

Abstract
While the balance between carbohydrates and fatty acids for energy production appears to be crucial for cardiac homeostasis, much remains to be learned about the molecular mechanisms underlying this relationship. Given the reported benefits of cGMP signaling on the myocardium, we investigated the impact of its chronic activation on cardiac energy metabolism using mice overexpressing a constitutively active cytoplasmic guanylate cyclase (GC+/0) in cardiomyocytes. Ex vivo working GC+/0 heart perfusions with 13C-labeled substrates revealed an altered pattern of exogenous substrate fuel selection compared to controls, namely a 38±9% lower contribution of exogenous fatty acids to acetyl-CoA formation, while that of carbohydrates remains unchanged despite a two-fold increase in glycolysis. The lower contribution of exogenous fatty acids to energy production is not associated with changes in energy demand or supply (contractile function, oxygen consumption, tissue acetyl-CoA or CoA levels, citric acid cycle flux rate) or in the regulation of β-oxidation (acetyl-CoA carboxylase activity, tissue malonyl-CoA levels). However, GC+/0 hearts show a two-fold increase in the incorporation of exogenous oleate into triglycerides. Furthermore, the following molecular data are consistent with a concomitant increase in triglyceride hydrolysis: (i) increased abundance of hormone sensitive lipase (HSL) protein (24±11%) and mRNA (22±4%) as well as (ii) several phosphorylation events related to HSL inhibitory (AMPK) and activation (ERK 1/2) sites, which should contribute to enhance its activity. These changes in exogenous fatty acid trafficking in GC+/0 hearts appear to be functionally relevant, as demonstrated by their resistance to fasting-induced triglyceride accumulation. While the documented metabolic profile of GC+/0 mouse hearts is partly reminiscent of hypertrophied hearts, the observed changes in lipid trafficking have not been previously documented, and may be part of the molecular mechanism underlying the benefits of cGMP signaling on the myocardium.
Keywords: Hormone sensitive lipase, Energy metabolism, Guanylate cyclase, Perfusion, Isotopes
1. Introduction
In the healthy adult heart, the concerted regulation of long chain fatty acid and carbohydrate metabolism ensures optimal energy production and, hence, cardiac homeostasis. Likewise, alterations in cardiac substrate metabolism are considered as independent determining factors that contribute to contractile dysfunction, to the heart's susceptibility to injury, and to progression from compensated left ventricular hypertrophy to cardiac failure [1,2]. However, much remains to be learned about the (patho)physiological significance of specific alterations in cardiac substrate utilization beyond their effect on ATP production. For example, while a shift from fatty acid towards carbohydrate utilization for energy production (a characteristic of the hypertrophied heart [1,2]) has been shown to be beneficial for ischemic and failing hearts [1,2], decreased fatty acid oxidation may also lead to potentially detrimental consequences such as intracellular lipid accumulation and its associated lipotoxic sequelae [3,4].
Molecular mechanisms regulating the balance between carbohydrate and fatty acid utilization for energy production or storage involve the participation of signaling pathways. Among the latter, AMP kinase (AMPK) and protein kinase B (Akt) have been the subject of active research [5–7]. Recently, we became interested in cyclic GMP (cGMP), a downstream effector of the nitric oxide (NO) and natriuretic peptide pathways, since cGMP exerts a host of cardioprotective effects [8,9]. In this regard, our group has developed a mouse transgenic model overexpressing the constitutively active catalytic fragment of the guanylate cyclase domain of the atrial natriuretic factor (ANF) receptor in a cardiomyocyte-specific manner (GC+/0) [10]. We have shown that expression of this transgene was accompanied by increased guanylate cyclase activity and cGMP concentration in isolated cardiomyocytes compared to non-transgenic littermates, and that it protects these mice against the hypertrophic effects of isoproterenol or abdominal aortic constriction [10]. Likewise, expression of the transgene improves the cardiac function of mice carrying mdx mutation of dystrophin, along with a increased of cGMP concentration in whole-heart extracts [11].
Interestingly, a number of studies have reported that NO or cGMP mimetics modulate energy metabolism in various tissues by influencing substrate selection for ATP production, expression of metabolic genes as well as genes of the nutrient signaling pathways [12–16]. However, there appears also to be a complex relationship between NO, the cGMP pathway and energy metabolism in the heart, which differs from that in the skeletal muscle and depends on many factors such as the level of myocardial activation of AMPK or contractility, as well as the (sub)cellular location of NO/cGMP production [17]. For example, myocardial glucose uptake or utilization are (i) enhanced following addition of NO synthase inhibitors [14] or in eNOS null mouse [18], and, conversely, (ii) decreased with addition of the cGMP analog 8-bromo-cGMP or of NO donors [15]. In contrast, a recent study shows that activation of the cGMP pathway contributes to the AMPK stimulation of glucose uptake in left ventricular papillary muscle [19]. Hence, much remains to be learned about the metabolic impact of enhanced cGMP signaling in cardiomyocytes.
To address this question, we used our previously described methodology of ex vivo working heart perfusion with 13C-labeled substrates [20] to measure simultaneously various hemodynamic and metabolic flux parameters in our GC+/0 transgenic mice. This approach allows for detailed and simultaneous measurements of the dynamics of cardiac energy substrate metabolism, information which is not accessible from static measurements of mRNA or protein expression. Our isotopic data demonstrate substantial differences in substrate selection for energy production as well as in lipid partitioning between β-oxidation and esterification between control and GC+/0 mice hearts. Additional molecular data are consistent with a concomitant increase in triglyceride (TG) hydrolysis.
2. Experimental procedures
2.1. Materials and animal model
Sources of chemicals, biological products, and 13C-substrates have been reported previously [20–26]. Antibodies against the phosphorylated forms of hormone-sensitive lipase (HSL) and extracellular-regulated kinase (ERK) forms 1 and 2 were purchased from Cell Signaling Technologies (Danvers, USA), and total HSL antibody was obtained from Cayman Chemicals (Ann Arbor, USA).
All procedures on the animals were approved by the local ethics committee in agreement with the guidelines of the Canadian Council on Animal Care. Our GC+/0 transgenic mice [10] have been backcrossed for at least 12 generations into the C57Bl/6J mouse strain. We used male transgenic and age-matched wild-type (WT) littermates mice at 12 to 13 weeks of age, all of which had similar body weights (25.7±0.7 vs 26.3± 0.6 g, respectively).
2.2. Working mouse heart perfusion
Mice were anesthetized (1 µL/g, i.p.) with a mixture of ketamine (100 mg/mL) and xylazine (20 mg/mL) and heparinized (5000 U/kg, i.p.) 15 min before surgery. The procedure for heart isolation and its ex vivo perfusion in the working mode has been previously described in detail [20]. The composition of the Krebs–Henseleit buffer (110 mM NaCl, 4.7 mM KCl, 2.1 mM CaCl2,0.24 mM KH2PO4, 0.48 mM K2HPO4, 0.48 mM Na2HPO4, 1.2 mM MgSO4, 25 mM NaHCO3, 0.1 mM EDTA) was modified to adjust free calcium levels (1.55±0.02 mM) and sodium concentration to a physiological value. The preload and afterload pressures were set at 15 and 50 mmHg, respectively. Myocardial oxygen consumption (MVO2; μmol/min), intracellular pH, rate pressure product (mm Hg · beats · min−1 · 10−3), cardiac power (mW), and cardiac efficiency (mW · μmol−1 · min−1) were calculated from previously reported equations [20].
