

Abstract
The peptidyl prolyl isomerase Pin1 has two domains that are considered to be its binding (WW) and catalytic (PPIase) domains, both of which interact with phosphorylated Ser/Thr-Pro motifs. This shared specificity might influence substrate selection, since many known Pin1 substrates have multiple sequentially close phosphoSer/Thr-Pro motifs, including the protein IRAK1. The IRAK1 undefined domain (UD) contains two sets of such neighboring motifs (Ser131/Ser144 and Ser163/Ser173), suggesting possible bivalent interactions with Pin1. Using a series of NMR titrations with 15N-labeled full-length Pin1 (Pin1-FL), PPIase, or WW domain and phosphopeptides representing the Ser131/Ser144 and Ser163/Ser173 regions of IRAK1-UD, bivalent interactions were investigated. Binding studies using singly-phosphorylated peptides showed that individual motifs displayed weak affinities (>100 μM) for Pin1-FL and each isolated domain. Analysis of dually phosphorylated peptides binding to Pin1-FL showed that inclusion of bivalent states was necessary to fit the data. The resulting complex model and fitted parameters were applied to predict the impact of bivalent states at low micromolar concentrations, demonstrating significant affinity enhancement for both dually phosphorylated peptides (3.5 μM and 24 μM for peptides based on the Ser131/Ser144 and Ser163/Ser173 regions, respectively). The complementary technique biolayer interferometry confirmed the predicted affinity enhancement for a representative set of singly and dually phosphorylated Ser131/Ser144 peptides at low micromolar concentrations, validating model predictions. These studies provide novel insights regarding the complexity of interactions between Pin1 and activated IRAK1, and more broadly suggest that phosphorylation of neighboring Ser/Thr-Pro motifs in proteins might provide competitive advantage at cellular concentrations for engaging with Pin1.
Keywords: bivalent interaction, Pin1, IRAK1, NMR titration, multi-state equilibrium
Graphical abstract
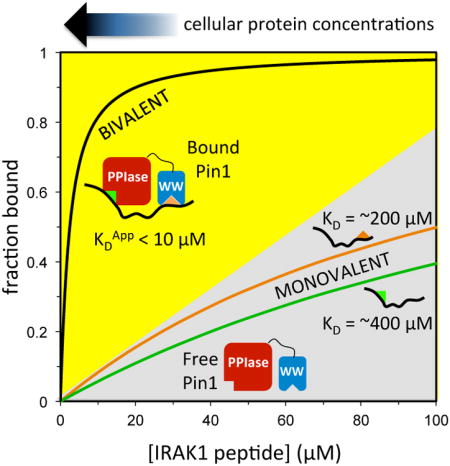
Bivalent interactions between Pin1 and phosphopeptides derived from substrate IRAK1 were investigated by NMR, and quantified using a multi-state model that allows prediction of binding at lower concentrations. Biolayer interferometry measurements confirmed the predicted affinity enhancement at micromolar concentrations. These studies suggest that phosphorylation of neighboring Ser/Thr-Pro motifs might impart competitive advantage for binding to Pin1 at cellular protein concentrations.
Introduction
The peptidyl prolyl isomerase Pin1 is a key regulator of diverse cellular processes in humans [1-3]. Pin1 has a substantial effect on neuronal development and survival [4, 5], and regulates the processing of amyloid precursor protein in cell culture and in mice [6, 7]. It is also important for telomere maintenance [8] and chromatin condensation [9]. The multitude of roles Pin1 plays is not surprising since it is unique in its ability to isomerize phosphorylated Ser-Pro or phosphorylated Thr-Pro (pS/pT-P) motifs, which are highly prevalent in eukaryotes, consitituting over a quarter of all protein phosphorylation sites [10-15].
Pin1 has two domains, a catalytic PPIase domain that performs interconversion between cis and trans pS/pT-P peptide bonds, and a type IV WW binding domain that binds to pS/pT-P motifs [16]. The selection of targets by Pin1 will be driven by the relative amounts and affinities of potential interaction partners, and by their phosphorylation states. The balance of specific kinase and phosphatase activities at the multitude of pS/pT-P motifs within the cell determines what substrates are available to Pin1 at any given moment.
A key factor in Pin1 substrate selection may be the number of pS/pT-P motifs in close spatial proximity in a potential substrate protein. In principle, the WW domain could anchor the enzyme at one pS/pT-P site, while the PPIase domain isomerizes a neighboring pS/pT-P site. This simultaneous, or bivalent, binding can theoretically increase the effective affinity by orders of magnitude [17-19]. Indeed, a PPIase inhibitor and a WW inhibitor linked using various lengths of polyproline helix bind Pin1 with higher affinity than either inhibitor alone [17]. This suggests that cellular Pin1 partners with multiple motifs may use a similar mechanism to enhance their interaction with Pin1. Many proteins implicated as Pin1 substrates are phosphorylated at multiple nearby pS/pT-Pro sites, including RNA polymerase II [20], tau [21], the transcription factor c-Jun [22], cdc25c [14], Notch [23] and IRAK1 [24].
IRAK1 is a kinase comprised of four domains: a death domain (DD), an undefined domain (UD), a kinase domain (KD) and a C-terminal domain (CTD) (Figure 1). IRAK1 plays a critical role in innate immunity in humans, specifically acting as part of the Toll-like receptor /Interlukein-1 receptor signaling cascades. Upon receptor stimulation, a multi-protein signaling complex assembles on the cytoplasmic side of the receptor. This signaling complex is initiated by receptor dimerization, which creates a platform for recruitment of MyD88, which in turn recruits IRAK4 leading to formation of a scaffold thought to be comprised of four MyD88 and four IRAK4 subunits [25]. This scaffold recruits and subsequently phosphorylates four IRAK1 molecules, leading to autophosphorylation of multiple Ser sites in the UD and activation of IRAK1, in a Pin1-dependent manner [24, 26]. Autophosphorylation of the IRAK1-UD results in six pS-P motifs, with two pairs of motifs (pSer131/pSer144 and pSer163/pSer173) separated by less than 12 residues (Figure 1). Intriguingly, these two IRAK1-UD regions are implicated in the mechanism of Pin1 activation of IRAK1 [24].
Figure 1. Diagram of the peptide sequences used, shown in context of full length IRAK1.

The synthetic peptides used in these experiments are aligned by shared phosphorylation sites. The separation between the sets of proximal sites used in the bivalent binding-capable peptides is shown with regards to a diagram of FL IRAK1 where the DD (in orange) is the death domain (residues 27-106), UD is the undefined domain (107-211), KD (in blue) is the kinase domain (212-521), and CTD is the C-terminal domain (521-691). The rectangles represent areas of predicted stable 3D structure and the lines are predicted intrinsically disordered regions.
Here, we have investigated potential bivalent interactions of Pin1 and the IRAK1-UD using NMR titration experiments with 15N-labeled Pin1 (full-length (FL), PPIase, or WW domain) and synthetic phosphopeptides (either singly or dually phosphorylated) representing the pSer133/pSer144 and pSer163/pSer173 regions of IRAK1-UD. Binding of singly-phosphorylated peptides to isolated Pin1 domains was first measured, then binding of singly-phosphorylated peptides to Pin1-FL, and finally binding of dually phosphorylated peptides to Pin1-FL. The affinities determined using singly phosphorylated peptides provided parameters for analysis of the complex binding interaction between dually phosphorylated peptides and Pin1-FL, and allowed bivalent binding of these peptides to be modeled. These interactions were quantified using NMR titration analysis, a powerful technique to investigate multi-state equilibria of complex protein/ligand interactions. We employed a novel combination of VCell modelling software [27] in conjunction with in-house Matlab code to perform simulations and to fit the NMR titration data. To validate model predictions, biolayer interferometry (BLI) was used to compare binding of singly and dually phosphorylated representative peptides at low micromolar concentrations. The resulting affinities were in excellent agreement with model predictions for the pair of peptides employed. Overall, our approach allowed detailed investigation of the multi-state equilibrium that fully describes the interaction of a two-domain protein with a two-motif ligand, where both domains can interact with both motifs. The results of these studies support the influence of allostery in Pin1 binding, provide evidence for bivalent binding of Pin1 to natural targets in the IRAK1-UD, and suggest the importance of bivalent interactions for Pin1 target selection in the cellular milieu.
Results and Discussion
Isolated PPIase and WW domains display a range of binding affinities for singly phosphorylated IRAK1 UD-derived peptides
To provide a foundation for investigating bivalent interactions between Pin1 and IRAK1-UD, we first performed a series of NMR-detected titration experiments using isolated 15N-labeled PPIase and WW domains and unlabeled, singly phosphorylated peptides representing the regions of interest in IRAK1-UD (Figure 1). Notably, wild type PPIase, for which the complete thermodynamic cycle has been determined [28], rather than a catalytically dead mutant [29] is employed to avoid altering the native binding function. For example, the catalytically dead mutant C113D results in reduced binding affinity to substrate [30] and K63 has been implicated in binding to the phosphate group of substrates [31]. The KD values for WW binding to p131, p144 and p173 peptides were previously published [24], and binding of p173 to the PPIase domain in full length Pin1 was extremely weak (vide infra) so was not investigated for the isolated PPIase domain. For the four remaining domain/peptide interactions (WW binding to p163, and PPIase binding to p131, p144, and p163), the 15N-1H HSQC spectrum for a constant concentration of each 15N-labeled domain was used to monitor chemical shift changes of selected peaks as a function of peptide concentration to produce binding curves (Figure 2&3).
Figure 2. Goodness of fit of bimolecular model to data for titrations of isolated Pin1 domains with singly phosphorylated IRAK1-UD peptides.

Binding curves are shown, where lines are calculated curves and the points are the normalized mean of the chemical shift perturbation of the binding of different IRAK1-UD-derived peptides to isolated domains of Pin1. The WW domain is represented by blue and PPIase is represented by red. Chemical shift perturbations were normalized by dividing the measured perturbation by the final chemical shift perturbation for each residue. The x-axis is the concentration of the individual peptide in mM and the y-axis is the mean of the normalized chemical shift perturbation of selected residues (see methods section). Error bars are the SEM of the individual residues. Plots are of A) WW domain binding to p163 (residues used were S18, R21, V22, W34e, W34, and E35 in the proton dimension and S18, R21, V22, Y23, Y24, W34, and E35 in the nitrogen dimension, providing n=13), B) PPIase domain binding to p131 (residues used were H59, R68, R69, S114, K117, L122, R127, G128, and M130 in the proton dimension, and H59, V62, R68, R69, S114, K117, L122, R127, G128, and M130 in the nitrogen dimension, providing n=19), C) PPIase domain binding to p144 (residues used were V62, R68, R69, S114, S115, A116, L122, G128, and Q129 in both dimensions, providing n=18), and D) PPIase domain binding to p163 (residues used were H59, L61, V62, R69, S115, A116, A118, R119, R127 and Q129 in both dimensions, providing n=20).
Figure 3. The simultaneous fits of isolated domains of Pin1 titrated with singly phosphorylated IRAK1-UD derived peptides shown in terms of chemical shift perturbation of selected residues in individual dimensions.

