

Abstract
The innate immune response of airway epithelial cells to aeroallergen initiates the development of T cell responses that are central to allergic inflammation. Although proteinase allergens induce the expression of interleukin 25 we show that epithelial matrix metalloproteinase 7 (MMP7) was expressed in asthma and was required for maximal activity of IL-25 in promoting T helper type 2 cell differentiation. Allergen-challenged Mmp7−/− mice showed reduced airway hyperreactivity, allergic inflammatory cytokine production and increased expression of retinal dehydrogenase (RALDH)-1. Inhibition of RALDH-1 restored the asthma phenotype in Mmp7−/− mice and inhibited lung T regulatory cell responses while exogenous administration of retinoic acid attenuated the asthma phenotype. Thus, MMP7 coordinates allergic lung inflammation by activating IL-25 while simultaneously inhibiting retinoid-dependent T regulatory cell development.
Proximal and distal airway epithelial cells directly communicate with the outside environment and thus their response to inhaled allergens could play an integral part in the initiation of allergic lung inflammation. Identification of the critical epithelial-derived mediators that drive allergic lung responses is at an early stage, but is essential to a complete understanding of the molecular mechanism(s) underlying diseases such as asthma. In prior studies, we demonstrated that, upon inhalation, proteinase allergens (e.g. aeroallergens with active proteinase property) rapidly induce a powerful inflammatory response that initiates the recruitment of allergic immune cells into the lung1,2. While the role of cytokines expressed in the airway epithelium in asthma has been extensively studied3,4, the mechanism(s) by which allergens initiate airway TH2 cell differentiation is poorly understood. Interleukin 25 (IL-25, also known as IL-17E) is a member of the IL-17 family of cytokines that is produced primarily in the airway epithelium in response to proteinase allergens5,6. Transgenic expression of human and mouse IL-25 induces TH2 immune responses marked by pronounced lung eosinophilia, mucus hyperproduction and increased expression of IL-4, IL-5, IL-13 and CCL11 (eotaxin 1) in the airway7,8. Neutralization of IL-25 by blocking antibodies has been shown to reverse airway hyperreactivity in a mouse model of asthma9. Moreover, inability to develop a strong TH2 biased immune response was shown in IL-25-deficient mice, which failed to clear gut parasitic infections with Nippostrongylus brasiliensis and Trichuris muris10. These studies suggest a critical role for epithelial cells in providing the IL-25 signals that may be required for effective TH2 responses. Although the role of IL-25 in allergic lung disease is well known, a broader understanding of how IL-25 interacts with other airway epithelial factors to coordinately regulate allergic responses is needed.
Matrix metalloproteinases (MMPs) are zinc-dependent enzymes that are induced in response to a large number of stimuli. Although MMPs were originally shown to mediate turnover of extracellular matrix molecules, it is now clear that they control diverse biological processes unrelated to matrix degradation11. Although the exact role for MMPs in chronic models of lung inflammation is debated, it is clear that several members of this family are acutely induced either locally or systemically at the onset of allergen challenge12–16. MMP2 and MMP9 are not required for development of allergic lung disease, but play key roles in the clearance of inflammatory cells from lung parenchyma by establishing the necessary trans-epithelial chemokine gradients15,16. However, while neither MMP2 nor MMP9 is expressed in the airway epithelium17, MMP7 (also known as matrilysin; http://www.signaling-gateway.org/molecule/query?afcsid=A001477), an epithelial cell specific MMP, is induced in the lung and the gut during inflammation18,19. MMP7 has been shown to modify pro-α-defensin, Fas-L and tumor necrosis factor (TNF) in the gut during defense against enteric pathogens, but its role in allergic inflammation has yet to be explored19,20.