Working mouse hearts were perfused for 30 min with a semi-recirculating modified Krebs–Henseleit solution containing physiological concentrations of substrates (11 mM glucose, 0.8 nM insulin, 50 µM carnitine, 5 nM epinephrine, 1.5 mM lactate, 0.2 mM pyruvate, and 0.4 mM oleate bound to 3% albumin). For any given perfusion, one of the unlabeled substrates was replaced by its corresponding labeled substrate, i.e. either: [U-13C18]oleate (25% initial molar percent enrichment (MPE)), [U-13C6]glucose (25% initial MPE), and [U-13C3]lactate/[U-13C3]pyruvate (100% initial MPE).
Throughout the perfusion, influent and effluent perfusates were collected at regular intervals to document lactate dehydrogenase (LDH) release rates (every 5 min), the oxygen and carbon dioxide partial pressures (at 10 and 20 min) and the lactate and pyruvate efflux rates (at 30 min). Subsequent to each perfusion period, hearts were freeze-clamped with metal tongs chilled in liquid nitrogen and weighed. There were no significant differences in the wet weight of perfused hearts between groups (data not shown). All samples were stored at −80 °C until further analysis.
2.3. Tissue processing
2.3.1. Flux measurements
Our previously published studies [20,23] provide (i) definitions of the 13C terminology and detailed descriptions for the measurements by gas chromatography-mass spectrometry (GCMS; Hewlett-Packard 6890 N gas chromatograph coupled to a 5973N mass spectrometer) of (i) the 13C-enrichment of citric acid cycle (CAC) intermediates and related metabolites (citrate, OAA moiety of citrate, succinate, fumarate and pyruvate) necessary for calculations of flux ratios relevant to substrate selection for citrate synthesis, and (ii) other metabolites (lactate and pyruvate) used to determine the glycolytic flux, as well as for (iii) the calculation of the absolute CAC flux rates from oxygen consumption rates and the stoichiometric relationship between oxygen consumption and citrate formation from β-oxidation and pyruvate decarboxylation. Briefly, GCMS data are expressed as MPE. Mass isotopomers of metabolites containing 1 to n 13C-atoms are identified as Mi with i=1, 2, … n, and the absolute MPE of individual 13C-labeled mass isotopomers (Mi) of a given metabolite are calculated as follows: MPE (Mi)=% AMi /[AM + ΣAMi], where AM and AMi represent the peak areas from ion chromatograms corrected for natural abundance, corresponding to unlabeled (M) and 13C-labeled (Mi) mass isotopomers, respectively. Metabolic flux ratios, which reflect the contribution of exogenous fatty acids (oleate) and of carbohydrates (lactate, pyruvate and glucose) to acetyl-CoA and/or to oxaloacetate (OAA) formation for citrate synthesis, are calculated from the MPE in Mi isotopomers of the acetyl (ACCIT) and oxaloacetate moiety of citrate (OAACIT; corrected for the formation of M3 OAA from CAC metabolism of citrate isotopomers) and expressed relative to citrate synthase (CS): (i) pyruvate decarboxylation (PDC)=M2 ACCIT/M3 pyruvate, (ii) oleate oxidation (OLE)=M2 ACCIT/M18 oleate, and (iii) pyruvate carboxylase (PC)=M3 OAACIT/M3 pyruvate. Finally, the contribution of other substrates (OS), most likely endogenous TGs, to the formation of acetyl-CoA is evaluated by calculating the OS/CS ratio, which equals 1 − (PDC/CS+OLE/CS).
2.3.2. Quantification and 13C-enrichment of triglycerides
Fatty acids from heart tissue triglycerides (TG) were analyzed by GCMS as their methyl ester (FAME) derivative. Briefly, tissue was pulverized under liquid nitrogen and spiked with a labeled external standard ([2H33]heptadecanoic acid). Total tissue lipids were extracted by a modified Folch method [27] in 20-fold weight-to-volume ratio of chloroform/methanol (2:1), containing 0.1% butylated hydroxytoluene (BHT), overnight at 4 °C [28]. Homogenates were then filtered through gauze, dried under nitrogen gas and resuspended in detergent for quantification. TGs were quantified enzymatically with a commercial kit (GPO Trinder; Sigma, USA). Triolein, dissolved in chloroform/methanol and processed similarly to samples, was used as a standard [29]. For measurements of 13C-enrichment in oleate of triglycerides, the dried lipid extracts were resuspended in 100 µl of hexane/chloroform/methanol (95:3:2). Lipid classes were separated using a solid phase extraction aminopropyl column (Varian, Harbor City, USA) according to the method of Ruiz [30]. Samples were applied on a column previously activated with 7.5 ml of hexane and TGs eluted with 5 ml of chloroform stabilized with pentene. The TG fraction was dried, resuspended in 2 ml methanol/hexane (4:1) containing 0.004% BHT, 200 µl acetyl chloride was added and then samples were heated to 80 °C for 1 h to yield FAMEs according to a modification of the method described by Lepage and Roy [31]. The remaining acetyl chloride was neutralized with 5 ml 6% potassium carbonate and the upper hexane phase, containing FAMEs, was collected and analyzed by GCMS on an Agilent Technologies HP-5 column (50 m, 0.2 mm ID, 0.5 µm film thickness) using high-purity helium as the carrier gas at a constant flow-rate of 0.7 ml/min and under the following conditions: 180 °C for 3 min, increased by 5°C/min until 295 °C, then by 25°C/min until 325 °C. At the end of each run, the temperature was kept at 325°C for 4 min to clean the column. Oleate was analyzed in selected ion monitoring mode and the molecular and major ions were quantified for both unlabelled (ions at m/z 296 and 264) and [U-13C18]oleate (ions at m/z 314 and 282) at a retention time of 20.66 min.
2.4. Abundance and phosphorylation status of regulators of TG hydrolysis
2.4.1. Immunoblotting
The abundance and phosphorylation of several proteins were evaluated by Western blot analyses by loading samples from each group in a random fashion and analyzing the corresponding signals in a blinded manner. Tissue extraction, electrophoresis conditions and immunoblotting on nitrocellulose membranes (0.22 µm) were performed as described previously [26,32]. The membranes were probed with antibodies against total αAMPK or phospho-αAMPK (Thr-172) [32], or with commercial antisera against either phospho-HSL (Ser-565), phospho-HSL (Ser-660), total HSL, phospho-ERK1/2, total ERK1/2, or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Immunoreactive bands were quantified by digitizing the ECL signal using a VersaDoc 4000 Gel Imaging System and analyzed using Quantity One software (Bio-Rad, Hercules, USA).
2.4.2. ACC activity
Powdered heart tissue was homogenized in 0.05 M Tris, 0.25 M mannitol, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 5 mM Na pyrophosphate, 10% glycerol, pH 7.5 and protease inhibitor cocktail (4:1 buffer-to-tissue ratio), then centrifuged for 10 min at 800 ×g. ACC assays were based on acetyl-CoA- dependent 14CO2 fixation as described previously [33,34].
2.4.3. Malonyl-CoA and CoASH quantification
Malonyl-CoA was measured as previously described [33]. Briefly, [3H]acetyl-CoA was converted by fatty acid synthase into petroleum ether-soluble material in proportion to the malonyl-CoA content of the sample. Assays were calibrated using exogenous malonyl-CoA as an internal standard. CoASH was measured by a recycling assay using CoASH internal standards [34].