The x-axis represents the amount of peptide titrated in and the y-axis is the chemical shift perturbations. The points represent the data and the lines represent the two-state fit of said data. The WW residues are shown as blue and the PPIase residues are in red. A) selected residues in the proton dimension (S18 filled◊, R21 unfilled□, V22 filledΔ, W34 *, W34e ×, E35 filled ○) for WW domain titrated with p163. B) selected residues in the nitrogen dimension (S18 filled◊, R21 filled□, V22 filledΔ, Y23 ×, Y24 *, W34 filled ○, E35 +) for WW domain titrated with p163 C) selected residues in the proton dimension (H59 filled◊, R68 filled□, R69 filledΔ, S114 ×, K117 *, L122 filled ○, R127 +, G128 -, M130—) for PPIase domain titrated with p131 D) selected residues in the nitrogen dimension (H59 filled◊, V62 filled□, R68 filledΔ, R69 ×, S114 *, K117 filled ○, L122 +, R127 -, G128 —, M130 filled◊) for PPIase domain titrated with p131 E) selected residues in the proton dimension (V62 filled◊, R68 filled□, R69 filledΔ, S114 ×, S115 *, A116 filled ○, L122 +, G128 -, Q129 —) for PPIase domain titrated with p144 F) selected residues in the nitrogen dimension (V62 filled◊, R68 filled□, R69 filledΔ, S114 ×, S115 *, A116 filled ○, L122 +, G128 -, Q129 —) for PPIase domain titrated with p144 G) selected residues in the proton dimension (H59 filled◊, L61 unfilled□, V62 filledΔ, R69 ×, S115 *, A116 filled ○, A118 +, R119 small unfilled□, R127 —, Q129 unfilled◊) for PPIase domain titrated with p163 H) selected residues in the nitrogen dimension (H59 filled◊, L61 filled□, V62 filledΔ, R69 ×, S115 *, A116 filled ○, A118 +, R119 -, R127 —, Q129 unfilled◊) for PPIase domain titrated with p163.
For the group of selected peaks in a given titration, the observed residue-specific chemical shift changes (relative to apo) in the15N and 1H dimensions (ΔδiN,obs and ΔδiH,obs for residue i, respectively) were globally fit to the standard bimolecular reaction scheme (Figure 4A). For this model, fitted parameters for a given domain/peptide interaction consist of the global KD and residue-specific bound chemical shifts, ΔδiN,bound and ΔδiH,bound. Experimental errors in ΔδiN,obs and ΔδiH,obs were estimated by two methods as described in Materials and Methods, yielding σΔδN = 0.028 ppm and σΔδH = 0.0042 ppm (based on peaks insensitive to ligand addition) and σΔδN = 0.0205 ppm and σΔδH = 0.00407 ppm (based on the assumption that the bimolecular model is appropriate). The close agreement between these two sets of errors supports the use of the bimolecular model for each isolated domain. The corresponding set of errors, σΔδN = 0.0205 ppm and σΔδH = 0.00407 ppm, which are smaller and thereby provide a more stringent test of goodness of fit to a given model, were used here and for the more complex binding equilibria investigated (vide infra).
Figure 4. Models for interactions of monovalent and bivalent peptides with Pin1.
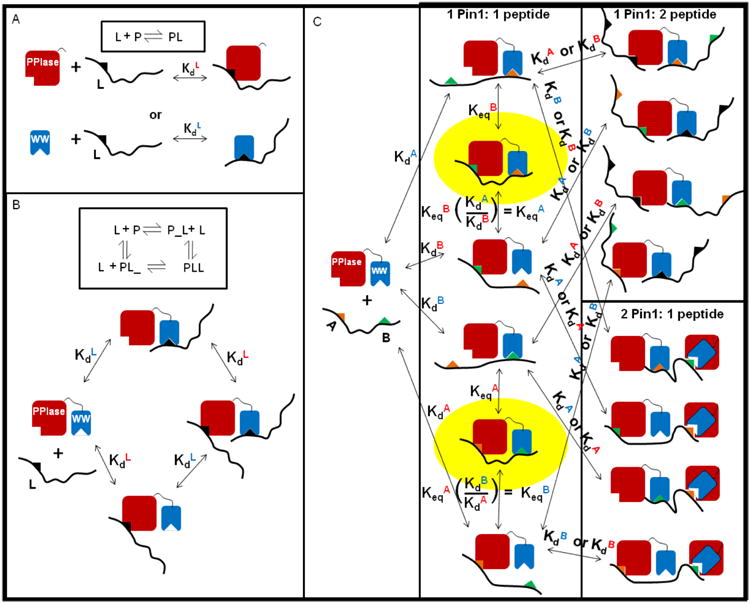
A) Simple bimolecular binding equilibrium, [L] + [P] ↔ [LP], where P is either isolated WW or PPIase domain and L is a singly phosphorylated (monovalent) peptide. B) The four-state model for Pin1-FL (P in diagram) binding to a monovalent peptide (L) in a 1:1 fashion bound to either the WW domain (PL_) or the PPIase domain (P_L) or in a 2:1 fashion with two peptides bound to Pin1, one in each domain (PLL). C) Multi-state model for Pin1-FL binding to a bivalent peptide where A (orange triangle) is the N-terminal site (either p131 or p163) and B (green triangle) is the C-terminal site (either p144 or p173). For simplicity, black triangles represent either site and the overlaid binding sites on Pin1 represent binding to either domain.
The resulting affinities for each singly phosphorylated peptide binding to the individual domains range between 120 to nearly 700 μM (Table 1), indicating that these individual pS-P motifs would not be expected to be significant Pin1 binding partners at low to submicromolar concentrations, as might be found in cells. These reported affinities are “apparent” binding constants (KDapp) that do not distinguish the isomer state of the ligand. Although the intrinsic binding constants for each isomer can be determined [32], the focus of the present study is on bivalent binding, for which KDapp provides the necessary measure of the total bound domain for given conditions. Notably, p131 and p163 both display tighter binding to the PPIase catalytic domain than to the WW binding domain. This is unusual, since for Pin1 the WW domain typically displays higher substrate affinity while the PPIase domain is a classical enzyme with weaker binding to substrates [33]. While the consensus Pin1 binding sequence Pintide [11] displays appreciable binding to the PPIase domain (KD = 86 μM), this peptide library-derived sequence binds even more strongly to the WW domain (KD = 44 μM) [33]. In the context of the isolated Pin1 domains, the p131 and p163 peptides are to our knowledge the first natural Pin1 substrate sequences to display greater preference for the PPIase domain than to the WW domain.
Table 1. Binding affinities for the interaction of isolated Pin1 domains and IRAK1-UD derived singly phosphorylated peptides.
| Peptide | WW KD (μM) | Proton χ2red | Nitrogen χ2red | PPIase KD (μM) | Proton χ2red | Nitrogen χ2red |
|---|---|---|---|---|---|---|
| p131 | 220 ± 15* | 161 ± 31 | 0.53 | 0.86 | ||
| p144 | 120 ± 12* | 689 ± 149 | 0.92 | 1.05 | ||
| p163 | 385 ± 96 | 0.97 | 0.61 | 230 ± 42 | 0.90 | 0.61 |
| p173 | 260 ± 75* | n.d. |
Previously reported values [24].
PPIase and WW domains in full length Pin1 display distinct binding affinities to singly phosphorylated, IRAK1 UD-derived peptides when compared with isolated domains
NMR titration experiments were next performed using full-length 15N-Pin1 (Pin1-FL) and the four singly-phosphorylated IRAK1 peptides (Figure 1). These measurements investigate whether linking the WW and PPIase domains alters binding affinity for these IRAK1 peptides, and provide necessary parameters for analyzing titrations with dually phosphorylated peptides (vide infra). Since each domain can in principle bind to the same singly phosphorylated peptide, titration data was analyzed using a four-state model (Figure 4B). This model includes two apparent binding constants, KDWW,app and KDPPIase,app. As NMR allows direct observation of residues in each domain, it is uniquely suited for determining domain-specific affinities in multi-domain proteins such as Pin1. Residues for each domain were selected based on their sensitivity to binding, as reflected by peak movement. Binding curves (Figure 5&6) for selected residues were simultaneously (i.e. globally) fit (proton and nitrogen dimensions were fit separately) to obtain KDWW,app and KDPPIase,app (Table 2), as well as the residue-specific bound chemical shifts, ΔδiN,bound and ΔδiH,bound, for each domain.
Figure 5. Goodness of fit of four-state model to data for titrations of Pin1-FL with singly phosphorylated IRAK1-UD peptides.

In these binding curves, datapoints represent the normalized mean of the observed chemical shift perturbations across selected domain-specific residues in Pin1-FL induced by titration with a given peptide, and lines are the resulting fits to the four-state model. Chemical shift perturbations were normalized by dividing the measured or calculated perturbation by the fitted Δδij,bound for each residue i. For Pin1-FL titrations, WW domain data are denoted by blue open diamonds (nitrogen dimension) and blue + (proton dimension), and PPIase domain data by red open squares (nitrogen dimension) and red × (proton dimension). Dashed lines represent the fits in the nitrogen dimension and solid lines represent the corresponding fits in the proton dimension. The error bars are the SEM of the individual residues used in each dimension. Plots of Pin1-FL binding to A) p131, where selected residues for the proton dimension were F25, Q33, W34, W34e, and E35 in the WW domain (n=5) and H59, R69, S114, S115, R127, G128, Q129, and M130 in the PPIase domain (n=8). Selected residues for the nitrogen dimension were Y23, Y24, Q33, W34, W34e, and E35 in the WW domain (n=6) and V62, R69, S114, A116, R127, G128, Q129, and M130 in the PPIase domain (n=8). B) p144, where selected residues in the proton dimension were Y24, F25, W34, W34e, and E35 in the WW domain (n=5) and H59, V62, R69, S114, S115, A116, G128, and Q129 in the PPIase domain (n=8). In the nitrogen dimension, selected residues were Y23, Y24, F25, Q33 W34, W34e in the WW domain (n=6) and V62, R69, S114, S115, A116, R127, G128, and Q129 in the PPIase domain (n=8). C) p163, where selected residues in the proton dimension were Y23, Y24, W34, and W34e in the WW domain (n=4) and H59, V61, V62, R69, A116, K117, A118, G128, and Q129 in the PPIase domain (n=9). In the nitrogen dimension, selected residues were Y23, Y24, W34, and E35 in the WW domain (n=4) and H59, V62, R69, S115, A116, A118, R127, Q129 and K132 in the PPIase domain (n=9). D) p173, where selected residues in the proton dimension were F25, W34, and W34e in the WW domain (n=3) as well as S114 and S115 in the PPIase domain (n=2). In the nitrogen dimension, selected residues were Y23, Y24, F25, W34, and E35 in the WW domain (n=5) and R68, Q129, and M130 in the PPIase domain (n=3).
Figure 6. The simultaneous fits of intact Pin1 with singly phosphorylated IRAK1-UD derived peptides shown in terms of chemical shift perturbation of selected residues in individual dimensions.