In this study, we examined how airway epithelial IL-25 coordinates proteinase-dependent allergic lung disease in the context of other potentially relevant airway epithelial-derived factors including MMP7. We show that in response to proteinase active allergen or recombinant IL-25, mouse airway epithelial cells expressed MMP. Activation of MMP7 was critical for IL-25 function because MMP7-cleaved IL-25 enhanced TH2 cell differentiation. Furthermore, we show that humans with chronic asthma also expressed MMP7 and IL-25 in their distal airspaces and in response to ragweed extract, a potent allergenic stimuli, patients with a history of allergy significantly increased secretion of nasal MMP7. In the absence of MMP7, mice showed attenuated allergic responses to proteinase allergen challenge and enhanced expression of retinal dehydrogenase-1 (RALDH-1), a rate-limiting enzyme for retinoic acid production. Moreover Mmp7−/− mice showed an increase in the number of regulatory T cells in the lung parenchyma. Together, these results support a role for MMP7 as a proinflammatory mediator that specifically enhances the function of IL-25 that is necessary for robust TH2 responses. Finally, we demonstrate a tolerogenic mechanism initiated by airway epithelial cells in response to proteinase allergen challenge in which RALDH-1 is induced to promote immunosuppressive T regulatory cells.
Results
MMP7 mediate increase function of IL-25
We previously showed that MMP2 and MMP9 are not expressed in airway epithelial cells following allergen challenge17. In contrast, a fungal-derived proteinase allergen strongly enhanced expression of airway epithelial MMP7 (Fig. 1a). Whereas induction of MMP2 and MMP9 was IL-13 dependent15, MMP7 induction was independent of IL-13 as equivalent induction of MMP7 was observed in allergen-challenged mice deficient in STAT6, the principal mediator of IL-13 effects in the lung (data not shown). Because MMP7 is known to cleave and activate cytokines in the gut13, we next determined if MMP7 modifies cytokines that are critical for initiating allergic lung responses. In addition to IL-25, IL-13 and thymic stromal lymphopoietin (TSLP) are critical participants in the molecular pathways underlying allergic lung disease5,21. We found that while recombinant (r)IL-13, and rTSLP were not substrates for activated MMP7, under the same conditions rIL-25 was cleaved at multiple sites (Fig. 1b, Supplementary Table 1 and Supplementary Fig. 1 online). To determine the functional activity of cleaved IL-25, we examined the response of naive spleen and lymph node cells after activation by T cell receptor crosslinking in vitro. We found that the presence of MMP7-cleaved rIL-25 (hereafter called rIL-25′C), induced significantly more secretion of TH2 cytokines (IL-4, IL-5 and IL-13), than native rIL-25 and showed enhanced binding to IL-17RB-Fc fusion protein in vitro (Fig. 1c,d; Supplementary Fig. 2a,b online). Indeed, native IL-25 had slight effect on IL-4 secretion, but rIL-25′C enhanced IL-4 secretion 10-fold, suggesting that the major active form of IL-25 is IL-25′C. No effect of rIL-25 or rIL-25′C on induction of interferon-γ (IFN-γ), a canonical TH1 cytokine, was seen (Fig. 1c). Collectively these data indicate that IL-25 is a substrate for MMP7 and that cleavage of IL-25 by MMP7 is necessary to drive robust TH2 responses.
Figure 1. MMP7 is induced in allergic inflammation and modulates IL-25 function.
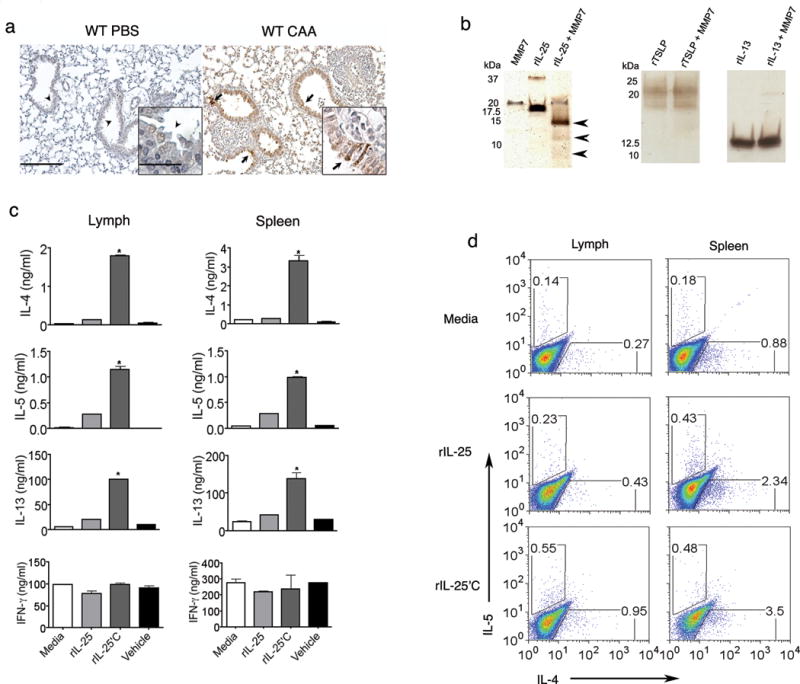