2.4.4. Gene expression analysis
Analyses of changes in gene expression were carried out in hearts freeze-clamped from mice sacrificed in the afternoon (light phase). RNAwas extracted using standard methods and analyzed using reverse transcription followed by real-time quantitative polymerase chain reaction, as described previously [35]. We measured the level of expression of several genes involved in either (i) fatty acid metabolism [namely medium-chain acyl-CoA dehydrogenase (mcad), long-chain acyl-CoA dehydrogenase (lcad), cd36/fatty acid transporter (cd36), fatty acid transport protein (fatp), mitochondrial carnitine palmitoyltransferase 1 (mcpt1), malonyl-CoA dehydrogenase (mcd), long-chain acyl-CoA synthetase (acsl), adipose TG lipase (atgl), diacylglycerol acyltransferase-2 (dgat2) and hormone sensitive lipase (hsl)], or (ii) glucose metabolism [namely glucose transporter 1 (glut1), glucose transporter 4 (glut4), pyruvate dehydrogenase kinase-4 (pdk4) and pyruvate carboxylase (pcx)]. The sequences of the primer and the TaqMan probes for most of the above genes have been previously published [24,25,36,37]. For hsl, the sequences were as follows: forward: 5′-GCGCTGGAGGAGTGTTTTT-3′; reverse: 5′-TGTCCCCTGCAAGGCATAT-3′; probe: 5′-FAM-TCTCCAGTTGAACCAAGCAGGTCACA-TAMRA-3′. For all genes, transcript levels were normalized to total RNA content (as measured by UV spectrophotometry).
2.5. Statistical analysis
Data are expressed as means±SEM. Statistical significance was reached at P ≤ 0.05 using an unpaired t-test, a one-way ANOVA followed by post-hoc Bonferroni-corrected comparison tests.
3. Results
3.1. Perfused GC+/0 hearts show decreased contribution of exogenous oleate to acetyl-CoA and enhanced glycolysis
Upon ex vivo perfusion at a physiological afterload of 50 mmHg with a buffer containing a mixture of substrates and hormones mimicking the in vivo milieu in the fed state (11 mM glucose, 1.5 mM lactate, 0.2 mM pyruvate, 0.4 mM oleate bound to 3% albumin, and 0.8 nM insulin), hearts from both groups maintained similar values for the various functional and physiological parameters over the entire 30-min perfusion period (Table 1). However, LDH release rate (an index of membrane integrity) was significantly decreased in the GC+/0 hearts (Fig. 1).
Table 1.
Functional and physiological parameters of isolated working heart from control WT and GC+/0 mice
| Parameters | WT | GC+/0 | |
|---|---|---|---|
| Heart rate (beats/min) | 447±1 | 429±2 | |
| LVSP (mm Hg) | 87±1 | 92±1 | |
| LVEDP (mm Hg) | 14.8±0.1 | 14.9±0.1 | |
| +dP/dt (mm Hg · s−1) | 4302±35 | 4579±32 | |
| −dP/dt (mm Hg · s−1) | 3582±25 | 3470±31 | |
| Rate pressure product (mm Hg · beats · min−1 · 10−3) | 31819±226 | 32480±167 | |
| Cardiac output (ml/min) | 8.01±0.05 | 7.82±0.05 | |
| Aortic flow (ml/min) | 4.31±0.10 | 4.57±0.07 | |
| Coronary flow (ml/min) | 3.33±0.04 | 3.24±0.02 | |
| Cardiac power (mW) | 1.66±0.10 | 1.67±0.05 | |
| MVO2 (μmol/min) | 1.93±0.11 | 1.71±0.10 | |
|
|
1.01±0.04 | 1.13±0.06 | |
| pHi | 7.37±0.00 | 7.38±0.01 |
Data are means±SEM of 13–17 heart perfusion experiments. Values shown represent averages for the entire perfusion period. MVO2 and pHi were calculated from pO2 and pCO2 values determined in influent and effluent perfusate collected between 15 and 20 min. LVSP, left ventricular systolic pressure; LVEDP, left ventricular end-diastolic pressure; MVO2, oxygen consumption; pHi, intracellular pH.
Fig. 1.
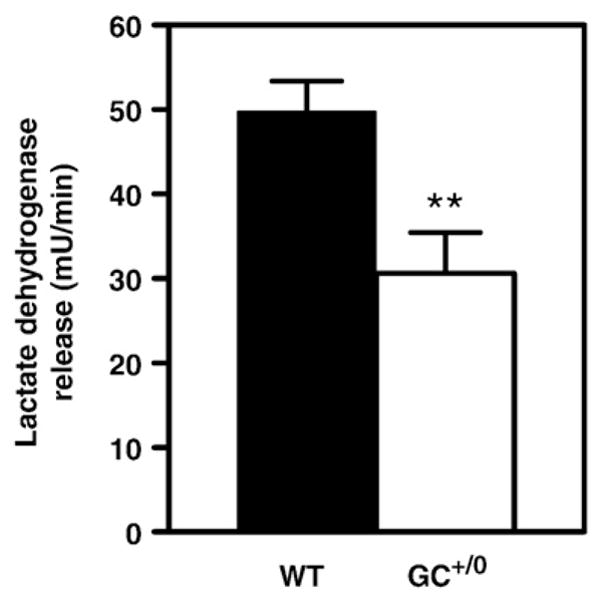
Index of membrane integrity in isolated hearts from control WTand GC+/0 mice. Data are means±SEM of 13–15 heart perfusion experiments. Values shown represent averages over the entire perfusion period. Lactate dehydrogenase release rates of control (WT) mice (solid bars) and hearts from mice overexpressing guanylate cyclase in a cardiomyocyte-specific manner (GC+/0) mice (open bars) were determined enzymatically by spectrophotometric method in effluent perfusates collected every 5 min. **P < 0.01 GC+/0 vs control WT mouse hearts.
In these hearts, the contribution of exogenous substrates to mitochondrial acetyl-CoA formation for citrate synthesis was evaluated using various 13C-labeled substrates. Compared to controls, GC+/0 hearts perfused with [U-13C18]oleate (initial MPE=25%) showed lower values for (i) the MPE M2 of the acetyl moiety of citrate (9.4±2.0 vs 12.4±1, P < 0.05) and, accordingly, (ii) the flux ratio OLE/CS (Table 2), thus demonstrating a significantly decreased formation of acetyl-CoA for citrate synthesis from exogenous fatty acid β-oxidation. In contrast, perfusions with [U-13C6]glucose (initial MPE=25%) or [U-13C3]lactate plus [U-13C3]pyruvate (initial MPE=100%) demonstrated a similar percent contribution of individual carbohydrates to tissue pyruvate formation in GC+/0 and control hearts (Table 2). Furthermore, the contribution of carbohydrates to acetyl-CoA formation for citrate synthesis via mitochondrial pyruvate decarboxylation (i.e. the PDC/CS flux ratio) was also similar in both groups (Table 2). Finally, the OS/CS flux ratio, which reflects the contribution of other sources (essentially endogenous TGs), was calculated to be 17.7±5.2% in GC+/0 hearts, compared to a negligible contribution in WT (−8.6%±5.9%) (P < 0.05).
Table 2.
Metabolic parameters of GC+/0 hearts and their WT littermates
| Parameters | WT | GC+/0 |
|---|---|---|
| OLE/CS | 0.49±0.03 | 0.31±0.05* |
| PDC/CS | 0.59±0.07 | 0.52±0.04 |
| PC/CS | 0.05±0.01 | 0.04±0.01 |
| Contribution of carbohydrates to tissue pyruvate formation (%) | ||
| Glucose | 32±4% | 33±2% |
| Pyruvate+lactate | 54±3% | 54±1% |
| Others | 18±4% | 13±3% |
| Malonyl-CoA concentration (nmol/gww) | 1.16±0.04 | 1.23±0.07 |
| ACC activity (μmol min−1 gww−1) | 0.76±0.12 | 0.83±0.09 |
| [Acetyl-CoA]-to-[CoA] ratio (arbitrary units) | 0.0559±0.0054 | 0.0580±0.0073 |
| Citric acid cycle flux rate (μmol min−1 gww−1) | 2.85±0.20 | 3.11±0.08 |
Data are means±SEM of 3–8 heart perfusion experiments. The Experimental procedures provide details about the (i) determinations of flux ratios, which reflect the contribution exogenous fatty acids (oleate) and of carbohydrates (lactate, pyruvate and glucose) to acetyl-CoA and or oxaloacetate formation for citrate synthesis (CS), and (ii) calculation of citric acid cycle flux rate, and (iii) determinations of levels of CoA derivatives and acetyl-CoA carboxylase (ACC) activity. OLE: oleate oxidation; PDC: pyruvate decarboxylation; PC: pyruvate carboxylation.