The x-axis is the concentration of peptide titrated in and the y-axis is the chemical shift perturbation. Blue represents WW domain residues while red represents PPIase residues. The symbols are the experimental data and the lines are the simultaneous fits. A) selected residues in the proton dimension (F25 unfilled□, Q33 filled◊, W34 filled ○, W34e ×, E35 filledΔ, H59 filled○, R69 ×, S114 -, S115 *, R127 ×, G128 filledΔ, Q129 filled□, and M130 —) for Pin1 titrated with p131. B) selected residues in the nitrogen dimension (Y23 filledΔ, Y24 ×, Q33 *, W34 filled ○, W34e +, E35 -, V62 —, R69 filled◊, S114 unfilled□, A116 filledΔ, R127 ×, G128 unfilled ○, Q129 filled ○, M130 +) for Pin1 titrated with p131. C) selected residues in the proton dimension (Y24 *, F25 filled ○, W34 +, W34e -, E35 —, H59 filled◊, V62 -, R69 filled□, S114 filledΔ, S115 *, A116 ×, G128 +, Q129 filled ○) for Pin1 titrated with p144. D) selected residues in the nitrogen dimension (Y23 filled ○, Y24 +, F25 -, Q33 —, W34 filled◊, W34e filled□, V62 *, R69 unfilled◊, S114 filled□, S115 ×, A116 filled◊, R127 -, G128 filledΔ, Q129 filled ○) for Pin1 titrated with p144. E) selected residues in the proton dimension (Y23 filled□, Y24 filled ○, W34 filledΔ, W34e filled◊, H59 unfilledΔ, L61 ×, V62 *, R69 unfilled□, A116 unfilled ○, K117 —, A118 unfilled◊, G128 -, Q129 +) for Pin1 titrated with p163. F) selected residues in the nitrogen dimension (Y23 *, Y24 filled ○, W34 filled◊, E35 -, H59 —, V62 -, R69 filled◊, S115 filled□, A116 unfilledΔ, A118 ×, R127 filled ○, Q129 filled◊, K132 *) for Pin1 titrated with p163. G) selected residues in the proton dimension (F25 +, W34 -, W34e —, E35 filled◊, S114 filled□, S115 filled ○) for Pin1 titrated with p173 H) selected residues in the nitrogen dimension (Y23 —, Y24 filled◊, F25 filled□, W34 filledΔ, E35 ×, R68 unfilledΔ, Q129 unfilled ○, M130 +) for Pin1 titrated with p173.
Table 2. Binding affinities for the interaction of Pin1-FL and IRAK1-UD derived singly phosphorylated peptides.
| Peptide | WW KD (μM) | PPIase KD (μM) | χ2red | |||
|---|---|---|---|---|---|---|
| Proton | Nitrogen | Proton | Nitrogen | Proton | Nitrogen | |
| p131 | 145 ± 11 | 151 ± 6 | 424 ± 24 | 427 ± 28 | 1.17 | 2.25 |
| p144 | 257 ± 10 | 255 ± 17 | 414 ± 23 | 470 ± 33 | 1.56 | 0.71 |
| p163 | 233 ± 27 | 279 ± 26 | 185 ± 15 | 157 ± 8 | 1.17 | 0.5 |
| p173 | 215 ± 19 | 186 ± 25 | 681 ± 178 | 611 ± 236 | 2.18 | 0.6 |
Interestingly, several significant differences are observed in the resulting affinities of these singly phosphorylated peptides compared with their binding to the isolated PPIase and WW domains (Tables 1 and 2). For p131, linking the WW and PPIase domain in Pin1-FL significantly weakens binding to the PPIase domain, while slightly enhancing binding to the WW domain. Conversely, for p144, linking the two domains strengthens affinity to the PPIase domain and weakens affinity to the WW domain. For p163, Pin1-FL provides a binding enhancement for both domains relative to their isolated forms, and further accentuates the anomalous preference of this site for the PPIase domain. Finally, for p173, the PPIase residues of Pin1-FL display very little chemical shift perturbation upon titration (reflecting very weak binding), while WW binding affinity is approximately the same as for the isolated WW domain. Due to the weak binding affinity observed for the p173 peptide with PPIase in Pin1-FL, the isolated PPIase domain titration with p173 peptide was not performed (vide supra). For three of the four specific pS-P motifs examined, the intrinsic binding affinities display sensitivity to the presence of both Pin1 domains, suggesting allosteric interactions between domains. Notably, the one pS-P motif that displayed no evidence for allostery only interacted with the WW domain, suggesting the possible dominance of a PPIase-initiated allosteric mechanism.
Several prior studies support the presence of allosteric interactions between the PPIase and WW domains of Pin1. In terms of the PPIase domain catalytic function, its affinity and isomerization rate depend on the presence of the WW domain [34, 35]. Interestingly, numerous x-ray crystal structures of Pin1 reveal a clear interaction interface between the PPIase and WW domains [16, 33, 36-41]. In solution, interdomain coupling, which was quantified in terms of global tumbling of each domain, is enhanced in a ligand-dependent manner [42, 43]. Moreover, an allosteric conduit connecting the PPIase catalytic site to the interdomain interface in Pin1 has been elucidated through methyl group and backbone dynamics studies [35, 44].
Here, we also observe clear evidence for interdomain communication, not only in the altered affinities resulting from linking the PPIase and WW domains, but also in chemical shift changes induced by binding of singly phosphorylated ligands to isolated PPIase and WW domains. For example, titrations of isolated PPIase domain with each of the p131 and p144 peptides show chemical shift perturbations (Figure 7A) in the previously defined conduit residues [35, 44]. The p131 and p144 peptides (only 11 residues long) should avert non-specific encounters between extended segments of bound peptide and remote regions of the PPIase domain. For a given conduit residue, chemical shift changes induced by both ligands lie along a linear trajectory, with p131 imparting greater effect than p144 (Figure 7A, right column), suggesting a ligand-dependent shift in the conformational ensemble sampled by the dynamic conduit [44, 45]. In contrast, catalytic site residue peaks move in entirely different directions in response to addition of p131 versus p144 (Figure 7A, left column), reflecting the distinct chemical features of p131 and p144 peptides (Figure 1). Strikingly, the linear relationship we observe for the A140 and L141 peaks when isolated PPIase domain is apo, saturated with p131, or saturated with p144 (Fig. 7A) is consistent with the linear alignment observed for these same peaks in intact Pin1, Pin1-I28A, Pin1W34A, and Pin1 saturated with pCdc25C peptide [46]. The persistence of these linear trajectories of interface residue peaks observed here in the PPIase domain is fully consistent with the detailed and robust work that describes the role of these residues in intradomain and interdomain allostery in intact Pin1 [46]. Our observation that binding of p131 to the WW domain is stronger in Pin1-FL suggests that the dynamic PPIase conduit might positively regulate this interaction. Since p144 binding to the WW domain in Pin1-FL is weaker than in the isolated WW domain, this suggests that the effect of the allosteric conduit is highly ligand-specific.
Figure 7. Difference between peak movements attributable to interdomain interactions versus protein-peptide binding induced by titration with p131 and p144.

Regions extracted from 15N-1H HSQC spectra of Pin1-FL in apo (red), the most saturated spectrum from titration with p131 (blue), and the most saturated spectrum from titration with p144 (green). A) PPIase residues in either the binding site or implicated in conduit that informs on allostery[44, 45]. B) WW residues in either the binding site or at the interface between the two domains.
A similar analysis from the perspective of the isolated WW domain shows the same trend of linear chemical shift perturbations induced by p131 and p144 peptide binding for residues in the interdomain interface (Figure 7B). Remarkably, although p144 binds more tightly than p131 to the isolated WW domain, p131 again imparts larger chemical shift changes in interface residues at the highest peptide concentrations examined. Specifically, T29 and N30 that directly hydrogen bond to the PPIase domain in Pin1-FL, and N26 that hydrogen bonds to T29 and I28, undergo ligand-induced peak movements along the same direction in the isolated WW domain, with larger perturbations induced by p131. Since binding of p131 to the PPIase domain in Pin1-FL is significantly weaker than to the isolated PPIase domain (Tables 1 and 2), this suggests that the ligand-induced conformational shift at the WW domain interface region acts as a negative allosteric regulator of binding to the PPIase domain. The smaller p144-induced perturbation of interface residues correlates with the smaller weakening of PPIase affinity for this ligand in Pin1-FL (Tables 1 and 2), and is consistent with a negative allosteric mechanism.
Evidence for Pin1-FL bivalent binding to dually phosphorylated IRAK1-derived peptides
To investigate possible simultaneous binding of Pin1 to neighboring pS-P motifs, the two IRAK1-UD derived peptides, p131/p144 and p163/p173 (Figure 1), were used in titration experiments with 15N-Pin1-FL (Figure 8). For each dually phosphorylated peptide titration, either pS-P site can bind to either of the two Pin1 domains, generating a multi-state equilibrium (Figure 4C). This model accounts for all possible interaction states between Pin1-FL and a dually phosphorylated peptide, and includes two bivalent states and their associated equilibrium constants that govern their populations (Figure 4C). While simpler models have been previously employed for Pin1 binding to ligands containing two binding motifs [17, 21], these models do not account for the full complexity of the binding reaction scheme for a two-domain protein interacting with a two-motif ligand, where both domains can interact with both motifs.
Figure 8. Modeling of data for titrations of Pin1 FL with dually phosphorylated peptides.

The data points and corresponding globally fit or simulated binding curves (panels A – C and E – G) are color-coded by domain (blue = WW residues, red = PPIase residues), for titrations of Pin1-FL with p131/p144 (A – D) and with p163/173 (E – H), showing simulations without the bivalent states (A,E), fitting for the two global Keq values (B,F) and fitting for the two global Keq values and the the residue-specific bound chemical shift perturbations for the bivalent species (C,G). For p131/p144 plots (A-C), in the proton dimension Y24 is filled◊, F25 filled Δ, H59 +, S115 ×, R127 *, M130 open ◊; additionally in the nitrogen dimension, W34ε filled Δ, E35 filled ○, S114 *, A116 +, G128 -, and Q129 —). For p163/173 plots (E-G), in the proton dimension V22 is filled ◊, Y23 filled large □, Y24 filled Δ, W34ε filled small □, E35 filled ○, H59 +, L61 open Δ, V62 -, K117 ×, L122 *, Q129 —; additionally, in the nitrogen dimension V22 is filled ◊, Y23 filled □, W34 filled Δ, E35 filled ○, H59 +, V62 -, L122 *, and Q129 —. D) Equilibrium populations of the different types of species resulting from the fit in C, where species are grouped into: bivalent species (red), 2 pin1: 1 peptide species (blue), 1 Pin1:2 peptide species (green) and non-bivalent 1 Pin1:1 peptide species (purple). H) The equilibrium populations given different concentrations of peptide of the different types of species resulting from the fit where the species are grouped into: Bivalent species (red), 2 pin1: 1 peptide species (blue), 1 Pin1:2 peptide species (green) and non-bivalent 1 Pin1:1 peptide species (purple).
To assess whether accounting for bivalency is necessary, we first asked whether the observed p131/p144 and p163/p173 titration data could be adequately simulated using the KD and Δδij,bound values (where j = N or H) obtained from the titrations of Pin1-FL with singly-phosphorylated peptides (above), without invoking the bivalent states (i.e., we set both KeqA:WW and KeqB:WW values in the model to 107 and did not allow them to vary). These simulations (Figure 8A,E) and the resulting chi-square values (ranging from 9 to 75, Table 3) indicate that the p131/p144 and p163/p173 titration data are not adequately reproduced by the predicted distribution over all possible non-bivalent states (reduced chi-square values for an appropriate model are expected to be close to one).
