectroscopy) with 45% overall yield of 23. Increasing the amount of TEMPO to 3.0 equivalents completely eliminated the undesired cyclization (see SI) and provided the desired cross-coupling product 23 in 66% yield. After many experiments by changing reaction parameters and screening different copper sources (entries 8–15 of Table 1) the optimized condition was found to be 1.0 equiv of CuI, 1.0 equiv of ligand, 4.0 equiv of Cs2CO3, and 3.0 equiv of TEMPO. This combination furnished 89% yield of the desired cross-coupled product to the exclusion of the 7-membered byproduct as compared to (60–68%) with a palladium catalyst.21,25 This modified reaction condition was effective for both stereoisomers of the C-19 methyl substitution and in both the Na-H and Na-CH3 series (16, 18, 20 and, 22) as well. This permitted the application of this Cu-mediated cross-coupling process to access the key intermediates (17, 19, 21 and, 23) in gram-quantities toward all of the macroline/sarpagine alkaloids discussed above (Scheme 4). While TEMPO was initially chosen as a radical probe, the reason for its useful effect is under investigation.
Scheme 4.
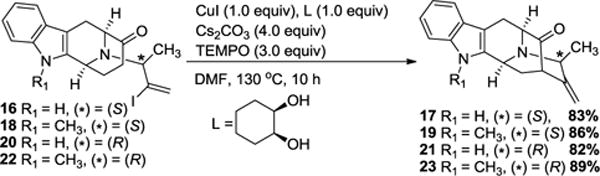
Access to the key pentacyclic ketone intermediates 18, 20, 22 and, 24 via the optimized conditions
Surprisingly, it was observed that the base Cs2CO3 alone in DMF (entry 2 of Table 1) could yield both of the products in an approximately 2 to 1 ratio. This observation indicated the possibility of another competing mechanism wherein the vinyl iodide (22) or intermediate underwent an E-2 like elimination or radical process to produce a terminal alkyne (31, in situ). This alkyne subsequently could undergo a 6-(enolendo)-exo-dig (process A in Scheme 5) which would produce the six-membered external alkene (23) as a major product and a 7-(enolendo)-endo-dig cyclization (process B in Scheme 5) to produce the seven-membered internal alkene (23′) as the minor product (Scheme 5). Both of these processes are allowed by Baldwin’s rules for ring closure.26,27 This hypothesis has been confirmed by stopping the reaction (at 3 hours) before completion. The alkyne 31 was detected along with traces of the cross-coupling products 23 and 23′.
Scheme 5.
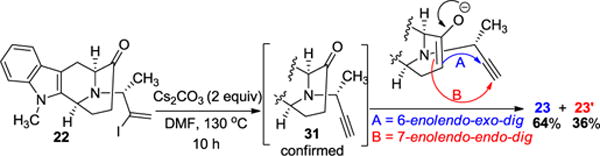
Possible mechanism for the observed base mediated cyclization (Table 1 entries 2 and 4)
The investigation of the competing mechanisms is now ongoing; however, gratifying excellent yields (82–89%) of only the desired six-membered ring were obtained in a stereospecific fashion with copper iodide (Scheme 4).
With the pentacyclic ketones in hand, in excellent yields, the total synthesis of a series of C-19 methyl substituted sarpagine macroline indole alkaloids was undertaken, which included the potent anticancer alkaloid, N(4)-methyltalpinine (10), as well as the leishmanicidal base (9), macrocarpines A–G (1–7), and, deoxyperaksine (13). Herein is reported the first total synthesis of the Na-H bearing macroline indole alkaloids macrocarpine D (4) and E (5) (Scheme 6, 7).
Scheme 6.
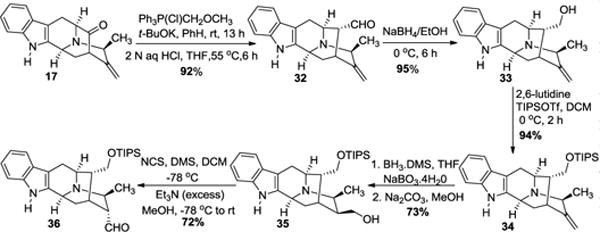
Toward macrocarpine D (4) and E (5) from the key intermediate 17
Scheme 7.