P < 0.05.
We also assessed rates of cytosolic glycolysis from the release rates of [U-13C3]lactate and [U-13C3]pyruvate in hearts perfused with [U-13C6]glucose. Both lactate and pyruvate release rates were significantly increased in GC+/0 hearts when compared to WT (Fig. 2A), although their production ratio, which reflects the cytosolic redox [NAD+]-to-[NADH] ratio, was unchanged (Fig. 2B).
Fig. 2.

Lactate and pyruvate production rate and the lactate-to-pyruvate production ratio assessed in isolated working hearts from control WT and GC+/0 mice. Data are means±SEM of 4 heart perfusion experiments. A. Lactate and pyruvate release rates were calculated from the product of coronary flow rates and concentration differences in the influent and effluent perfusates of [U-13C3]lactate (open bars) and [U-13C3]pyruvate (solid bars) in hearts perfused with [U-13C6]glucose as determined by gas chromatography coupled to mass spectrometry (GCMS) and enzymatic assays. B. Lactate-to-pyruvate ratio, in arbitrary units, is expressed as the release rate of lactate divided by the release rate of pyruvate in WT hearts (solid bars) and GC+/0 hearts (open bars). *P < 0.05 and **P < 0.01 GC+/0 vs WT mouse hearts.
The observed alterations in exogenous fatty acid β-oxidation and glycolysis in GC+/0 hearts could not be readily explained by changes in gene expression, since the mRNA levels of key enzymes related to the metabolism of either carbohydrates (glut1, glut4, pdk4, pcx) or fatty acids (mcad, lcad, cd36, fatp, mcpt1, mcd and acsl) were similar in both groups (as assessed by real-time RT-PCR; Suppl. Table 1).
3.2. Lack of alteration in mechanisms regulating β-oxidation in perfused GC+/0 hearts
We investigated other mechanisms involved in β-oxidation regulation, including: (i) the concentration of malonyl-CoA, which potently inhibits carnitine palmitoyltransferase I (CPT-I), an enzyme required for entry of long chain fatty acyl groups into mitochondria; (ii) the activity of ACC, the enzyme responsible for malonyl-CoA synthesis, and (iii) the [acetyl-CoA]-to-[CoA] ratio [38]. As reported in Table 2, none of the measured values for these metabolic parameters were different between GC+/0 and WT hearts. Furthermore, the fact that similar values were obtained in both groups for the [acetyl-CoA]-to-[CoA] ratio and the CAC flux rate, indicates that energy supply from substrate oxidation is not affected by the expression of the transgene.
3.3. Perfused GC+/0 hearts shuttle more exogenous 13C-labeled oleate into TGs
Next, we evaluated whether the decreased contribution of exogenous [U-13C18]oleate to acetyl-CoA formation observed in perfused GC+/0 hearts could result from its preferential partitioning into TG synthesis. While there were no differences in TG content before and after perfusion of GC+/0 hearts (4.92± 1.5 vs 4.20±1.76 μmol/gww; NS), we found a significant 2.5-fold increase in the MPE of the oleate moiety of TGs in GC+/0 when compared to WT hearts (Fig. 3A). As reflected by a labeling ratio of ~1.0 between the MPE of oleate in TGs and that of the acetyl moiety of citrate (Fig. 3B) in perfused GC+/0 hearts, there is an equal partitioning of [U-13C18]oleate between its two primary metabolic fates, namely esterification and β-oxidation, respectively. This contrasts with WT hearts, where the amounts of exogenous oleate that were shuttled into TG stores are about 3 times lower than those used for β-oxidation (Fig. 3B).
Fig. 3.

Incorporation of exogenous oleate into triglyceride stores of isolated working hearts from control WT and GC+/0 mice. Data are means±SEM of 5 heart perfusion experiments. The 13C-enrichment of oleate in triglycerides was assessed in hearts following a 30-min perfusion in the working mode with [U-13C18]oleate by GCMS analysis of fatty acid methyl esters. Data are expressed as (A) absolute molar percent enrichment (MPE) or (B) relative to the MPE of the acetyl moiety of citrate. *P < 0.05 GC+/0 vs WT hearts.
3.4. GC+/0 hearts exhibit alterations in factors influencing TG lipolysis
The increased incorporation of exogenous oleate into TGs in GC+/0 hearts was paradoxical given the fact that total TG content was not altered. This paradox could be explained if GC+/0 hearts exhibited a concomitant increase in TG hydrolysis. The finding that the 17±11% OS/CS flux ratio in GC+/0 hearts was greater than the negligible values measured in WT suggested that this was indeed a likely possibility. Consequently, we tested whether factors known to influence TG turnover, namely HSL and AMPK [39,40], were affected by expression of the transgene. Compared to controls, GC+/0 hearts had ~60% higher levels of HSL protein (Fig. 4A) and increased mRNA concentration (Suppl. Table 1), but we found no difference in the transcript levels of other enzymes involved in TG metabolism, namely atgl and dgat2 (Suppl. Table 1). In addition to changes in HSL abundance, we also detected a 53.4±6.1% decrease in the phosphorylation of HSL at Ser-565 in GC+/0 versus WT hearts (Fig. 4B). Ser-565 is a known target of AMPK, and phosphorylation of this residue inhibits HSL activity [41]. In keeping with its decreased phosphorylation in GC+/0 hearts, we found that phosphorylation of AMPK at Thr-172 (which results in its activation) was significantly decreased by 36.7±13.4% in GC+/0 hearts compared to their WT counterparts (Fig. 4C). Phosphorylation at Ser-660 (which activates HSL activity [41]) was not changed (data not shown). Finally, ERK1/2 are other kinases known to activate HSL via phosphorylation of Ser-600 [41]. The phosphorylation of ERK1 at its activation site, was significantly increased by 64± 18% in GC+/0 hearts, while phosphorylation of ERK2 showed a trend towards an increase (Fig. 4D). Note that for HSL abundance and phophorylation levels as well as for ERK1/2 phosphorylation levels, we have obtained the same results twice with different animals.
Fig. 4.

Mechanisms regulating lipolysis: levels and phosphorylation status of hormone sensitive lipase, AMP-activated kinase and extracellular signal regulated kinases 1/2 in WT and GC+/0 mouse hearts. Data are means±SEM of 4–5 (A, B and D) and 8 (C) freeze-clamped hearts of WT (solid bars) and GC+/0 (open bars) mouse hearts. Representative immunoblots using: (A) hormone-sensitive lipase (HSL) protein normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), (B) anti-phospho-HSL (Ser-565) with anti-HSL serving as loading controls for total HSL protein total, (C) anti-phospho-αAMP-activated kinase (αAMPK; Thr-172) antibodies with anti-αAMPK antibodies serving as loading controls for total αAMPK protein, and (D) anti-phospho extracellular signal regulated kinases 1/2 (ERK1/2) with anti-ERK1/2 serving as loading controls for total ERK 1/2 protein. Densitometry of phosphorylated protein to total protein ratios from experiments performed on tissue homogenates extracts is shown. *P < 0.05 and **P < 0.01 GC+/0 vs WT hearts.