Table 3. Assessment of the need for inclusion of bivalent states in fitting of NMR data from titrations of 15N-Pin1-FL with dually phosphorylated ligands.
| Peptide | Simulation (no bivalent) | Fitted KeqA:WW, KeqB:WW | Fitted KeqA:WW, KeqB:WW and Δδij,bound for bivalent states | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| χ2red |
|
|
χ2red |
|
|
χ2red | ||||
| p131/p144 | Proton | 34 | 0.12 | 0.08 | 24 | 0.02 | 0.43 | 1.25 | ||
| Nitrogen | 75 | 0.25 | 0.10 | 12 | 0.35 | 0.02 | 1.55 | |||
| p163/p173 | Proton | 9 | 100 | 0.32 | 3.67 | 0.35 | 0.29 | 1.30 | ||
| Nitrogen | 18 | 0.44 | 0.13 | 1.93 | 0.98 | 0.41 | 1.29 | |||
A is the most N-terminal phosphorylated site on the peptide, B is the C-terminal site.
We next asked whether adding the two possible bivalent states to the model and fitting for their equilibrium constants could improve the fit. For simplicity, the bivalent state Δδij,bound values were held fixed at their corresponding values determined from Pin1-FL titrations with singly phosphorylated peptides (e.g., WW domain bound to p131, etc.). For this model, only KeqA:WW and KeqB:WW values (one independent Keq for each bivalent orientation) were fit. This approach yielded significant improvements (Figure 6B,F), but the reduced chi-square values (now ranging from 1.9 to 24, Table 3), still did not indicate sufficiently good fits.
As a logical next step to improve the fits, the Δδij,bound values of the bivalent species were added as fitted parameters. Values of Δδij,bound for the bivalent species might be expected to differ from the those determined from Pin1-FL titrations with singly phosphorylated peptides, since a bivalent interaction might require compromise in one or both interaction interface(s) and/or might induce bivalent-specific allosteric changes. For this model, the fitted parameters include KeqA:WW, KeqB:WW and, for each domain-specific residue i, two Δδij,bound values corresponding to each of the two bivalent species. The resulting fits (Figure 8C,G) all yielded reduced chi-square values < 1.6, indicating adequate fits to this model (Table 3). In general, the fitted Δδij,bound values for p131/p144 varied significantly from the corresponding values determined by titrations of Pin1-FL with monovalent ligands (Figure 9A-D), while p163/p173 values were in closer agreement to those values (Figure 9E-H) as expected based on the better fits of p163/p173 with the simpler model (Figure 8F). For p131/p144, fitting of the proton and nitrogen dimensions yielded opposite orientation preferences, indicating an insensitivity of the fit to orientation for this peptide. Regardless, the population of bivalently bound species is dominant in fits of data from both proton and nitrogen dimensions (Figure 8D). For p163/p173, fits in both proton and nitrogen dimensions show a preference for the orientation in which p173 is bound to WW and p163 is bound to PPIase, and bivalent populations are dominant at lower concentrations (Figure 8H).
Figure 9. Comparison between fitted bound Δδ values for bivalent and corresponding non-bivalent species of Pin1-FL.

Residue-specific bound Δδ values (Δδij,bound) obtained by fitting of data from Pin1-FL titrated with dually phosphorylated peptides (green bars) and the corresponding fitted values from fitting of data from Pin1-FL titrated with singly phosphorylated peptides (orange bars) are shown for the residues used in the bivalent fits shown in Fig. 6C and 6G. The AB orientation is the species with WW bound to the first site (pS131-P132 or pS163-P164) and PPIase bound to the second site (pS144-P145 or pS173-P174). The BA orientation is the species with WW bound to the second site (pS144-P145 or pS173-P174) and PPIase bound to the first site (pS131-P132 or pS163-P164). A-D) Comparisons of fitted proton and nitrogen Δδ values obtained for p131/p144, p131, and p144. E-G) Comparisons of fitted proton and nitrogen Δδ values obtained for p163/p173, p163, and p173.
These results show that, for both bivalent ligands studied here, accounting for the bivalently bound species is required for obtaining adequate fits of the titration data, and that the influence of these states is dominant at low concentrations. Importantly, this model provides predictive power for exploring the competitive advantage achieved by bivalent interactions at micromolar concentrations as might be found in cells.
Model predicts that bivalent interactions for the p131/p144 and p163/p173 peptides impart competitive advantage in the cell
The overall goal of this study was to gain insight into the potential competitive advantage of a Pin1 substrate with two neighboring pS/pT-P sites. To compare the effective affinity of dually versus singly phosphorylated peptides at approximate cellular concentrations, the binding models (Figure 4B,C) and the corresponding best-fit parameters (Tables 1, 2, and 3) were used to simulate Pin1-FL binding to each pair of neighboring sites. In these simulations, the total fraction bound is the sum of all bound Pin1 species divided by total Pin1 (∼0.5 μM [47]), and the peptide concentration was varied between 0.01 μM and 100 μM. For the p131/p144 peptide, the capacity for bivalent binding imparts a marked advantage in Pin1 binding compared with the singly phosphorylated p131 or p144 peptides (Figure 8A). At 20 μM peptide, the total fraction of bound Pin1 is 0.85 for the p131/p144 peptide, which corresponds to an equivalent bimolecular interaction KD value at these conditions of 3.5 μM (with corresponding change in Gibbs free energy of ΔGequiv = -7.49 kcal/mol). Similarly, for the p163/p173 peptide at the same conditions, the total fraction of bound Pin1 is 0.45 (Figure 10B) and the equivalent bimolecular interaction KD value is 24 μM (ΔGequiv = -6.34 kcal/mol), representing a significant gain over the corresponding affinities of the p163 and p173 monovalent peptides. These simulations suggest that each of these bivalent IRAK1-UD sequences could effectively compete with other biological Pin1 substrates in the cell [21, 33, 48].
Figure 10. Prediction of significance of bivalent binding at low (micromolar) concentrations.

Simulation of fraction of Pin1-FL bound (sum of all species including bound Pin1 in the system) when 0.5 μM Pin1-FL is titrated with various IRAK1-UD derived, phosphorylated peptides using the models from Figure 3. A) Comparison of predicted fraction bound for Pin1-FL with IRAK1 where S131-P132 is phosphorylated (green), S144-P145 is phosphorylated (purple), or both S131-P132 and S144-P145 are phosphorylated (orange and red). The KD and Keq values were taken from the fits of titrations with p131, p144 (where in both cases the simulation in the proton dimension is recapitulated with the nitrogen dimension), and the nitrogen (orange) and proton (red) dimension based fits of the p131/p144 titration. B) Comparison of calculated fraction bound for Pin1-FL and phosphorylated S163-P164 (green), pS173-P174 (purple), or pS163-P164 and pS173-P174, where the parameters used are based on fits of the p163, p173 (where in both cases the simulation in the proton dimension is recapitulated with the nitrogen dimension), and p163/p173 titrations (nitrogen dimension based parameters yielded the solid line simulation and the proton dimension yielded the short dashed line.
Validation of model predictions at low concentrations using biolayer interferometry
To test the predictive power of our model for Pin1 binding to bivalent substrates at low concentrations (on the order of physiologic), we conducted biolayer iterferometry assays [49, 50] with Pin1-FL and varying concentrations of either p144 or p131/p144. This pair of peptides is expected to be representative of the effect of having either one or two binding sites on a given IRAK1-UD derived peptide. Use of the short p144 in particular and its associated dually phosphorylated peptide avoids potential long range encounters that could occur between the extended portions of a long singly phosphorylated peptide (e.g. p163) and Pin1-FL. Biolayer interferometry also allows us to probe binding at low micromolar concentrations (not feasible using NMR) in order to compare the effective binding of singly or dually phosphorylated peptides. The data from biolayer interferometry allows direct observation of koff, which is concentration independent, and kon, which is dependent on the concentration of the binding partner (i.e the IRAK1-UD derived peptides) in solution (i.e., the amount of Pin1 loaded on a given biosensor does not need to be known). As KD = koff/kon, it is possible to obtain the effective KD of the system using the 1:1 model available in the Kinetic Analysis module in the Octet Data Acquisition software (ForteBio).
As is clearly visualized in Figure 11, Pin1 binding to p131/p144 is distinct while binding p144 at the same concentration of peptide is not measurable. Even if binding were compared by concentration of phosphorylated binding sites (i.e. 80μM of p144 is equivalent to 40μM of p131/p144), binding is still only detected for the dually phosphorylated peptide. Our model predicts dominance of the bivalently-bound species at low micromolar concentrations (Fig. 8D, E); hence, as expected, the biolayer interferometry data for the p131/p144 peptide could be fit to a simple two-state model. The resulting binding affinity, 1 μM for the interaction of Pin1-FL and p131/p144, is notably close to the effective KD predicted by our complex computational model that was developed based on analysis of NMR titrations at higher concentrations. The agreement between measurements using the complimentary technique of biolayer interferometry and predictions using our NMR-derived model demonstrates the predictive power of our model. Of note is the slow off rate observed via biolayer interferometry, which is consistent with the macroscopic nature of the measurement where any of the many bound states in the multi-state equilibrium (Fig. 4) are observed as bound. In contrast, the fast exchange observed via NMR reflects the microscopic exchange occurring at each of the two binding interfaces. Thus, the apparently disparate off rates obtained from the two techniques primarily reflect the different levels at which the bound state is sensed, although concentrating Pin1 on a two-dimensional surface could promote a slower macroscopic off rate due to rebinding.
Figure 11. Biolayer Interferometry demonstrates bivalent advantage of IRAK1-Pin1 interaction at low micromolar concentrations.