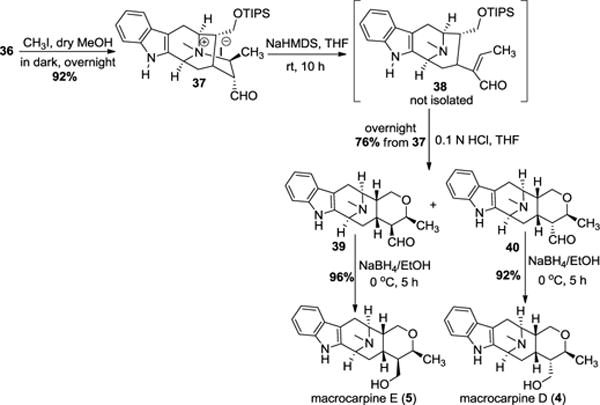
Total synthesis of macrocarpine D (4) and E (5)
The pentacyclic ketone (17) was subjected to a one-carbon homologation via Wittig olefination using methoxymethyl trip-henylphosphonium chloride and potassium tert-butoxide in benzene to furnish the enol ether, which was hydrolyzed without purification to aldehyde (32) under acidic conditions (Scheme 6). The aldehyde was isolated in the stable α position even in the presence of the C-19 β-methyl group. The aldehyde (32) was reduced to alcohol (33) with sodium borohydride in ethanol and subsequently protected with a TIPS group to give the silyl ether (34). The alkene (34) was subjected to hydroboration (borane dimethyl sulfide) and Kabalka oxidation (NaBO3) to provide primary alcohol (35) in 73% yield. Oxidation of the primary alcohol under Corey-Kim conditions at −78 °C produced a mixture of α and β-aldehydes with the α isomer as the major product. This mixture was epimerized entirely to the α-aldehyde (36) with Et3N in methanol added to the mixture.
Quaternization of the Nb-group with iodomethane in methanol gave the iodide salt (37, Scheme 7). The quaternary ammonium salt underwent retro-Michael ring opening in the presence of NaHMDS in THF to produce the α,β–unsaturated aldehyde (38) in 78% yield similar to the work first reported by LeQuesne.5
The TIPS group was removed by heating 38 under mild acidic conditions in THF. The so formed alcohol added in Michael fashion to the α,β-unsaturated aldehyde to produce (39) and (40) as an epimeric mixture of aldehydes with the β-methyl group. Each of these could be isolated by silica gel flash chromatography. The desired aldehydes (39) and (40) upon reduction with sodium borohydride in ethanol gave macrocarpine E (5) and macrocarpine D (4) in 96% and 92% yield, respectively. Spectroscopic data and optical rotations of the synthetic products are in complete agreement with natural macrocarpine D & E. The total synthesis of the other alkaloids of interest is ongoing.
In summary, the first total synthesis of macrocarpine D (4) and E (5) has been accomplished via a key copper-mediated cross-coupling process toward the important intermediates. This general strategy enables one to access all of the indole alkaloids of the same class with stereospecific incorporation of the important β (or α)-methyl function at C-19. Replacement of the palladium catalyst with CuI provides a much more useful and cheaper method since the copper catalyst, even at stoichiometric amounts, is much cheaper than catalytic palladium (see SI for a comparative price table). More importantly, accessing 17 in 83% yield with copper compared to 60% with catalytic palladium serves as an example of replacement of palladium in this enolate-mediated process which makes it much more useful for others, especially in the pharmaceutical industry.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the National Institutes of Health (MH096463; HL118561) and the Shimadzu Analytical Laboratory of Southeastern Wisconsin.