3.5. GC+/0 hearts are resistant to fasting-induced TG accumulation
To assess the significance of the documented changes in exogenous long chain fatty acid handling by GC+/0 hearts, WT and GC+/0 mice were subjected to a 24-h fast, a condition that increases myocardial TG levels [42]. In contrast to WT hearts, hearts from fasted GC+/0 mice did not show the expected increase in TGs in response to fasting (Fig. 5A). We also assessed the abundance of total HSL and its phosphorylation status at the AMPK-dependent Ser-565 inhibitory site in fasted animals. Similar to hearts from fed animals shown in Fig. 4, total HSL was greater and phospho-HSL at Ser-565 was lower in GC+/0 than WT mice. However, the differences in total HSL between the two groups were amplified after fasting, since total HSL was ~4.6 fold higher in GC+/0 than in their WT counterparts (as opposed to the 62% increase we had detected in fed animals.
Fig. 5.

Effect of fasting on triglyceride levels, the abundance of HSL and its inhibitory phosphorylation at Ser-565 in hearts of WT and GC+/0 mouse hearts. Data are means±SEM of 3-5 freeze-clamped hearts. (A) Triglyceride content of WT (solid bars) and GC+/0 (open bars) mouse hearts, either fed or following a 24-h fast (fasted). (B) HSL protein, normalized to GAPDH, and (C) phosphorylation levels of HSL (Ser-565), normalized against total HSL protein, in fasted WT and GC+/0 mouse hearts. Western blot analyses were as described in Fig. 4. *P < 0.05 GC+/0 vs WT hearts.
4. Discussion
While multiple studies have shown that enhanced cGMP signaling exerts numerous cardioprotective effects [9–11,43], little is known about its impact on the heart's substrate utilization for energy and storage. Collectively, our results demonstrate that enhanced cGMP signaling within cardiomyo-cytes modifies exogenous substrate utilization, including fatty acid partitioning between β-oxidation (energy production) and esterification to TGs (for storage), in the absence of changes in cardiac function.
Specifically, we initially found that the contribution of exogenous oleate to acetyl-CoA formation was significantly decreased in GC+/0 mouse hearts, while that of carbohydrates was unchanged. Despite this decrease in the OLE/CS flux ratio, the levels of acetyl-CoA and free CoA, the calculated CAC flux rate, the MVO2 and the contractile function in perfused GC+/0 mouse hearts all remained similar to their WT counterparts. This suggested that the myocardial energy status was unaffected and that the decreased contribution of exogenous oleate to β-oxidation did not necessarily mean that total fatty acid β-oxidation was decreased. Accordingly, we found no change in the tissue level of malonyl-CoA (a known inhibitor of β-oxidation [44]) and, likewise, in the activity of ACC. In contrast, we found that the incorporation of exogenous 13C-labeled oleate into TGs was increased almost 3-fold in perfused GC+/0 mouse hearts.
Given that the TG content remained constant during perfusion in GC+/0 mouse hearts, we reasoned that the increased incorporation of exogenous oleate into TGs was compensated by a matching increase in the mobilization of unlabeled long chain fatty acid from endogenous TG stores (i.e. turnover), which could subsequently undergo β-oxidation to acetyl-CoA. This possibility is very likely given that: (i) the OS/CS flux ratio was greater in GC+/0 hearts than in their WT counterparts; (ii) the abundance of HSL protein and mRNA was also greater in the hearts of GC+/0 mice; and (iii) several phosphorylation events occurring in the hearts of GC+/0 mice should further contribute to enhance the activity of HSL. For instance, phosphorylation of HSL at the Ser-565 site (which inhibits the activity of this enzyme [45]) is decreased in GC+/0 hearts. Phosphorylation of Ser-565 is the mechanism by which AMPK (a major regulator of lipolysis [46]) inhibits HSL activity [45]. Accordingly, phosphorylation of AMPK at Thr-172 (an event that activates AMPK activity) was significantly lower in the hearts of GC+/0 mice. Of note, ANF has been shown to induce lipolysis in human adipose tissue precisely by activating HSL via the cGMP-dependent inactivation of AMPK [16,45]. HSL activity can be regulated via the phosphorylation of other of its amino acids. We found no changes in phosphorylation of HSL at Ser-660, a site by which protein kinase A (PKA) activates HSL. In contrast, phospho-ERK 1 was increased in GC+/0 hearts compared to their WT counterparts. Activated ERK 1/2 can enhance the activity of HSL via phosphorylation of Ser-600 [41] (for which phospho-specific antibodies are unfortunately not available). Phosphorylation of ERK1 in GC+/0 hearts is also compatible with several reports indicating that the cGMP signaling pathway is a strong activator of ERK1/2 [47–49], this event having even been reported as necessary for ANF-mediated cardioprotection [49].
Collectively, our isotopic and molecular data support the notion that enhanced cGMP signaling in cardiomyocytes promotes both TG synthesis and hydrolysis concomitantly. While an in depth mechanistic study of the link between cGMP and the observed effects was beyond the scope of this study, our results support the following mechanistic scheme in GC+/0 mouse heart: enhanced cGMP signaling phosphorylates (most likely via cGMP-dependent kinase) and activates ERK1/2, which subsequently phosphorylates and activates HSL, an event that is expected to promote TG hydrolysis. The activity of HSL in the heart has been estimated (assuming 200 mg protein per g heart wet weight) to be ~ 7 nmol min−1 gww−1 under basal condition [42]. Although we do not know up to which levels cardiac HSL activity can be increased to by stimulating condition, it has been shown in white adipose tissue that its activity can be increased up to ~15-fold [40]. Thus, assuming that cardiac HSL activity could vary from ~7 to 105 nmol min−1 gww−1, these values are within the range of the maximal rate of acetyl-CoA formation from endogenous TG-derived fatty acids, which we estimated to be ~ ~64 nmol min−1 gww−1 (from the OS/CS flux ratio and CAC flux rate assuming that endogenously formed palmitate is the only substrate contributing to this flux). On this basis, HSL could be responsible, at least in part, and in addition to other players (such as adipose TG lipase) of hydrolyzing sufficient amounts of endogenous TG hydrolysis to provide fatty acids to mitochondrial β-oxidation. It is also noteworthy that, at least in liver and skeletal muscle, decreased AMPK activity would be expected to increase glycerol-3-phosphate acyltransferase 1 activity, the first enzyme involved in TG synthesis [50,51]. This may suggest a mechanism for the increased [U-13C18]oleate incorporation into TG in GC+/0 mouse heart.
Similarly to what has been reported in transgenic mice with heart-specific overexpression of HSL[42] (and contrary to control mice), fasting did not increase myocardial TG accumulation in GC+/0 mice (Fig. 5A). This effect is likely to be the functional consequence of cGMP-dependent modulation of HSL activity. Indeed, our molecular data indicate that food removal decreased the abundance of HSL protein (two-fold) while increasing its inhibition by Ser-565 (1.5-fold) in WT hearts but not in their GC+/0 counterparts (from Figs. 4 and 5). The importance of lipolysis for normal cardiac homeostasis has recently been emphasized by studies in transgenic mice lacking enzymes involved in this process. For example, hearts lacking adipose TG lipase develop severe cardiac dysfunction and altered energy metabolism [52]. One of the potential benefits of increased TG synthesis/hydrolysis appears to be increased compartmentalization of lipids in order to reduce their cardiotoxic effects [53]. However, additional studies are necessary to test whether increased intracellular turnover of long chain fatty acids is by itself sufficient to confer protection against non-metabolic insults, such as pressure-overload hypertrophy.