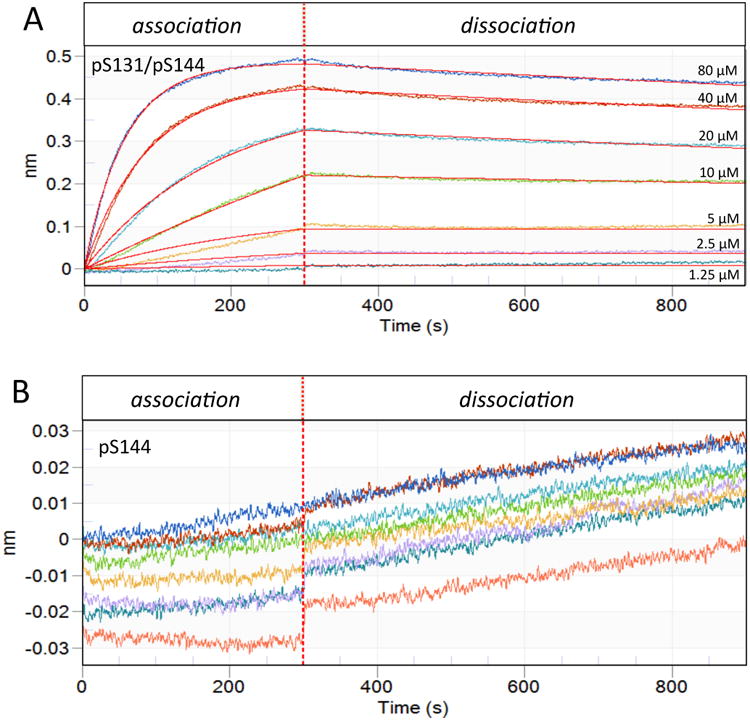
Pin1 was immobilized on the biosensor tip and dipped into peptide-containing well (association) then buffer only well (dissociation). The X-axis is the residence time in each well and the Y-axis denotes the change in wavelength upon binding. The vertical dotted line represents transfer of the biosensor between the association phase (containing peptide) and the dissociation phase (lacking peptide). A) Association-dissociation curves for p131/p144 peptide concentrations of 80, 40, 20, 10, 5, 2.5 and 1.25 μM (as marked, also color-coded) were fit to a simple bimolecular binding model (red lines). B) Association-dissociation curves for p144 peptide concentrations as in A (with same color-coding), plotted on a vertical scale amplified by an order of magnitude relative to A. Binding was insufficient for curve fitting. Since analysis could not be performed, the control spectrum is also shown (salmon).
Potential consequences of Pin1 bivalent interactions
Avidity enhancement of Pin1 binding to linked targets has been observed previously using composite peptides comprised of inhibitors of each domain separated by a peptide-based linker that included four residues (or mimics) plus a variable number (three, five, or six) of sequential proline residues [17]. Avidity enhancement occurred in all cases, but was greatest when five prolines were utilized [17]. This suggests a preferred linker length of ten residues between the corresponding pS/pT residues of neighboring motifs. Relative to this inferred optimal linker, our p163/p173 peptide has one fewer linker residue and contains four sequential prolines, while p131/p144 peptide has two additional linker residues and lacks any proline.
To compare the relative linkage contributions for our two bivalent IRAK1-UD peptides with the composite peptides comprised of linked inhibitors, we employed the effective concentration, Ceff, that reflects the concentration of one pS-P site once the other pS-P site is bound [17]. This value is obtained from Ceff = exp(-ΔGS/RT) [17], where ΔGS is the entropy barrier for bivalent binding and is the difference between the sum of free energies for the monovalent binding events (ΔGA +ΔGB) and the bivalent free energy (ΔGequiv), or ΔGS = (ΔGA +ΔGB) –ΔGequiv [18]. For the two IRAK1-UD bivalent peptides studied here, ΔGequiv was determined at the same Pin1 concentration as was used for the composite peptide measurements (20 μM, [17]), and ΔGA and ΔGB were obtained from the domain-specific monovalent ligand affinities for Pin1-FL, yielding Ceff values in the millimolar range: 21 mM and 35 mM for the two orientations of p131/p144, and 1.4 mM and 6.8 mM for the two orientations of p163/p173. For comparison, the reported Ceff values for the composite peptides ranged from 0.58 mM to 2.07 mM [17]. Hence, although the affinities of the monovalent and bivalent IRAK1-UD peptides are comparatively weaker, the bivalent IRAK1-UD peptides achieve more of the additive free energies of binding than the composite inhibitor-based peptides [17], resulting in greater effective concentrations of one pS-P site once the other pS-P site is bound. Our demonstration here of significant avidity enhancement in two IRAK1-derived bivalent sequences provides new insights regarding hyperphosphorylated sequences and their competitive advantage for interactions with Pin1.
The bivalent binding advantage demonstrated here for these IRAK1-UD substrates raises the question of how this might impact the velocity of the Pin1 catalytic reaction. The reversible Pin1 reaction is represented as,
where C=cis, T=trans, and E=Pin1. The equilibrium assumption for this reversible reaction (koffC ≫ kcatCT and koffT ≫ kcatTC) allows the Michaelis Menten velocities to be expressed as
Since the on-enzyme catalytic rates kcatCT and kcatTC are constants, the ratio of bivalent (subscript biv) to monovalent (subscript mv) reaction velocities is then given by
| Eq. 1 |
and similarly,
| Eq. 2 |
Equations 1 and 2 show that the bivalent binding advantage will increase the reaction velocity compared to the monovalent case under conditions where [EC]mv and [ET]mv are below their maximum values of [EC]mv = [E]tot/(1+kcatCT/ kcatTC) and [ET]mv = [E]tot/(1+kcatTC/ kcatCT). At the relatively high concentrations typical in NMR samples, the monovalent and bivalent velocities are expected to be similar since E is approximately saturated. Under such enzyme-saturating conditions, the concentration of free substrate, [C] or [T], will also be similar between bivalent and monovalent cases (for equal substrate concentrations), yielding similar values of kex (since kCT = vCT/[C], kTC = vTC/[T], and kex = kCT + kTC). Thus, it is important to note that even in cases where bivalency does not significantly alter the exchange rates measured by NMR exchange spectroscopy, significant acceleration of bivalent substrate catalysis can occur at concentrations below enzyme-saturating conditions, as expected in the biological environment.
Based on our model, the differences in population between Pin1-bound bivalent and monovalent IRAK1-UD peptides would be negligible at the high peptide and comparatively low Pin1 concentrations typically used for NMR studies of isomerase activity. Indeed, NMR exchange measurements at 1-3 mM peptide and 5-10 μM Pin1 showed no significant enhancement of catalysis for bivalent as compared to monovalent versions of pS-P sites in these IRAK1-UD peptides (data not shown), as anticipated due to the saturation of Pin1 in both cases. Importantly, Pin1 catalytic function and three of the four IRAK1-UD pS/T-P motifs studied here (S131, S144 and S173) were found to be essential for activation of IRAK-1 in dendritic cells [24], suggesting that bivalent advantage might translate into acceleration of reaction velocities at cellular concentrations. Investigation of the impact on Pin1 catalysis of these IRAK1-UD bivalent substrates relative to their monovalent counterparts will require measurement of bond-specific isomerization rates at peptide and Pin1 concentrations at or below ∼50 μM. This would be highly challenging for NMR exchange spectroscopy, where quantitative analysis of weak crosspeaks is required, and is beyond the scope of the present work.
Our findings on the potential impact of bivalent binding advantage have broader implications for Pin1 catalytic function beyond IRAK1. Recent work on a bivalent Pin1 substrate indicates that Pin1 catalysis of the pS1126-P site in the metabotrophic glutamate receptor V (mGluR5) plays a central role in dopamine-dependent plasticity [51]. While Pin1 kcat values determined by NMR exchange spectroscopy are similar for the pS1126-P peptide bond in singly and dually phosphorylated mGluR5 (pS1126-P and pT1123-PP-pS1126-P, respectively, Table 4), tight binding is attributed to the WW domain interacting with the pT1123-P site [51]. As presented above, this implies that the Pin1-catalyzed reaction velocity should be higher for the bivalent versus monovalent substrate at biological concentrations. Another example of a bivalent target where Pin1-catalyzed isomerization influences biological function is separase, a protein essential in chromosome segregation [52]. Separase binds to securin in the trans conformation and Cdk1-cyclin B1 in the cis conformation of the pS1126-P1127 peptide bond, and conformational exchange is regulated via a bivalent interaction with Pin1 that additionally involves pS1153-P1154 [52]. Again, one site (pS1126-P1127) is isomerized and the other (pS1153-P1154) contributes to binding (see Table 4), suggesting that bivalency accelerates the reaction at low biological concentrations. In contrast, studies of bivalent and monovalent forms of the AT180 epitope of tau showed that bivalent binding enhancement does not occur for the tandem motifs in this sequence (pT231-PPKpS235P, Table 4), Pin1 catalysis of the pT231-P peptide bond is not observed by NMR, and that Pin1 binding to this epitope is facilitated by the WW domain binding to the pT231-P site [21, 53, 54]. Recent in vitro studies have questioned the role of Pin1-catalyzed cis/trans isomerization in regulating the function and pathology of tau via the pT231-P site has been questioned [53, 55-57], which have been documented in vitro and in vivo and even in human diseases brains using various approaches including cis and trans conformation-specific antibodies [29, 34, 58, 59]. Notably, numerous additional proteins with multi-phosphorylated sequences have been implicated as interacting with Pin1 (Table 4), suggesting broad importance of bivalency in the cellular functions of Pin1.
Table 4. Multi-phosphorylated sequences implicated as interacting with Pin1.
| Name | Sequence | References |
|---|---|---|
| IRAK1-UD | 127AEAWpSPRKLPSSASTFLpSPAFPGSQTHSGPELGLVPpSPASLWPPPPpSPAPSSTK180 | [24] |
| MGLUR5 | 1159ELVALpTPPpSPFRDSV1173 | [51] |
| Separase | 1122STNSpSPVLKTKPQPIPNFLSHSPTCDCSLCApSPVLTA1158 | [52] |
| Tau | 210SRpTPSLPpTPPTREPKKVAVVRpTPPKpSPS237 | [21, 53, 62] |
| Cdc25c | 46PRpTPVGKFLGDSANLSILSGGpTPK69 | [14, 33, 44] |
| RNA Pol II | (CTD repeat) YSPTpSPSYSPTpSPSYSPTpSPS | [33] |
| Notch | 2121pSPQLHGAPLGGpTPTLpSPP2138 | [23] |
| Nanog | 51VpSPLPSSMDL LIQDpSPDSSTpSPK73 | [63] |
| Raf-1 | 285SESApSPSALSSpSPNNL300 | [64] |
| Myt1 | 411ApTPPGpSPPCS418;440LpSPEAVLARTVGSTSpTPRSRCpTP462 | [33] |
| CK2-α1 | 342GSpTPVSSANMMSGISSVPpTPpSPLGPLAGpSPVI373 | [65] |
| SMRT | 1461QGpTPLKYDTGASTTGSKKHDVRSLIGpSPGR1490 | [66] |
| c-Jun | 58SDLLTpSPDVGLLKLApSPEL76 | [22] |
| c-Fos | 322PLCpTPVVTCpTPSC334 | [67] |
| STAT3 | 712CVpTPTTCSNTIDLPMpSPRTLD732 | [68] |
| Smad3 | 177PEpTPPP182;202AGpSPNLpSPN PMpSPA215 | [69, 70] |
| PML | 515KAVpSPP HLDGPPpSPRS530 | [71] |
Potential Pin1 recognition motifs are shown in bold, with phosphoserine (pS) and phosphothreonine (pT) in red.
In summary, we have quantified the enhanced affinity of Pin1 interacting with pairs of neighboring pS/T-P motifs in the IRAK1-UD, and developed a model for the multi-state system that offers predictive power for envisioning the functional impact of bivalent Pin1:substrate interactions at cellular protein concentrations. Model-based predictions of the bivalent advantage expected at physiological protein concentrations were validated at low micromolar concentrations using the independent and complementrary technique of biolayer interferometry. The work presented here provides a framework for understanding the potential regulatory roles of hyperphosphorylation of Pin1 substrates, in terms of both their interactions with and catalysis by Pin1.