(a) Detection of MMP7 in lung of PBS treated (C57BL/6), and mice immunized with CAA by immunohistochemistry (arrows indicate MMP-7 expression; insets show 40× magnification of airway). Bars indicate 100 μm and 25 μm bar. (b) Recombinant (r)IL-25 (5 μg), rIL-13 (5 μg) and rTSLP (5 μg) were incubated for 2 h at 37 °C with APMA-activated MMP7 (0.5 μg), and were resolved on 16.5% Tricine gel followed by detection using silver stain (arrows indicate bands corresponding to fragments of rIL-25 that were cleaved by MMP7). (c) Naïve lymph node cells (left) and spleen cells (right) isolated from C57BL/6 mice, were stimulated with 2 μg/ml of plate-bound anti-CD3 in the presence of equal amount of native rIL-25 (250 ng/ml) or MMP7 cleaved rIL-25 (IL-25′C; 250 ng/ml). On day 2, cell culture-supernatants from each condition were examined for the concentration of IL-4, IL-5, IL-13 and IFN-γ by ELISA. Media alone and APMA containing buffer that was used to activate MMP-7 (vehicle) were used as controls (n=3; data represent mean values ± SD; representative of three different experiments).*P< 0.05 relative to rIL-25, using the one-way ANOVA test. (d) Representative flow cytometry data of lymphocytes, following three days of culture under the same conditions as described in (c). Cells were restimulated with 500 ng/ml ionomycin and 50 ng/ml PMA in the presence of GolgiStop for 5 h, and the percentage of cells expressing IL-4 and IL-5 were detected by intracytoplasmic staining. Data is representative of 3 independent experiments.
Attenuated asthma phenotype in Mmp7−/− mice
The increased potency of rIL-25′C in activating TH2 cells in vitro raised the possibility of an essential role for MMP7 in mediating allergic inflammation in vivo. To determine this, we compared the phenotype of Mmp7−/− with wild-type mice in response to the proteinase allergen extracted from fungi (complete aspergillus allergen; CAA), a powerful allergen that we have previously used to elicit allergic lung disease in mice1. This airway inflammation model is characterized by a predominant eosinophil influx and airway obstruction due to enhanced glycoprotein secretion, as well as increased airway hyperactivity to secondary challenge with acetylcholine (Ach)22,23. As compared to wild-type mice, Mmp7−/− mice immunized with CAA showed reduced airway hyperreactivity (AHR) as assessed by both reduction in respiratory system resistance (RRS) in response to multiple Ach doses and an increase in the provocative concentration of Ach required to elicit a 200% change in RRS from baseline (PC200). Moreover, Mmp7−/− mice secreted less bronchoalveolar lavage (BAL) fluid glycoproteins following CAA challenge (Fig. 2a–c).
Figure 2. MMP-7 null mice show reduced asthma phenotype.

Age and sex matched wild-type (WT), (n = 4) and Mmp7−/− mice (n = 4) were immunized intranasally with CAA or PBS for every 4 days for a total of five times and 24 h after the last immunization, mice were assessed for (a,b) AHR is shown as dose response curve to acetylcholine and using PC 200, and (c) Glycoproteins in BAL detected by ELISA. (d) Eosinophil cell counts from BAL were determined from the same groups of mice. *P < 0.05 relative to PBS challenged WT and **P < 0.05 relative to CAA immunized WT mice using one way ANOVA and t-test. Data represent mean values ± SD (e) Representative photomicrographs of bronchovascular bundles (n=4 per group) stained with H&E. Bar indicates 100 μm. (f,g) WT and MMP7 null mice were given intranasal PBS or rIL-25 (5 μg) twice daily for 3 days, and 24 h following the last dose, total cell and eosinophil count (f) and IL-4, and IL-5 (g) were assessed in the BAL fluid. (Data represent mean values ± SD; representative of 3 independent experiments; n=4 per group; *P < 0.05 relative to WT mice treated with rIL-25).