Footnotes
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and spectral data for 17, 19, 21, 23 and 23′ as well as spectral data and 1H and 13C comparison tables between natural and synthetic 4 and 5 (pdf)
X-ray crystallographic data for 19, 22, 23, and 23′ (cif)
References
- 1.Khyade MS, Kasote DM, Vaikos NPJ. J Ethnopharmacol. 2014;153:1. doi: 10.1016/j.jep.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 2.Ziegler J, Facchini PJ. Annu Rev Plant Biol. 2008;59:735. doi: 10.1146/annurev.arplant.59.032607.092730. [DOI] [PubMed] [Google Scholar]
- 3.Cordell GA, Quinn‐Beattie ML, Farnsworth NR. Phytother Res. 2001;15:183. doi: 10.1002/ptr.890. [DOI] [PubMed] [Google Scholar]
- 4.Namjoshi OA, Cook JM. In: The Alkaloids: Chemistry and Biology. Knӧlker H-J, editor. Vol. 76. Academic Press; San Diego, CA: 2016. p. 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garnick RL, Le Quesne PW. J Am Chem Soc. 1978;100:4213. [Google Scholar]
- 6.Elderfield RC, Gilman RE. Phytochemistry. 1972;11:339. [Google Scholar]
- 7.Kishi T, Hesse M, Gemenden C, Taylor W, Schmid H. Helv Chim Acta. 1965;48:1349. doi: 10.1002/hlca.19650480615. [DOI] [PubMed] [Google Scholar]
- 8.Kam T-S, Choo Y-M, Komiyama K. Tetrahedron. 2004;60:3957. [Google Scholar]
- 9.Lim S-H, Low Y-Y, Sinniah SK, Yong K-T, Sim K-S, Kam T-S. Phytochemistry. 2014;98:204. doi: 10.1016/j.phytochem.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 10.Tan S-J, Lim J-L, Low Y-Y, Sim K-S, Lim S-H, Kam T-S. J Nat Prod. 2014;77:2068. doi: 10.1021/np500439u. [DOI] [PubMed] [Google Scholar]
- 11.Lounasmaa M, Hanhinen P, Westersund M. In: The Alkaloids: Chemistry and Biology. Cordell GA, editor. Vol. 52. Academic Press; San Diego, CA: 1999. p. 103. [Google Scholar]
- 12.Pan L, Terrazas C, Acuña UM, Ninh TN, Chai H, de Blanco EJC, Soejarto DD, Satoskar AR, Kinghorn AD. Phytochem Lett. 2014;10:54. doi: 10.1016/j.phytol.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan ZM, Hesse M, Schmid H. Helv Chim Acta. 1967;50:1002. doi: 10.1002/hlca.19670500403. [DOI] [PubMed] [Google Scholar]
- 14.Iwu M. Planta Med. 1982;45:105. doi: 10.1055/s-2007-971256. [DOI] [PubMed] [Google Scholar]
- 15.Ullmann F, Bielecki J. Ber Dtsch Chem Ges. 1901;34:2174. [Google Scholar]
- 16.Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054. doi: 10.1021/cr8002505. [DOI] [PubMed] [Google Scholar]
- 17.Beletskaya IP, Cheprakov AV. Coord Chem Rev. 2004;248:2337. [Google Scholar]
- 18.Wang T, Cook JM. Org Lett. 2000;2:2057. doi: 10.1021/ol000095+. [DOI] [PubMed] [Google Scholar]
- 19.Zhao S, Liao X, Wang T, Flippen-Anderson J, Cook JM. J Org Chem. 2003;68:6279. doi: 10.1021/jo030055u. [DOI] [PubMed] [Google Scholar]
- 20.Edwankar CR, Edwankar RV, Deschamps JR, Cook JM. Angew Chem, Int Ed. 2012;51:11762. doi: 10.1002/anie.201206015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwankar RV, Edwankar CR, Deschamps JR, Cook JM. J Org Chem. 2014;79:10030. doi: 10.1021/jo5016163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin W, Kabir MS, Wang Z, Rallapalli SK, Ma J, Cook JM. J Org Chem. 2010;75:3339. doi: 10.1021/jo100279w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu P, Wang T, Li J, Cook JM. J Org Chem. 2000;65:3173. doi: 10.1021/jo000126e. [DOI] [PubMed] [Google Scholar]
- 24.Wright PJ, English AM. J Am Chem Soc. 2003;125:8655. doi: 10.1021/ja0291888. [DOI] [PubMed] [Google Scholar]
- 25.Edwankar RV, Edwankar CR, Deschamps J, Cook JM. Org Lett. 2011;13:5216. doi: 10.1021/ol202101p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baldwin JE, Lusch MJ. Tetrahedron. 1982;38:2939. [Google Scholar]
- 27.Baldwin J. Chem Soc, Chem Commun. 1976:734. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.