Materials & Methods
Pin1 expression and purification
Pin1 was expressed using either an ampicillin-resistant pGEX vector with a GST tag and thrombin cut site as used previously [29] [11] or a newly constructed kanamycin resistant pET28a vector with a 6×-His tag and a TEV protease cut site. Both vectors express full length Pin1 (residues 1-163) that after cleavage from the affinity tag has either 20 (pGEX) or two (pET28a) additional N-terminal residues. Importantly, both constructs give rise to essentially identical NMR spectra for peaks arising from Pin1 residues, allowing them to be used interchangeably. GST-Pin1 was expressed in BL21 star E. coli [Invitrogen] cells grown in 1 L of either LB [Beckton Dickenson and Company] or M9 minimal media (using 19 mM 15NH4Cl [Isotec] with 100 μg/mL ampicillin [Fisher]) at 37°C. Expression was induced at an OD600 ∼0.6, with 1 mM IPTG [CalBioChem] and growth was continued at 15°C for ∼20 hours. Cells were pelleted and resuspended in lysis buffer (25 mM HEPES [Fisher, sodium salt], 150 mM NaCl [Mallinckrodt Baker], 10 mM Imidazole [Alfa Aesar], pH 7.5, 100 μL protease inhibitor [Sigma], 1 mM DTT [Gold Biotechnology], and 2 mM EDTA [Fisher]). Cells were then lysed by freezing and thawing on ice, adding 1 mg/ml lysozyme [EMD Millipore], and sonicating (10 cycles). Lysed cells were centrifuged at 23,000 × g and the supernatant was filtered with a 0.8-μm syringe filter (Corning). Filtrate was then applied to a glutathione sepharose column [GE Health Sciences] and washed with 20 bed volumes wash buffer (25 mM HEPES, 150 mM NaCl, 10 mM Imidazole, pH 7.5). The column was then equilibrated with cleavage buffer (20 mM Tris-HCl [Mallinckrodt Baker], 150 mM NaCl, 2.5 mM CaCl2 [Fisher], pH 8.4), and 5 units of biotinylated thrombin [Novagen] were added. Cleavage of the Pin1 protein from the GST tag was performed while tumbling overnight at room temperature. The biotinylated thrombin was removed from the eluted Pin1 protein solution with streptavidin agarose beads [Novagen] and Pin1 was dialyzed into NMR buffer (10 mM HEPES, 10 mM NaCl, 1 mM TCEP [Thermo Scientific], pH 6.8). NMR samples also included 5 mM NaN3 [Fisher]. Protein was concentrated with VivaSpin-20 5000 MWC centrifugal concentrators [GE Health Sciences]. The concentration of Pin1 was measured by the UV absorbance spectrum, using the theoretical extinction coefficient at 280 nm of 21220 cm-1M-1. Protein purity was verified by SDS-PAGE. The pH of buffers and samples was adjusted using HCl [Mallinckrodt Baker] and NaOH [Sigma].
The 6×His-Pin1 construct was expressed and affinity purified as previously described for 6×His-PPIase [28]. The protein was dialyzed into NMR buffer, and 5mMNaN3 was added. The construct for the PPIase domain of Pin1, comprised of residues 46-163, (also in a pET28a vector with kanamycin resistance, a 6×-His tag and a TEV cleavage site) was purified using the same procedure. This protocol was also utilized in the preparation of WW domain (residues 1-50) as previously described [32].
IRAK1 Peptides
All peptides (Figure 1) were purchased from the Tufts University Core Facility and had unblocked ends, except p131/p144 which was purchased in the acetyl-peptide-amide form. Peptides were dissolved in NMR buffer, and their pH was adjusted with NaOH to 6.9.
NMR spectroscopy
NMR experiments were performed at a sample temperature of 25°C on a Varian Inova 600 MHz spectrometer equipped with a (H,C,N) Z-axis gradient probe. For each titration point, a 15N-1H fast HSQC spectrum [60] was recorded with a spectral width of 8 kHz in the proton dimension (total of 2048 complex data points) and either 1.77 kHz or 2.0 kHz in the nitrogen dimension (total of 256 complex data points). Spectra were processed and analyzed using the software tools nmrPipe, nmrDraw [61], and Sparky (T.D. Goddard and D.G. Kneller, University of California, San Fransisco). Data were apodized using a shifted sine bell function and zero filled prior to Fourier transformation. Peak positions were measured using the peak detection modules of Sparky.
Titration of isolated Pin1 domains
All NMR titration experiments were performed using the reverse titration method, where titration begins with the sample containing the highest ligand concentration and subsequent samples are prepared by mixing a portion of the previous sample with a stock solution of equal protein concentration. In this manner, the protein concentration remains constant and the ligand is sequentially diluted. Isolated WW and PPIase domains were individually titrated with one of three peptides derived from IRAK1-UD (p131, p144, and p163). For titration of WW with p163, WW concentration was 0.4 mM and p163 concentrations were 2.3, 1.1, 0.71, 0.37, 0.20, and 0 mM. For titration of PPIase with p131, PPIase concentration was 0.16mM and p131 concentrations were 7.6, 3.8, 1.9, 0.63, 0.21 or 0 mM. For titration of PPIase with p144, PPIase concentration was 0.22mM and p144 concentrations were 8.1, 4.05, 2.02, 1.01, 0.51, 0.25, 0.13 and 0 mM. For titration of PPIase with p163, PPIase concentration was 0.20mM and p163 concetrations were 3.8, 1.9, 0.96, 0.48, 0.24, 0.12, 0.06, 0.03 and 0 mM.
Each titration was analyzed using a representative selection of residues. Criteria for residue choice were: the largest observed chemical shift change (relative to apo) must reach a threshold value (ΔδiN,obs ≥ 0.1 ppm for nitrogen, ΔδiH,obs ≥ 0.0154 ppm for proton); the titration data must produce a reasonable binding curve; and the residue must be within the vicinity of the established binding site of its resident domain. Commonly accepted binding site residues are within WW domain residues 22-25 and 34-35, and within PPIase domain residues 57-63, 68-9, 113-118, and 128-130 [44]. Since the observed residue-specific chemical shift changes in the15N and 1H dimensions (ΔδiN,obs and ΔδiH,obs) were treated separately, residue choice can differ for the two dimensions.
The experimental data for each isolated domain was fit to a simple bimolecular binding model (Figure 4A) using Excel 2007 and the Solver Add-in. Both dimensions were simultaneously fit to produce one global KD for each peptide, where values (in thse nitrogen dimension) were multiplied by 0.154 to appropriately scale their contribution to the fit. The uncertainty in the global KD value obtained for each titration was approximated by individually fitting the corresponding selected residues, and determining the standard deviation of the resulting distribution of KD values.
Errors in ΔδiN,obs and ΔiδH,obs, required for evaluating the goodness of fit of a given model to the data, were estimated in two ways. First, the positions of peaks insensitive to ligand addition were monitored in spectra across the titration series to estimate the average uncertainty in peak location. The average standard deviation of these changes in each dimension were taken as the chemical shift uncertainty, σδN and σδH, for all peaks. Standard error propagation was applied to obtain the corresponding errors for the chemical shift differences relative to apo, σΔδN and σΔδH. Additionally, assuming the bimolecular model is valid for each individual domain, the average error values that produced reduced chi-square values of 1.0 for the best fits of the individual domain titration data in each dimension were determined. Since smaller errors produce more conservative assessments of model complexity, the smallest resulting set of values was used for all model assessments.
Goodness of fit was assessed using the reduced chi-square, given by:
where Δδiobs and Δδicalc are the experimental and calculated chemical shift perturbations for residue i, the sum is over N residues used in the fit, σi is the error in Δδiobs, and ρ is the number (#) of degrees of freedom: ρ = (# titration pts * # of residues) − # fitted parameters −1.
Titration of full-length Pin1
All NMR titration experiments were performed using the reverse titration method as described above. Pin1-FL was titrated with each of six peptides derived from IRAK1-UD (p131, p144, p163, p173, p131/p144 or p163/p173). For titration with p131, Pin1-FL concentration was 0.2 mM and p131 concentrations were 2.6, 1.3, 0.65, 0.33, 0.16, 0.08, 0.04, and 0 mM. For titration with p144, Pin1-FL concentration was 0.25 mM, and p144 concentrations were 7.85, 2.82, 1.66, 0.76, 0.37, 0.21, 0.103 and 0 mM. For titration with p163, 0.1 mM Pin1-FL was titrated with 1.54, 0.77, 0.51, 0.26, 0.13, 0.064, 0.032 and 0 mM p163. For titration with p173, Pin1-FL concentration was 0.25 mM and p173 concentrations were 2.0, 1.5, 1.0, 0.5, 0.25, 0.12, 0.06 and 0 mM of p173. For titration with p131/p144, Pin1-FL concentration was 0.25 mM and p131/p144 concentrations were 2.36, 1.18, 0.59, 0.30, 0.15, 0.07, 0.04, and 0 mM. For titration with p163p173, Pin1-FL concentration was 0.2 mM and p163p173 concentrations were 2.64, 1.32, 0.66, 0.33, 0.17, 0.08, 0.06, 0.04, and 0 mM.
Residues were selected for use in model analysis and fitting based on the following criteria. For all Pin1-FL titrations, the largest values of ΔδiN,obs and ΔδiH,obs must reach a threshold value (ΔδiN,obs ≥ 0.1 ppm for nitrogen, ΔδiH,obs ≥ 0.0154 ppm for proton), peaks must move in the same direction in the isolated domain and Pin1-FL titrations, residues with peaks that overlap with other peaks during titration were excluded, and residues previously implicated in either the PPIase conduit or the interface between the PPIase and WW domains were excluded (to the extent possible given the small size of the WW domain). An emphasis was placed on selecting residues known to be important for binding and residues in close spatial proximity to the Ala-Pro dipeptide or the sulfate ion in the 1PIN.pdb crystal structure [16]. Additionally, for the dually phosphorylated peptide titrations, in order to allow for possible differentiation between different binding orientations, selected residues were required to display disparate bound chemical shifts for each binding motif. In some cases, residues selected for bivalent analysis were not previously selected in the monovalent analysis. For such residues, the residue-specific bound Δδij,bound values were obtained by using the previously determined global KD values (Table 2) and fitting for Δδij,bound.
Chemical shift perturbation data were analyzed separately for the nitrogen and proton dimensions, which enhanced sensitivity of peak trajectories to shifting populations among the multiple states during the titration and yielded independent measures of each KD. Titrations of Pin1-FL binding to a singly phosphorylated peptide were fit to an analytical model in which the peptide can either bind the PPIase domain or the WW domain with distinct affinities (figure 4B). The error in a given KD value is the standard deviation of a series of KD values calculated using Monte Carlo analysis, in which artificial noise is added to the data to generate 100 synthetic data sets, each of which are fit. The artificial noise was generated for each data point using the output of a normally distributed, random number generator scaled by σ (0.00407 ppm in the proton dimension and 0.0205 ppm in the nitrogen dimension). The initial guesses for parameters being fit were also allowed to vary randomly in a normal distribution about the determined values (Table 2), where the boundaries are set at half and twice the determined value.