Airway eosinophilia was also significantly reduced in Mmp7−/− CAA immunized mice (Fig. 2d). Because lung parenchymal eosinophil egression into the airways is impaired in Mmp2−/− and Mmp9−/− mice15,16, we determined lung parenchymal eosinophil numbers to rule out a similar defect in Mmp7−/− animals. Analysis of total number of lung parenchymal inflammatory cells showed fewer eosinophils in the lungs of CAA immunized Mmp7−/− as compared to wild-type mice (Fig. 2e, Supplementary Fig. 3 online), indicating that the decreased cell numbers in the airway was not due to reduced cell trafficking.
We turned next to testing whether MMP7-mediated modification of IL-25 was influential in determining allergic lung responses. rIL-25 was given intranasally to wild-type and Mmp7−/− mice and eosinophil recruitment was determined. In this assay, rIL-25 alone readily induced lung eosinophilia and de novo expression of MMP7 in airway epithelial cells of wild-type mice, but significantly fewer eosinophils were enumerated and reduced IL-4 and IL-5 concentrations were detected in BAL fluid of Mmp7−/− mice (Fig. 2f,g and Supplementary Fig. 4 online). Collectively, these findings support the concept that induction of MMP7 in response to allergens is critical for enhancing IL-25 function during initiation of the allergic immune response.
Mmp7−/− mice exhibit reduced Th2 responses
We next determined if the reduced AHR and BAL inflammatory cells seen in Mmp7−/− mice were secondary to decreased TH2 responses in Mmp7−/− mice. As expected, IL-4, IL-5, IL-13, IL-6 and TNF concentrations in BAL fluid of CAA-challenged Mmp7−/− mice were reduced as compared to wild-type mice (Fig. 3 a–c and Supplementary Fig. 5 online) while no significant differences were found in CCL3, IL-9 and IL-1β concentrations (data not shown). Consistent with reduced lung eosinophilia, we found a significant decrease in CCL11 expression in Mmp7−/− CAA-immunized mice compared to wild-type, but no significant differences were detected in CCL7 and CCL17 concentrations (Fig. 3d, and data not shown). Further, whole lung analysis of the RNA from immunized Mmp7−/− mice revealed reduced expression of Il25 compared to wild-type mice (Fig. 3e), suggesting that in addition to proteolytic modification of IL-25, activation and function of MMP7 in the lung is required for the transcriptional expression of Il-25 in this model of allergic airway disease.
Figure 3. Attenuated TH2 cytokines and chemokines in Mmp7−/− mice.

Mmp7−/− mice and age and sex matched wild-type (n=5 per group) mice immunized as described in Fig. 2 and 24 h after the last immunization, concentrations of (a) IL-4, (b) IL-5 and (c) IL-13 were measured in BAL fluid using Luminex. (d) Level of CCL11 in BAL fluid was measured by ELISA. (e) Expression of Il-25 mRNA in the lung of WT and Mmp7−/− CAA immunized mice was measured by real time RT-PCR. Data was normalized to actin. (Data represent mean values ± SD; representative of 3 independent experiments). *P < 0.05 relative to PBS challenged WT and **P < 0.05 relative to CAA immunized WT mice using one way ANOVA and t-test.
Increased retinoic acid in allergen challenged Mmp7−/−
To determine additional events that might regulate the airway epithelial response to allergen, we performed proteomic analyses of BAL fluid in wild-type and Mmp7−/− mice that were immunized with CAA17. Among the proteins that were significantly and differentially modulated in Mmp7−/− CAA-immunized mice (Supplementary Table 2 online), we found an increase in retinaldehyde dehydrogenase-1 (RALDH-1), (Fig. 4a,b and Supplementary Fig. 6 online). RALDH-1 is the rate-limiting enzyme for conversion of retinaldehyde to all-trans retinoic acid (ATRA) that plays a critical role in maintaining the tolerogenic environment of mucosal surfaces24. In support of our proteomic-based discovery of increased RALDH-1, BAL fluid analysis of Mmp7−/− CAA-immunized mice revealed an increase in ATRA concentration when compared to similarly treated wild-type mice (Fig. 4c).