Beyond the aforementioned changes in lipid trafficking in GC+/0 mouse hearts, our finding of enhanced lactate and pyruvate production suggests that glucose uptake and glycolysis are also increased by enhanced cGMP signaling in these hearts. This finding concurs with that of Li et al. [19] who reported a stimulation of glucose uptake by activation of cGMP signaling in heart papillary muscle. However, it contrasts with other studies reporting that glucose uptake is (i) increased following inhibition of NO synthase by either pharmacologic inhibitors or gene inactivation (which decrease the cardiac concentration of cGMP) [14,18] and, conversely, it is (ii) decreased using cGMP agonists or NO donors (which increase the cardiac concentration of cGMP) [15]. Several factors may explain these differences. Specifically, cGMP signaling in our particular model is increased chronically and specifically in cardiomyocytes, which excludes possible effects mediated by (i) cells other than cardiomyocytes and by (ii) NO, which exerts cGMP-independent metabolic effects such as inhibition of GAPDH through enhanced ADP-ribosylation [54], and of phosphofructokinase [55]. While the mechanism underlying the enhanced glycolysis in GC+/0 mouse hearts remains to be clarified, its potential benefit for the heart is discussed below.
Interestingly, the metabolic profile of GC+/0 mouse hearts (namely decreased β-oxidation of exogenous fatty acids, no change in carbohydrate oxidation and increased glycolysis) is partly reminiscent of that seen in hypertrophied hearts [56–58]. While it is still controversial whether a shift in exogenous substrate selection from fatty acid to carbohydrates is a compensatory or maladaptive mechanism, our finding of a similar shift in GC+/0 mouse hearts suggests that it is beneficial and cardioprotective. Similar shifts have also been reported in several other models of cardioprotection, including mechanically unloaded hearts [59] as well as the calcineurin-knockout mouse [60] or the glycogen synthase kinase-3 overexpressing mouse [61], where cardioprotection is accompanied by increased expression of ANF. However, two striking and distinctive features that we documented in our model are that: (i) TG synthesis is enhanced concomitantly with lipolysis; and (ii) the cytosolic redox state of the cells is conserved. To the best of our knowledge (and in contrast to the present study), previous reports have not assessed directly whether partitioning of exogenous fatty acids between β-oxidation and esterification is altered in hypertrophied hearts. Nonetheless, we had previously found (using a stable isotope approach in ex vivo perfusion) that endogenous sources (postulated at the time to be TGs) accounted for up to 20% of acetyl-CoA formation in hypertrophied hearts from spontaneously hypertensive rats, while it was 9±1% and negligible in control WKY and Wistar rat hearts, respectively [22].
With regards to the potential cardioprotective effects of cGMP signaling, an interesting peripheral observation of this study was that the expression of the transgene resulted in decreased release of LDH during ex vivo perfusion. While providing what are generally accepted to be physiological levels of workload, nutrients and calcium, the ex vivo working heart perfusion still constitutes a mild stress [21]. It is noteworthy, however, that the magnitude of the LDH release in hearts perfused under normoxia is marginal compared to that observed following reperfusion after ischemia or anoxia [62]. The cGMP-dependent reduction of LDH release may be independent of the aforementioned metabolic effects, since cGMP prevents cell death by combined effects on the mitochondrial transition pore, intracellular calcium regulation and stress-activated signaling [47,63,64]. Alternatively, increased availability of cytosolic glycolytically-derived ATP might improve ionic homeostasis, as multiple membrane-associated channels preferentially use glycolytically-derived ATP [65–69], and thus contribute in this fashion to improved membrane integrity.
In summary, this study provides direct evidence that cGMP signaling modulates cardiac energy metabolism in a cardiomyocyte-specific manner. Through an HSL-dependent activation of lipolysis, cGMP specifically acts on lipid compartmentalization. This may mediate some of its beneficial effects by favoring TG cycling and thereby preventing TG accumulation in cardiomyocytes, especially under conditions such as fasting where circulating long chain fatty acids are increased. Further investigations appear warranted to link the herein described effect of cGMP on lipid compartmentalization to its cardioprotective effects. This effect may be relevant to models of high fat feeding and diabetes where fatty acid-induced toxic effects has been reported [70–72], but might also contribute (at least in part) to the protective effects of cGMP against other cardiomyopathies including pressure overload and dystrophic hearts [10,11].
Supplementary Material
Acknowledgments
This work was presented at the at the Society Heart and Vascular Metabolism held in Semihamoo, in September 2006, and in Maastricht in June 2007, and at the World Congress of the International Society of Heart Research held in Bologna in June 2007. This study was supported by the Canadian Institutes of Health Research (grants #74460 to C.F.D. and C.D.R., #77791 to B.G.A. and #10865 to G.D.L.), by the Heart and Stroke Foundation of Canada (studentship to M.K.), and by the National Heart, Lung, and Blood Institute (grants #HL-074259 to MEY). B.G.A. is a senior scientist of the FRSQ.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.yjmcc.2008.05.012.
References
- 1.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005 Jul;85(3):1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 2.Neubauer S. The failing heart—an engine out of fuel. N Engl J Med. 2007 Mar 15;356(11):1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 3.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004 Nov;18(14):1692–700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 4.Borradaile NM, Schaffer JE. Lipotoxicity in the heart. Curr Hypertens Rep. 2005 Dec;7(6):412–7. doi: 10.1007/s11906-005-0035-y. [DOI] [PubMed] [Google Scholar]
- 5.Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol. 2006 Jul 1;574(Pt 1):95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Neill BT, Abel ED. Akt1 in the cardiovascular system: friend or foe? J Clin Invest. 2005 Aug;115(8):2059–64. doi: 10.1172/JCI25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem. 2006 Oct 27;281(43):32841–51. doi: 10.1074/jbc.M513087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costa AD, Garlid KD, West IC, Lincoln TM, Downey JM, Cohen MV, et al. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res. 2005 Aug 19;97(4):329–36. doi: 10.1161/01.RES.0000178451.08719.5b. [DOI] [PubMed] [Google Scholar]
- 9.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005 Feb;11(2):214–22. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 10.Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF. Expression of constitutively active guanylate cyclase in cardiomyocytes inhibits the hypertrophic effects of isoproterenol and aortic constriction on mouse hearts. J Biol Chem. 2003 Nov 28;278(48):47694–9. doi: 10.1074/jbc.M309661200. [DOI] [PubMed] [Google Scholar]