Pin1-FL binding to a dually phosphorylated peptide is more complex (Figure 4C), requiring numerical solution. Our method employed Virtual Cell modelling software [27] to generate the MATLAB code for numerical solution of the set of coupled ordinary differential equations that describe the model, yielding equilibrium populations for each state in the model. These populations were employed in fits of chemical shift perturbations, carried out in MATLAB using the nonlinear multivariable function fmincon. For these analyses, KD and Δδij,bound values for all non-bivalent states were set to their corresponding values obtained from Pin1-FL titrations with singly-phosphorylated peptides. For inclusion of bivalent states (Figure 4C, highlighted by yellow background), two independent equilibrium constants (Keq) are required to describe the closure of each monovalent precursor state into the corresponding bivalent state (Figure 4C), given by:
and
where the superscripts denote the relevant binding event (e.g. A:WW refers to the WW domain interacting with motif A, the N-terminal most motif in the dually-phosphorylated peptide). A set of simulations was carried out with KeqA:WW and KeqB:WW set at 107, so as to remove the pathway to either bivalent species, in order to determine if the bivalent species is necessary to fit the data. Reduced chi-square values were employed to assess the quality of all simulations and fits (using σΔδH = 0.00407 ppm and σΔδN = 0.0205 ppm, as determined in the isolated domain titrations).
Simulation of Pin1-FL interactions with IRAK1-UD motifs at low micromolar concentrations
Using VCell in conjunction with MATLAB and the fitted KD and Keq values, two models based on Figure 4B and 4C were constructed to simulate the bound population of Pin1 that would occur under biologically-relevant conditions where Pin1 is 0.5 μM [47], for varying concentrations and phosphorylation states of IRAK1.
Biolayer interferometry of Pin1 binding to singly or dually phosphorylated peptide
Biolayer interferometry (BLI) was performed at 25°C using an Octect RED (Rapid system Extended Detection) 96 well plate reader [FortéBio] and analyzed using the Octet Data Acquisition software to assess the interaction between Pin1-FL (with 6X-His tag) and either p144 or p131/p144. The assay was conducted in BLI buffer (10mM HEPES, 20mM NaCl, 0.1mM TCEP, pH 7.4) using Ni-NTA biosensors [FortéBio]. 10μg/mL of Pin1 in BLI buffer was simultaneously loaded onto eight independent biosensors over 600s (sufficient to reach equilibrium). The loaded biosensors were then quenched for 300s using BLI buffer with 10mM imidazole and 25μg/mL EZ-link biotin, limiting non-specific interactions with unbound Ni-NTA and strepavidin. The eight Pin1 loaded biosensors were then allowed to associate with varying concentrations of either the p144 or p131/p144 peptide (80 μM, 40 μM, 20 μM, 10 μM, 5 μM, 2.5 μM, and 1.25 μM peptide in BLI buffer) for 600s and dissociate in BLI buffer alone for another 600s. In order to control for non-specific binding of the peptides to the biosensor, the same assays were carried out for each peptide with biosensors lacking Pin1, where the biosensor loading step occurred in BLI buffer alone. These results were subtracted from the Pin1-loaded biosensor results to ensure that peptide binding to Pin1 was accurately measured. An effective KD for Pin1 interacting with each peptide was obtained using the 1:1 model available in the software provided with the Octet RED96 [FortéBio].
Acknowledgments
This research was supported by NSF grant MCB-1157806 (LKN), NIH grant 1R01-HL111430 (KPL), and NIH training grants T32GM007273 (MJR) and T32GM008267 (AIG). We thank Lucila Andrea Acevedo and Ross J. Resnick for helpful discussions and laboratory support. We thank Bob Dass (Scientist, FortéBio) and Erik Levy (Sales Manager, FortéBio) for their generous assistance that enabled our acquisition and analysis of BLI data.
Abbreviations
- pS/pT-P
phosphoSer/phosphoThr-Pro motif
- pS-P
phosphoSer-Pro binding motif
- IRAK1
Interleukin-1 receptor associated kinase-1
- UD
undefined domain
- KD
kinase domain
- CTD
C-terminal domain
- Pin1-FL
full length Pin1
- p131
phosphopeptide representing IRAK1 residues 126-135 phosphorylated at Ser131
- p144
phosphopeptide representing IRAK1 residues 140-149 phosphorylated at Ser144
- p163
phosphopeptide representing IRAK1 residues 157-180 phosphorylated at Ser163
- p173
phosphopeptide representing IRAK1 residues 157-180 phosphorylated at Ser173
- p131/p144
phosphopeptide representing IRAK1 residues 126-149 phosphorylated at Ser131 and Ser144
- p163/p173
phosphopeptide representing IRAK1 residues 157-180 phosphorylated at Ser163 and Ser173
- VCell
Virtual Cell software
- 15N-1H HSQC
15N-separated heteronuclear single quantum coherence spectroscopy
Footnotes
Author Contributions: MJR, AIG, JK and LKN planned experiments, MJR, JK and AIG performed experiments, MJR and JK analyzed data, AIG assisted with data analysis, JK contributed essential ideas to data analysis, KPL contributed reagents, and MJR, AIG, JK and LKN collaboratively wrote the paper.
Conflict of Interest: Dr. Lu declares that he is an inventor of Pin1 technology, which was licensed by BIDMC to Pinteon Therapeutics. Dr. Lu owns equity in, and consults for, Pinteon. His interests were reviewed and are managed by BIDMC in accordance with its conflict of interest policy.
Cited Literature
- 1.Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–16. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 2.Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, Xiao ZX. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419:849–53. doi: 10.1038/nature01116. [DOI] [PubMed] [Google Scholar]
- 3.Shaw PE. Peptidyl-prolyl cis/trans isomerases and transcription: is there a twist in the tail? EMBO Rep. 2007;8:40–5. doi: 10.1038/sj.embor.7400873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciarapica R, Methot L, Tang Y, Lo R, Dali R, Buscarlet M, Locatelli F, del Sal G, Rota R, Stifani S. Prolyl isomerase Pin1 and protein kinase HIPK2 cooperate to promote cortical neurogenesis by suppressing Groucho/TLE:Hes1-mediated inhibition of neuronal differentiation. Cell Death Differ. 2014;21:321–32. doi: 10.1038/cdd.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakamura K, Kosugi I, Lee DY, Hafner A, Sinclair DA, Ryo A, Lu KP. Prolyl isomerase Pin1 regulates neuronal differentiation via beta-catenin. Mol Cell Biol. 2012;32:2966–78. doi: 10.1128/MCB.05688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–34. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 7.Pastorino L, Ma SL, Balastik M, Huang P, Pandya D, Nicholson L, Lu KP. Alzheimer's disease-related loss of Pin1 function influences the intracellular localization and the processing of AbetaPP. J Alzheimers Dis. 2012;30:277–97. doi: 10.3233/JAD-2012-111259. [DOI] [PubMed] [Google Scholar]
- 8.Lee TH, Tun-Kyi A, Shi R, Lim J, Soohoo C, Finn G, Balastik M, Pastorino L, Wulf G, Zhou XZ, Lu KP. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat Cell Biol. 2009;11:97–105. doi: 10.1038/ncb1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu YX, Manley JL. The prolyl isomerase Pin1 functions in mitotic chromosome condensation. Mol Cell. 2007;26:287–300. doi: 10.1016/j.molcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 10.Lu Z, Hunter T. Prolyl isomerase Pin1 in cancer. Cell Res. 2014;24:1033–49. doi: 10.1038/cr.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 1997;278:1957–60. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 12.Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–41. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 13.Yeh ES, Means AR. PIN1, the cell cycle and cancer. Nat Rev Cancer. 2007;7:381–8. doi: 10.1038/nrc2107. [DOI] [PubMed] [Google Scholar]
- 14.Innes BT, Bailey ML, Brandl CJ, Shilton BH, Litchfield DW. Non-catalytic participation of the Pin1 peptidyl-prolyl isomerase domain in target binding. Front Physiol. 2013;4:18. doi: 10.3389/fphys.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–7. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 16.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–86. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 17.Daum S, Lucke C, Wildemann D, Schiene-Fischer C. On the benefit of bivalency in peptide ligand/pin1 interactions. Journal of molecular biology. 2007;374:147–61. doi: 10.1016/j.jmb.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 18.Jencks WP. On the attribution and additivity of binding energies. Proc Natl Acad Sci U S A. 1981;78:4046–50. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angewandte Chemie International Edition. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 20.Myers JK, Morris DP, Greenleaf AL, Oas TG. Phosphorylation of RNA polymerase II CTD fragments results in tight binding to the WW domain from the yeast prolyl isomerase Ess1. Biochemistry. 2001;40:8479–86. doi: 10.1021/bi0027884. [DOI] [PubMed] [Google Scholar]
- 21.Smet C, Wieruszeski JM, Buee L, Landrieu I, Lippens G. Regulation of Pin1 peptidyl-prolyl cis/trans isomerase activity by its WW binding module on a multi-phosphorylated peptide of Tau protein. FEBS Lett. 2005;579:4159–64. doi: 10.1016/j.febslet.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 22.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001;20:3459–72. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rustighi A, Tiberi L, Soldano A, Napoli M, Nuciforo P, Rosato A, Kaplan F, Capobianco A, Pece S, Di Fiore PP, Del Sal G. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat Cell Biol. 2009;11:133–42. doi: 10.1038/ncb1822. [DOI] [PubMed] [Google Scholar]
- 24.Tun-Kyi A, Finn G, Greenwood A, Nowak M, Lee TH, Asara JM, Tsokos GC, Fitzgerald K, Israel E, Li X, Exley M, Nicholson LK, Lu KP. Essential role for the prolyl isomerase Pin1 in Toll-like receptor signaling and type I interferon-mediated immunity. Nat Immunol. 2011;12:733–41. doi: 10.1038/ni.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–90. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. 2010;80:1981–91. doi: 10.1016/j.bcp.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 27.Dubitzky W, Wolkenhauer O, Cho KH, Yokota H, Moraru I. Encyclopedia of Systems Biology. Springer; New York: Virtual Cell (VCell) Modeling and Analysis Platform in; pp. 2342–2347. [Google Scholar]