Figure 4. Proteomics analysis of BAL fluid of WT and Mmp7−/− CAA immunized mice.

(a) Two-dimensional gel electrophoresis of pooled BAL fluid from wild-type (WT) (green, Cy 3) and Mmp7−/− (red, Cy 5) CAA immunized mice reveals differential processing of proteins (see Supplementary Table 2; n = 3 in each group) two independent experiments. Super-imposed green and red images highlight differences between WT and Mmp7−/− BAL proteins. Yellow indicates no change, red spots were more abundant in knockout, and green spots were more abundant in WT. Arrow indicated the location and abundance of Retinal dehydrogenase-1 (RALDH-1) protein in WT and Mmp7−/− CAA immunized mice. (b) Spot volumes were standardized and log transformed using DeCyder software, Biological Variation Analysis. Each point represents the spot volume from a pooled sample (n = 3), and graphically is shown as the mean value of RALDH-1 abundance. Data represent mean values ± SD; *P < 0.05 relative to saline challenged WT and **P < 0.05 relative to CAA immunized WT mice using t-test. (c) Representative chromatogram of ATRA absorbance unit (AU) in the BAL fluid (n = 3) as determined using HPLC) in WT and Mmp7−/− mice immunized with CAA or PBS (control) (Left, WT PBS control), middle, WT, and right panel Mmp7−/− mice immunized with CAA). (d) Total BAL cell count, (e) eosinophil cell count recovered from WT mice that were treated with liposomal ATRA (5 μg/grams body weight) 1 h before intranasal CAA immunization. Negative control group (PBS, liposomal ATRA; n = 5) and positive control group (CAA; n = 5) were used. Same groups of mice were used to measure (f) Il-25, and (g) Ccl11 mRNA expression in the lungs by real time RT-PCR, expression was normalized to actin and 18S respectively. (h) IL-4 and (i) IL-13 levels in BAL fluid were measured by Luminex. (Data represent mean values ± SD; representative of 3 independent experiments. *P < 0.05 relative to PBS challenged WT and **P < 0.05 relative to CAA immunized WT mice using one-way ANOVA test).
Please carry similar changes for remaining figures and supplementary items.
Next we determined if increased production of ATRA in the lung of Mmp7−/− mice might account, in part, for their attenuated response to allergen. Liposomal ATRA was delivered by respiratory aerosol to wild-type mice 1 hour before each immunization with CAA, after which lung allergic responses were determined. Compared to vehicle (empty liposome) aerosol, mice that received ATRA show an attenuated asthma phenotype as demonstrated by reduced total number of cells in BAL fluid, especially eosinophils, and decreased Il25 mRNA expression (Fig. 4d–f). Consistent with a decrease in inflammatory markers, we also detected less CCL11, IL-4 and IL-13, which act downstream of IL-25 signaling (Fig. 4g–i and Supplementary Fig. 7 online). These data confirm that ATRA exhibits potent anti-inflammatory activity when administered to the lung and that this activity can be enhanced through the inhibition of Il25 gene expression.
Epithelial RALDH-1 and ATRA induce T regulatory cells
We found that RALDH-1 protein is expressed in airway epithelial cells and alveolar macrophages of wild-type and Mmp7−/− mice immunized with CAA (Fig. 5a). Increased RALDH-1 expression and ATRA concentration has previously been linked to enhanced immune tolerance and increased production of inducible T regulatory cells25. Therefore, we determined if increased expression of RALDH-1 acts as a negative regulator in response to allergens and can increase the relative abundance of T regulatory cells in the lung of wild-type and Mmp7−/− mice following immunization with CAA. As compared to naive mice, wild-type and Mmp7−/− CAA immunized mice showed an increase in the relative abundance of CD25+FoxP3+ T regulatory cells in lungs (Fig. 5b and Supplementary Fig. 8 online). To further examine the functional role of RALDH-1 in inducing the production of T regulatory cells, we administered citral, an inhibitor of RALDH-1, to mice that were immunized with CAA26. Inhibition of retinoic acid synthesis by citral decreased the number of CD25+FoxP3+ T regulatory cells in the lung (Fig. 5b and Supplementary Fig. 9 online), suggesting a possible role for RALDH-1 and ATRA in the development of this inhibitory subset of lung T cells. Moreover, compared to CAA alone, wild-type and Mmp7−/− mice immunized with CAA and challenged with citral showed more BAL fluid eosinophils, IL-5 and CCL11 (Fig. 5c–e and data not shown).