- 11.Khairallah M, Khairallah RJ, Young ME, Allen BG, Gillis MA, Danialou G, et al. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc Natl Acad Sci U S A. 2008 May 13;105(19):7028–33. doi: 10.1073/pnas.0710595105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Recchia FA, McConnell PI, Loke KE, Xu X, Ochoa M, Hintze TH. Nitric oxide controls cardiac substrate utilization in the conscious dog. Cardiovasc Res. 1999 Nov;44(2):325–32. doi: 10.1016/s0008-6363(99)00245-x. [DOI] [PubMed] [Google Scholar]
- 13.Recchia FA, Osorio JC, Chandler MP, Xu X, Panchal AR, Lopaschuk GD, et al. Reduced synthesis of NO causes marked alterations in myocardial substrate metabolism in conscious dogs. Am J Physiol Endocrinol Metab. 2002 Jan;282(1):E197–206. doi: 10.1152/ajpendo.2002.282.1.E197. [DOI] [PubMed] [Google Scholar]
- 14.Depre C, Vanoverschelde JL, Goudemant JF, Mottet I, Hue L. Protection against ischemic injury by nonvasoactive concentrations of nitric oxide synthase inhibitors in the perfused rabbit heart. Circulation. 1995 Oct 1;92(7):1911–8. doi: 10.1161/01.cir.92.7.1911. [DOI] [PubMed] [Google Scholar]
- 15.Depre C, Gaussin V, Ponchaut S, Fischer Y, Vanoverschelde JL, Hue L. Inhibition of myocardial glucose uptake by cGMP. Am J Physiol. 1998 May;274(5 Pt 2):H1443–9. doi: 10.1152/ajpheart.1998.274.5.H1443. [DOI] [PubMed] [Google Scholar]
- 16.Sengenes C, Bouloumie A, Hauner H, Berlan M, Busse R, Lafontan M, et al. Involvement of a cGMP-dependent pathway in the natriuretic peptide-mediated hormone-sensitive lipase phosphorylation in human adipocytes. J Biol Chem. 2003 Dec 5;278(49):48617–26. doi: 10.1074/jbc.M303713200. [DOI] [PubMed] [Google Scholar]
- 17.Fischmeister R, Castro LR, bi-Gerges A, Rochais F, Jurevicius J, Leroy J, et al. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006 Oct 13;99(8):816–28. doi: 10.1161/01.RES.0000246118.98832.04. [DOI] [PubMed] [Google Scholar]
- 18.Tada H, Thompson CI, Recchia FA, Loke KE, Ochoa M, Smith CJ, et al. Myocardial glucose uptake is regulated by nitric oxide via endothelial nitric oxide synthase in Langendorff mouse heart. Circ Res. 2000 Feb 18;86(3):270–4. doi: 10.1161/01.res.86.3.270. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Hu X, Selvakumar P, Russell RR, III, Cushman SW, Holman GD, et al. Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle. Am J Physiol Endocrinol Metab. 2004 Nov;287(5):E834–41. doi: 10.1152/ajpendo.00234.2004. [DOI] [PubMed] [Google Scholar]
- 20.Khairallah M, Labarthe F, Bouchard B, Danialou G, Petrof BJ, Des Rosiers C. Profiling substrate fluxes in the isolated working mouse heart using 13C-labeled substrates: focusing on the origin and fate of pyruvate and citrate carbons. Am J Physiol Heart Circ Physiol. 2004 Apr;286(4):H1461–70. doi: 10.1152/ajpheart.00942.2003. [DOI] [PubMed] [Google Scholar]
- 21.Khairallah M, Khairallah R, Young ME, Dyck JR, Petrof BJ, Des Rosiers C. Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. J Mol Cell Cardiol. 2007 May 24; doi: 10.1016/j.yjmcc.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 22.Labarthe F, Khairallah M, Bouchard B, Stanley WC, Des Rosiers C. Fatty acid oxidation and its impact on response of spontaneously hypertensive rat hearts to an adrenergic stress: benefits of a medium-chain fatty acid. Am J Physiol Heart Circ Physiol. 2005 Mar;288(3):H1425–36. doi: 10.1152/ajpheart.00722.2004. [DOI] [PubMed] [Google Scholar]
- 23.Vincent G, Bouchard B, Khairallah M, Des Rosiers C. Differential modulation of citrate synthesis and release by fatty acids in perfused working rat hearts. Am J Physiol Heart Circ Physiol. 2004 Jan;286(1):H257–66. doi: 10.1152/ajpheart.00717.2003. [DOI] [PubMed] [Google Scholar]
- 24.Durgan DJ, Trexler NA, Egbejimi O, McElfresh TA, Suk HY, Petterson LE, et al. The circadian clock within the cardiomyocyte is essential for responsiveness of the heart to fatty acids. J Biol Chem. 2006 Aug 25;281( 34):24254–69. doi: 10.1074/jbc.M601704200. [DOI] [PubMed] [Google Scholar]
- 25.Young ME, Razeghi P, Cedars AM, Guthrie PH, Taegtmeyer H. Intrinsic diurnal variations in cardiac metabolism and contractile function. Circ Res. 2001 Dec 7;89(12):1199–208. doi: 10.1161/hh2401.100741. [DOI] [PubMed] [Google Scholar]
- 26.Boivin B, Villeneuve LR, Farhat N, Chevalier D, Allen BG. Sub-cellular distribution of endothelin signaling pathway components in ventricular myocytes and heart: lack of preformed caveolar signalosomes. J Mol Cell Cardiol. 2005 Apr;38(4):665–76. doi: 10.1016/j.yjmcc.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957 May;226( 1):497–509. [PubMed] [Google Scholar]
- 28.Ametaj BN, Bobe G, Lu Y, Young JW, Beitz DC. Effect of sample preparation, length of time, and sample size on quantification of total lipids from bovine liver. J Agric Food Chem. 2003 Apr 9;51(8):2105–10. doi: 10.1021/jf0259011. [DOI] [PubMed] [Google Scholar]
- 29.Roduit R, Masiello P, Wang SP, Li H, Mitchell GA, Prentki M. A role for hormone-sensitive lipase in glucose-stimulated insulin secretion: a study in hormone-sensitive lipase-deficient mice. Diabetes. 2001 Sep;50(9):1970–5. doi: 10.2337/diabetes.50.9.1970. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz J, Antequera T, Andres AI, Petron M, Muriel E. Improvement of a solid phase extraction method for analysis of lipid fractions in muscle foods. Analytica Chimica Acta. 2004 Aug 23;520(1–2):201–5. [Google Scholar]
- 31.Lepage G, Levy E, Ronco N, Smith L, Galeano N, Roy CC. Direct transesterification of plasma fatty acids for the diagnosis of essential fatty acid deficiency in cystic fibrosis. J Lipid Res. 1989 Oct;30(10):1483–90. [PubMed] [Google Scholar]
- 32.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004 Jul 30;279(31):32771–9. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- 33.Vavvas D, Apazidis A, Saha AK, Gamble J, Patel A, Kemp BE, et al. Contraction-induced changes in acetyl-CoA carboxylase and 5′-AMP-activated kinase in skeletal muscle. J Biol Chem. 1997 May 16;272(20):13255–61. doi: 10.1074/jbc.272.20.13255. [DOI] [PubMed] [Google Scholar]
- 34.Goodwin GW, Taegtmeyer H. Regulation of fatty acid oxidation of the heart by MCD and ACC during contractile stimulation. Am J Physiol. 1999 Oct;277(4 Pt 1):E772–7. doi: 10.1152/ajpendo.1999.277.4.E772. [DOI] [PubMed] [Google Scholar]
- 35.Lei B, Lionetti V, Young ME, Chandler MP, d’Agostino C, Kang E, et al. Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J Mol Cell Cardiol. 2004 Apr;36(4):567–76. doi: 10.1016/j.yjmcc.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 36.Dyck JR, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M, et al. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation. 2006 Oct 17;114(16):1721–8. doi: 10.1161/CIRCULATIONAHA.106.642009. [DOI] [PubMed] [Google Scholar]
- 37.Durgan DJ, Smith JK, Hotze MA, Egbejimi O, Cuthbert KD, Zaha VG, et al. Distinct transcriptional regulation of long-chain acyl-CoA synthetase isoforms and cytosolic thioesterase 1 in the rodent heart by fatty acids and insulin. Am J Physiol Heart Circ Physiol. 2006 Jun;290(6):H2480–97. doi: 10.1152/ajpheart.01344.2005. [DOI] [PubMed] [Google Scholar]
- 38.Kerbey AL, Randle PJ, Cooper RH, Whitehouse S, Pask HT, Denton RM. Regulation of pyruvate dehydrogenase in rat heart. Mechanism of regulation of proportions of dephosphorylated and phosphorylated enzyme by oxidation of fatty acids and ketone bodies and of effects of diabetes: role of coenzyme A, acetyl-coenzyme A and reduced and oxidized nicotinamide-adenine dinucleotide. Biochem J. 1976 Feb 15;154(2):327–48. doi: 10.1042/bj1540327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Small CA, Garton AJ, Yeaman SJ. The presence and role of hormone-sensitive lipase in heart muscle. Biochem J. 1989 Feb 15;258(1):67–72. doi: 10.1042/bj2580067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schweiger M, Schreiber R, Haemmerle G, Lass A, Fledelius C, Jacobsen P, et al. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J Biol Chem. 2006 Dec 29;281(52):40236–41. doi: 10.1074/jbc.M608048200. [DOI] [PubMed] [Google Scholar]