- 28.Greenwood AI, Rogals MJ, De S, Lu KP, Kovrigin EL, Nicholson LK. Complete determination of the Pin1 catalytic domain thermodynamic cycle by NMR lineshape analysis. J Biomol NMR. 2011;51:21–34. doi: 10.1007/s10858-011-9538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Kullertz G, Stark M, Fischer G, Lu KP. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6:873–83. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 30.Xu N, Tochio N, Wang J, Tamari Y, Uewaki J, Utsunomiya-Tate N, Igarashi K, Shiraki T, Kobayashi N, Tate S. The C113D mutation in human Pin1 causes allosteric structural changes in the phosphate binding pocket of the PPIase domain through the tug of war in the dual-histidine motif. Biochemistry. 2014;53:5568–78. doi: 10.1021/bi5007817. [DOI] [PubMed] [Google Scholar]
- 31.Behrsin CD, Bailey ML, Bateman KS, Hamilton KS, Wahl LM, Brandl CJ, Shilton BH, Litchfield DW. Functionally important residues in the peptidyl-prolyl isomerase Pin1 revealed by unigenic evolution. Journal of molecular biology. 2007;365:1143–62. doi: 10.1016/j.jmb.2006.10.078. [DOI] [PubMed] [Google Scholar]
- 32.De S, Greenwood AI, Rogals MJ, Kovrigin EL, Lu KP, Nicholson LK. Complete thermodynamic and kinetic characterization of the isomer-specific interaction between Pin1-WW domain and the amyloid precursor protein cytoplasmic tail phosphorylated at Thr668. Biochemistry. 2012;51:8583–96. doi: 10.1021/bi3008214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol. 2000;7:639–43. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 34.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–8. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 35.Namanja AT, Wang XDJ, Xu BL, Mercedes-Camacho AY, Wilson KA, Etzkorn FA, Peng JW. Stereospecific gating of functional motions in Pin1. P Natl Acad Sci USA. 2011;108:12289–12294. doi: 10.1073/pnas.1019382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Potter AJ, Ray S, Gueritz L, Nunns CL, Bryant CJ, Scrace SF, Matassova N, Baker L, Dokurno P, Robinson DA, Surgenor AE, Davis B, Murray JB, Richardson CM, Moore JD. Structure-guided design of alpha-amino acid-derived Pin1 inhibitors. Bioorg Med Chem Lett. 2010;20:586–90. doi: 10.1016/j.bmcl.2009.11.090. [DOI] [PubMed] [Google Scholar]
- 37.Potter A, Oldfield V, Nunns C, Fromont C, Ray S, Northfield CJ, Bryant CJ, Scrace SF, Robinson D, Matossova N, Baker L, Dokurno P, Surgenor AE, Davis B, Richardson CM, Murray JB, Moore JD. Discovery of cell-active phenyl-imidazole Pin1 inhibitors by structure-guided fragment evolution. Bioorg Med Chem Lett. 2010;20:6483–8. doi: 10.1016/j.bmcl.2010.09.063. [DOI] [PubMed] [Google Scholar]
- 38.Urusova DV, Shim JH, Kim DJ, Jung SK, Zykova TA, Carper A, Bode AM, Dong Z. Epigallocatechin-gallate suppresses tumorigenesis by directly targeting Pin1. Cancer Prev Res (Phila) 2011;4:1366–77. doi: 10.1158/1940-6207.CAPR-11-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen CH, Li W, Sultana R, You MH, Kondo A, Shahpasand K, Kim BM, Luo ML, Nechama M, Lin YM, Yao Y, Lee TH, Zhou XZ, Swomley AM, Allan Butterfield D, Zhang Y, Lu KP. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer's disease. Neurobiol Dis. 2015;76:13–23. doi: 10.1016/j.nbd.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang M, Wang XJ, Chen X, Bowman ME, Luo Y, Noel JP, Ellington AD, Etzkorn FA, Zhang Y. Structural and kinetic analysis of prolyl-isomerization/phosphorylation cross-talk in the CTD code. ACS Chem Biol. 2012;7:1462–70. doi: 10.1021/cb3000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Innes BT, Sowole MA, Gyenis L, Dubinsky M, Konermann L, Litchfield DW, Brandl CJ, Shilton BH. Peroxide-mediated oxidation and inhibition of the peptidyl-prolyl isomerase Pin1. Biochim Biophys Acta. 2015;1852:905–12. doi: 10.1016/j.bbadis.2014.12.025. [DOI] [PubMed] [Google Scholar]
- 42.Jacobs DM, Saxena K, Vogtherr M, Bernado P, Pons M, Fiebig KM. Peptide binding induces large scale changes in inter-domain mobility in human Pin1. J Biol Chem. 2003;278:26174–26182. doi: 10.1074/jbc.M300796200. [DOI] [PubMed] [Google Scholar]
- 43.VanWart AT, Eargle J, Luthey-Schulten Z, Amaro RE. Exploring Residue Component Contributions to Dynamical Network Models of Allostery. J Chem Theory Comput. 2012;8:2949–2961. doi: 10.1021/ct300377a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson KA, Bouchard JJ, Peng JW. Interdomain interactions support interdomain communication in human Pin1. Biochemistry. 2013;52:6968–81. doi: 10.1021/bi401057x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Namanja AT, Wang XJ, Xu B, Mercedes-Camacho AY, Wilson KA, Etzkorn FA, Peng JW. Stereospecific gating of functional motions in Pin1. Proc Natl Acad Sci U S A. 2011;108:12289–94. doi: 10.1073/pnas.1019382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Mahoney BJ, Zhang M, Zintsmaster JS, Peng JW. Negative Regulation of Peptidyl-Prolyl Isomerase Activity by Interdomain Contact in Human Pin1. Structure (London, England : 1993) 2015;23:2224–33. doi: 10.1016/j.str.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen M, Stukenberg PT, Kirschner MW, Lu KP. The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phosphoproteins. Genes Dev. 1998;12:706–20. doi: 10.1101/gad.12.5.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacobs DM, Saxena K, Vogtherr M, Bernado P, Pons M, Fiebig KM. Peptide binding induces large scale changes in inter-domain mobility in human Pin1. J Biol Chem. 2003;278:26174–82. doi: 10.1074/jbc.M300796200. [DOI] [PubMed] [Google Scholar]
- 49.Lea WA, O'Neil PT, Machen AJ, Naik S, Chaudhri T, McGinn-Straub W, Tischer A, Auton MT, Burns JR, Baldwin MR, Khar KR, Karanicolas J, Fisher MT. Chaperonin-Based Biolayer Interferometry To Assess the Kinetic Stability of Metastable, Aggregation-Prone Proteins. Biochemistry. 2016 doi: 10.1021/acs.biochem.6b00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah NB, Duncan TM. Bio-layer Interferometry for Measuring Kinetics of Protein-protein Interactions and Allosteric Ligand Effects. Journal of Visualized Experiments : JoVE. 2014:51383. doi: 10.3791/51383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park JM, Hu JH, Milshteyn A, Zhang PW, Moore CG, Park S, Datko MC, Domingo RD, Reyes CM, Wang XJ, Etzkorn FA, Xiao B, Szumlinski KK, Kern D, Linden DJ, Worley PF. A prolyl-isomerase mediates dopamine-dependent plasticity and cocaine motor sensitization. Cell. 2013;154:637–50. doi: 10.1016/j.cell.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hellmuth S, Rata S, Brown A, Heidmann S, Novak B, Stemmann O. Human chromosome segregation involves multi-layered regulation of separase by the peptidyl-prolyl-isomerase Pin1. Mol Cell. 2015;58:495–506. doi: 10.1016/j.molcel.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 53.Eichner T, Kutter S, Labeikovsky W, Buosi V, Kern D. Molecular Mechanism of Pin1-Tau Recognition and Catalysis. Journal of molecular biology. 2016;428:1760–75. doi: 10.1016/j.jmb.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 54.Smet C, Sambo AV, Wieruszeski JM, Leroy A, Landrieu I, Buee L, Lippens G. The peptidyl prolyl cis/trans-isomerase Pin1 recognizes the phospho-Thr212-Pro213 site on Tau. Biochemistry. 2004;43:2032–40. doi: 10.1021/bi035479x. [DOI] [PubMed] [Google Scholar]
- 55.Ahuja P, Cantrelle FX, Huvent I, Hanoulle X, Lopez J, Smet C, Wieruszeski JM, Landrieu I, Lippens G. Proline Conformation in a Functional Tau Fragment. Journal of molecular biology. 2016;428:79–91. doi: 10.1016/j.jmb.2015.11.023. [DOI] [PubMed] [Google Scholar]
- 56.Kamah A, Cantrelle FX, Huvent I, Giustiniani J, Guillemeau K, Byrne C, Jacquot Y, Landrieu I, Baulieu EE, Smet C, Chambraud B, Lippens G. Isomerization and Oligomerization of Truncated and Mutated Tau Forms by FKBP52 are Independent Processes. Journal of molecular biology. 2016;428:1080–90. doi: 10.1016/j.jmb.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 57.Kutter S, Eichner T, Deaconescu AM, Kern D. Regulation of Microtubule Assembly by Tau and not by Pin1. Journal of molecular biology. 2016;428:1742–59. doi: 10.1016/j.jmb.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 58.Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y, Wei S, Luo ML, Albayram O, Huang P, Rotenberg A, Ryo A, Goldstein LE, Pascual-Leone A, McKee AC, Meehan W, Zhou XZ, Lu KP. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523:431–6. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell. 2012;149:232–44. doi: 10.1016/j.cell.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mori S, Abeygunawardana C, Johnson MO, van Zijl PC. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J Magn Reson B. 1995;108:94–8. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 61.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 62.Ahuja P, Cantrelle FX, Huvent I, Hanoulle X, Lopez J, Smet C, Wieruszeski JM, Landrieu I, Lippens G. Proline Conformation in a Functional Tau Fragment. J Mol Biol. 2016;428:79–91. doi: 10.1016/j.jmb.2015.11.023. [DOI] [PubMed] [Google Scholar]
- 63.Moretto-Zita M, Jin H, Shen Z, Zhao T, Briggs SP, Xu Y. Phosphorylation stabilizes Nanog by promoting its interaction with Pin1. Proc Natl Acad Sci U S A. 2010;107:13312–7. doi: 10.1073/pnas.1005847107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–24. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 65.Messenger MM, Saulnier RB, Gilchrist AD, Diamond P, Gorbsky GJ, Litchfield DW. Interactions between protein kinase CK2 and Pin1. Evidence for phosphorylation-dependent interactions. J Biol Chem. 2002;277:23054–64. doi: 10.1074/jbc.M200111200. [DOI] [PubMed] [Google Scholar]
- 66.Stanya KJ, Liu Y, Means AR, Kao HY. Cdk2 and Pin1 negatively regulate the transcriptional corepressor SMRT. J Cell Biol. 2008;183:49–61. doi: 10.1083/jcb.200806172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Monje P, Hernandez-Losa J, Lyons RJ, Castellone MD, Gutkind JS. Regulation of the transcriptional activity of c-Fos by ERK. A novel role for the prolyl isomerase PIN1. J Biol Chem. 2005;280:35081–4. doi: 10.1074/jbc.C500353200. [DOI] [PubMed] [Google Scholar]
- 68.Lufei C, Koh TH, Uchida T, Cao X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene. 2007;26:7656–64. doi: 10.1038/sj.onc.1210567. [DOI] [PubMed] [Google Scholar]
- 69.Aragon E, Goerner N, Zaromytidou AI, Xi Q, Escobedo A, Massague J, Macias MJ. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 2011;25:1275–88. doi: 10.1101/gad.2060811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsuura I, Chiang KN, Lai CY, He D, Wang G, Ramkumar R, Uchida T, Ryo A, Lu K, Liu F. Pin1 promotes transforming growth factor-beta-induced migration and invasion. J Biol Chem. 2009;285:1754–64. doi: 10.1074/jbc.M109.063826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reineke EL, Lam M, Liu Q, Liu Y, Stanya KJ, Chang KS, Means AR, Kao HY. Degradation of the tumor suppressor PML by Pin1 contributes to the cancer phenotype of breast cancer MDA-MB-231 cells. Mol Cell Biol. 2008;28:997–1006. doi: 10.1128/MCB.01848-07. [DOI] [PMC free article] [PubMed] [Google Scholar]