Figure 5. Epithelial expression of RALDH-1 in response to allergens initiates a negative regulatory response.

a) RALDH-1 expression was detected using immune histochemistry in lung sections of WT and Mmp7−/− mice immunized intranasally with CAA or PBS (arrows indicate RALDH-1 expression; representative of two independent experiments). b) Representative flow figures show the abundance of T regulatory cells in lungs of WT and MMP7 null CAA or sham (PBS) immunized mice that were stained with anti-CD4, -CD25 and -FoxP3 (n=4 in each group, two independent experiments). Abundance of T regulatory cells were also determined from WT and Mmp7−/− mice that were given citral, 1 hour before CAA immunization. Intranasal administration of the same concentration of CAA was used controls. c) Total cell count, d) Eosinophil count, and e) IL-5 concentration in the BAL of mice treated in experimental conditions described in (b). Intranasal administration of citral and CAA were used as negative and positive controls respectively. (n=4 mice per group; data represent mean values ± SD; representative of 3 independent experiments. *P< 0.05 relative to CAA challenged WT and **P< 0.05 relative to CAA challenged MMP7 null mice using t-test).
MMP7 expression in response to allergen in human asthma
To address the relevance of MMP7 expression in human asthma, we studied human bronchial biopsy specimens from volunteers with and without asthma (non-asthmatic controls). Interestingly, while IL-25 was detected in airway epithelia of control and asthmatic subjects and therefore appeared to be constitutively present, expression of MMP7 was exclusively found in the airway of asthmatics (Fig. 6a). Consistent with these findings, analysis of nasal washings collected from 7 volunteers with allergic rhinitis 30 min and 5 hours following intranasal challenge with ragweed (a potent proteinase allergen), showed a significant increase in MMP7 secretion in response to ragweed, but not normal saline control (Fig. 6b and data not shown). We could not detect secretion of IL-25 in nasal washes. Nonetheless, these data suggest that constitutive IL-25 was activated by inducible expression of MMP7 during allergen challenge of the human airway.
Figure 6. MMP7 and IL-25 expression in human asthma.

a) Representative serial photomicrograph of human bronchial biopsies samples from a non-asthmatic (top panels) and an asthmatic (bottom panels) individual, stained with anti-MMP7 (left panels) or anti-IL-25 (right panels). Arrows point to positive immunoreactivity and arrowhead shows negative expression of MMP7 in non-asthmatic airway. Data represent mean values ± SD; n=5 in each group. b) Nasal washings obtained from seven atopic/allergic volunteers challenged with graded ragweed (RW) or saline (data not shown) intranasaly (see methods for detail), were used to detect MMP7 responses at each time point using ELISA (R&D). *P< 0.05 relative to 30min challenged RW using two-tailed paired t-test
Discussion
We have shown how proteinase allergens simultaneously activate pro- and anti-inflammatory programs in airway epithelial cells. Allergens enhance expression of MMP7 in airway epithelial cells to maximize allergic lung inflammation; conversely, in the absence of MMP7 mice develop an attenuated asthmatic phenotype. The mechanisms responsible for decreased allergic inflammation in Mmp7−/− mice appear to be in part related to reduced expression of IL-25 that has been shown to be critical in initiating robust TH2 responses5,7,27,28. We found that in addition to modulating Il25 gene expression, MMP7 mediated IL-25 cleavage that was required for most of its biological activity in allergic inflammation.