- 41.Holm C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem Soc Trans. 2003 Dec;31(Pt 6):1120–4. doi: 10.1042/bst0311120. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki J, Shen WJ, Nelson BD, Patel S, Veerkamp JH, Selwood SP, et al. Absence of cardiac lipid accumulation in transgenic mice with heart-specific HSL overexpression. Am J Physiol Endocrinol Metab. 2001 Oct;281(4):E857–66. doi: 10.1152/ajpendo.2001.281.4.E857. [DOI] [PubMed] [Google Scholar]
- 43.Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Invest. 1998 Feb 15;101(4):812–8. doi: 10.1172/JCI119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuthbert KD, Dyck JR. Malonyl-CoA decarboxylase is a major regulator of myocardial fatty acid oxidation. Curr Hypertens Rep. 2005 Dec;7(6):407–11. doi: 10.1007/s11906-005-0034-z. [DOI] [PubMed] [Google Scholar]
- 45.Garton AJ, Campbell DG, Carling D, Hardie DG, Colbran RJ, Yeaman SJ. Phosphorylation of bovine hormone-sensitive lipase by the AMP-activated protein kinase. A possible antilipolytic mechanism. Eur J Biochem. 1989 Jan 15;179(1):249–54. doi: 10.1111/j.1432-1033.1989.tb14548.x. [DOI] [PubMed] [Google Scholar]
- 46.Yeaman SJ. Hormone-sensitive lipase—new roles for an old enzyme. Biochem J. 2004 Apr 1;379(Pt 1):11–22. doi: 10.1042/BJ20031811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Das A, Smolenski A, Lohmann SM, Kukreja RC. Cyclic GMP-dependent protein kinase Ialpha attenuates necrosis and apoptosis following ischemia/reoxygenation in adult cardiomyocyte. J Biol Chem. 2006 Dec 15;281(50):38644–52. doi: 10.1074/jbc.M606142200. [DOI] [PubMed] [Google Scholar]
- 48.Parenti A, Morbidelli L, Cui XL, Douglas JG, Hood JD, Granger HJ, et al. Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase1/2 activation in postcapillary endothelium. J Biol Chem. 1998 Feb 13;273(7):4220–6. doi: 10.1074/jbc.273.7.4220. [DOI] [PubMed] [Google Scholar]
- 49.Silberbach M, Gorenc T, Hershberger RE, Stork PJ, Steyger PS, Roberts CT., Jr Extracellular signal-regulated protein kinase activation is required for the anti-hypertrophic effect of atrial natriuretic factor in neonatal rat ventricular myocytes. J Biol Chem. 1999 Aug 27;274(35):24858–64. doi: 10.1074/jbc.274.35.24858. [DOI] [PubMed] [Google Scholar]
- 50.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004 Mar;43(2):134–76. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 51.Muoio DM, Seefeld K, Witters LA, Coleman RA. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem J. 1999 Mar 15;338(Pt 3):783–91. [PMC free article] [PubMed] [Google Scholar]
- 52.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006 May 5;312(5774):734–7. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 53.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003 Mar 18;100(6):3077–82. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dimmeler S, Lottspeich F, Brune B. Nitric oxide causes ADP-ribosylation and inhibition of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1992 Aug 25;267(24):16771–4. [PubMed] [Google Scholar]
- 55.Tsuura Y, Ishida H, Shinomura T, Nishimura M, Seino Y. Endogenous nitric oxide inhibits glucose-induced insulin secretion by suppression of phosphofructokinase activity in pancreatic islets. Biochem Biophys Res Commun. 1998 Nov 9;252(1):34–8. doi: 10.1006/bbrc.1998.9601. [DOI] [PubMed] [Google Scholar]
- 56.Allard MF, Wambolt RB, Longnus SL, Grist M, Lydell CP, Parsons HL, et al. Hypertrophied rat hearts are less responsive to the metabolic and functional effects of insulin. Am J Physiol Endocrinol Metab. 2000 Sep;279(3):E487–93. doi: 10.1152/ajpendo.2000.279.3.E487. [DOI] [PubMed] [Google Scholar]
- 57.Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, et al. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension. 2004 Nov;44(5):662–7. doi: 10.1161/01.HYP.0000144292.69599.0c. [DOI] [PubMed] [Google Scholar]
- 58.Lydell CP, Chan A, Wambolt RB, Sambandam N, Parsons H, Bondy GP, et al. Pyruvate dehydrogenase and the regulation of glucose oxidation in hypertrophied rat hearts. Cardiovasc Res. 2002 Mar;53(4):841–51. doi: 10.1016/s0008-6363(01)00560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Depre C, Shipley GL, Chen W, Han Q, Doenst T, Moore ML, et al. Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat Med. 1998 Nov;4(11):1269–75. doi: 10.1038/3253. [DOI] [PubMed] [Google Scholar]
- 60.Bueno OF, Wilkins BJ, Tymitz KM, Glascock BJ, Kimball TF, Lorenz JN, et al. Impaired cardiac hypertrophic response in Calcineurin Abeta -deficient mice. Proc Natl Acad Sci U S A. 2002 Apr 2;99(7):4586–91. doi: 10.1073/pnas.072647999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, et al. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2002 Jan 22;99(2):907–12. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marshall T, Williams J, Williams KM. Electrophoresis of human serum proteins following acute myocardial infarction. Biochem Soc Trans. 1994 Aug;22(3):312S. doi: 10.1042/bst022312s. [DOI] [PubMed] [Google Scholar]
- 63.Abdallah Y, Gkatzoflia A, Pieper H, Zoga E, Walther S, Kasseckert S, et al. Mechanism of cGMP-mediated protection in a cellular model of myocardial reperfusion injury. Cardiovasc Res. 2005 Apr 1;66(1):123–31. doi: 10.1016/j.cardiores.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 64.Monastyrskaya E, Folarin N, Malyshev I, Green C, Andreeva L. Application of the nitric oxide donor SNAP to cardiomyocytes in culture provides protection against oxidative stress. Nitric Oxide. 2002 Sep;7(2):127–31. doi: 10.1016/s1089-8603(02)00107-6. [DOI] [PubMed] [Google Scholar]
- 65.Weiss JN, Lamp ST. Cardiac ATP-sensitive K+ channels. Evidence for preferential regulation by glycolysis. J Gen Physiol. 1989 Nov;94(5):911–35. doi: 10.1085/jgp.94.5.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aasum E, Lathrop DA, Henden T, Sundset R, Larsen TS. The role of glycolysis in myocardial calcium control. J Mol Cell Cardiol. 1998 Sep;30(9):1703–12. doi: 10.1006/jmcc.1998.0732. [DOI] [PubMed] [Google Scholar]
- 67.Xu KY, Zweier JL, Becker LC. Functional coupling between glycolysis and sarcoplasmic reticulum Ca2+ transport. Circ Res. 1995 Jul;77(1):88–97. doi: 10.1161/01.res.77.1.88. [DOI] [PubMed] [Google Scholar]
- 68.Glitsch HG, Tappe A. The Na+/K+ pump of cardiac Purkinje cells is preferentially fuelled by glycolytic ATP production. Pflugers Arch. 1993 Jan;422(4):380–5. doi: 10.1007/BF00374294. [DOI] [PubMed] [Google Scholar]
- 69.Paul RJ, Hardin CD, Raeymaekers L, Wuytack F, Casteels R. Preferential support of Ca2+ uptake in smooth muscle plasma membrane vesicles by an endogenous glycolytic cascade. FASEB J. 1989 Sep;3(11):2298–301. doi: 10.1096/fasebj.3.11.2528493. [DOI] [PubMed] [Google Scholar]
- 70.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 2000 Feb 15;97(4):1784–9. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002 Jan;109(1):121–30. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Unger RH, Orci L. Lipotoxic diseases of nonadipose tissues in obesity. Int J Obes Relat Metab Disord. 2000 Nov;24(Suppl 4):S28–32. doi: 10.1038/sj.ijo.0801498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.