Enhanced expression of MMP family members is a frequent manifestation of active or chronic inflammatory process, but the precise role of MMPs in these conditions remains poorly understood13. Previous studies have documented that proteinase allergens are capable of inducing an innate immune response that further modulates the adaptive immune response during allergic inflammation1,2. We show here airway epithelial expression of MMP7 was critical for the development of asthma-like disease, a finding that is in sharp contrast to the functions MMP2 and MMP9, two gelatinases that are also up regulated in the same model, as absence of these enzymes led to exaggerated lung allergic inflammation15–17. Further we show that MMP7 was expressed in the distal lung parenchyma of untreated asthmatics, and that in response to a proteinase allergen derived from ragweed, MMP7 expression was significantly induced. Although secreted amounts of IL-25 under the same conditions were below the detection limit of our ELISA assay, we found IL-25 expressed in the distal lung space in normal and asthmatic volunteers. Collectively, these findings show the temporal relation between exposure to allergens and induction of MMP7 in human allergic disease and may implicate activation of IL-25 by MMP7 under allergen-exposed conditions. It is important to note that during allergic inflammation many endogenous proteinases including members of the serine and cysteine family are abundantly present in the airway as we and others have shown1,29. However the unique finding that MMP7 was required for amplification of TH2 responses emphasizes the non-redundant roles that MMPs may play in the course of inflammation. The specificity of this response was exemplified by the specific proteolysis IL-25 by MMP7, but not other allergy-related cytokines such as TSLP and IL-13. Similarly, MMP7-mediated proteolytic modification of defensins, apoptotic ligands, and cytokines has been shown to play important role in diverse biological functions such as innate immunity in the gut to cancer cell metatasis19,30–32. The findings that rIL-25 induced expression of lung MMP7 and intranasal administration of rIL-25 to Mmp7−/− mice only weakly induced allergic inflammation strongly suggests that bioactivity of IL-25 is largely dependent on proteolysis by MMP7. These data further suggest the existence of a feedback loop wherein IL-25 upregulates lung MMP7 expression followed by cleavage and activation of IL-25 that further enhances both MMP7 expression and allergic lung disease.
Although regulation of gene expression has been studied for other members of the IL-17 family, little is known about IL-25 regulation12. We found that while little to no RALDH-1 was present in the airways of naive mice, in response to a proteinase allergens, this enzyme was expressed prominently in the airway epithelial cells of both wild-type and Mmp7−/− mice. Based on high throughput proteomic analysis we found excess RALDH-1 enzyme in immunized Mmp7−/− mice that was functionally relevant because it resulted in increased BAL fluid concentrations of ATRA and reduced allergic inflammation. The role of ATRA in the regulation of adaptive immunity, especially TH2 responses, is controversial. For example, IL-4-induced eotaxin production, eosinophil lineage commitment, and IL-5R expression were all strongly inhibited by addition of ATRA in vitro33,34. Furthermore, retinoic acid in the presence of TGF-β1 inhibited STAT6 binding to the Foxp3 promoter and effectively inhibited IL-4 signaling in T cells35. In other models of allergic lung disease, deficiency in vitamin A resulted in a decrease in muscarinic M2 receptor expression, which resulted in increased AHR36. In contrast, intranasal administration of high concentrations of ATRA (>1500 μg), failed to alter TH2 responses in another asthma model37 and vitamin A deficiency has been shown to attenuate TH2 cytokine production in mice38. In our study, allergen challenged mice that received 100 μg of ATRA showed a robust reduction in allergic inflammation and attenuation of the asthmatic phenotype. Such divergent findings are not surprising because of the known idiosyncrasies of retinoid that can act as hormones and depending on the physiological or pharmaceutical context, mediate diverse effects on cells of the immune system39.
Previous studies have implicated a role for ATRA in the development and differentiation of inducible T regulatory cells, an anti-inflammatory subset of T cells that play a significant role in homeostasis of lymphocytes in different organs40. In particular, dendritic cells in the lamina propria were found to provide ATRA that was critical for differentiation and homing of T regulatory cells to the intestinal mucosa41,42. Although we did not identify the cellular origin of ATRA, we found RALDH-1, the rate-limiting enzyme that converts retinaldehyde to ATRA, was up-regulated in response to allergic stimulation in the airway epithelium, implicating a possible site for the production of ATRA in the lung. Consistent with the requirement of ATRA for differentiation of T regulatory cells in vivo25,41,43, we found an increase numbers of CD25+ Foxp3+ T cells in the lung of Mmp7−/− mice immunized with CAA. Moreover we found that inhibition of retinoic acid synthesis by citral inhibited T regulatory cell development. The increased percentage of regulatory T cells following allergen challenge in both wild-type and Mmp7−/− mice suggested that RALDH-1 is a negative regulator of allergic inflammation, a plausible protective response regulated by airway epithelial cells that we found to be enhanced in the absence of MMP7. What remains unclear is the mechanism responsible for increased expression and function of RALDH-1 in the lung in the absence of MMP7. Our finding of RALDH-1 expression in airway epithelial cells and macrophages in response to allergen challenge is in agreement with prior reports implicating an anti-inflammatory role for these cells in allergic inflammation44. Furthermore, depletion of alveolar macrophages resulted in an exaggerated allergic response that could be inhibited by adoptive transfer of alveolar macrophages in Brown Norway rats45. Our data suggest that alveolar macrophage mediated anti-inflammatory responses during allergic inflammation might be operating through RALDH-1, a concept that is further consistent with tolerogenic role of RALDH-1 in gut lamina propria41,42.
In summary, we have described a pro-inflammatory role of MMP7 that is activated in response to proteinase allergens and involves proteolytic modification of IL-25 expression and function. In this model, we found activation of RALDH-1 to be downstream of allergen-induced inflammation and that MMP7 actively inhibits the function of this enzyme in vivo. At least in part, this unique mechanism involves inhibition of T regulatory cells that inhibit lung inflammation. Our findings thus substantially expand our understanding of the regulation of IL-25, a critical innate cytokine in allergic responses, and further identify pro- and anti-inflammatory targets that can be explored for therapeutic benefit.
Methods
Mice
Mmp7−/− mice nine generations backcrossed with C57BL/6 were kindly provided by P. Woo Park (Children’s Hospital, Harvard School of Medicine), and were bred in the Association for Assessment and Accreditation of Laboratory Animal Care (AALAC)-accredited transgenic animal facility at Baylor College of Medicine. All experiments were preformed in accordance with protocol approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine. Mmp7−/− mice were genotyped using standard PCR techniques (Neo R: 5′-TTGAGCCTGGCGAACAGT-3′, Neo F: 5′-TGGATTGCACGCAGGTTCT-3′, WT F: 5′-ACTTACCTCGGATCGTAGTG-3′, WT R: 5′-GTCCAGTACTCATCCTTGTC-3′). Wild-type C57BL/6 mice, and/or heterozygote littermates were used as controls and were purchased from the Baylor Center for Comparative Medicine facility. Stat6−/− mice (BALB/c background) and Il13−/− mice (C57BL/6 background) were purchased from Jackson Laboratories and bred 10–12 generations onto BALB/c background.
Experimental model of asthma
Complete aspergillus allergen (CAA) comprised of 1 mg/ml of Aspergillus oryzae proteinase (Sigma) and 0.5 mg/ml of OVA (Sigma) in 50 μl volume, was administered intranasally every four days for a total of five times, as described before1,16. In some experiments, rIL-25 (5 μg in 50 μl of PBS) were administered intranasally twice daily for 3 days. In some experiments, liposomal preparation of all transretnoic acid (ATRA) or citral was given by aerosol 1 h prior to CAA challenges. Briefly, liposome formulation of ATRA (Sigma) and citral (Tokyo Chemical Industry Chemical Co.) were made in dilauroylphosphatidylcholine (DLPC) (Avanti Polar Lipid Inc.) as we have previously described46. Aerosol was generated from an Aerotech II nebulizer (CIS USA; flow rate 10 L/Min) and was discharged from an opening to the entry orifice. Following a dose titration study that determined efficacy of aerosol delivery, mice received a total concentration of 5 μg/gram of body weight (approximately 100 μg) of ATRA, 5 mg of citral, or vehicle control (empty liposomes) over 1 h before each of the CAA challenges.
Experimental ragweed allergen challenge in human volunteers
Nasal provocation with ragweed pollen extract was performed as previously described47. The Institutional Review Board at University of Texas Medical Branch reviewed and approved the protocol and consent forms. Briefly, seven otherwise healthy volunteers with symptoms of seasonal allergic rhinitis (congestion, nasal discharge, sneezing) and positive prick skin test to ragweed extract were recruited for this study. Volunteers received intranasal saline challenges delivered with a nasal spray bottle on one day. On another day, the same volunteers received a graded intranasal challenge with ragweed (saline, 0.33, 1, 3, 9, 27 μg/ml Amb a 1 equivalent in ragweed, (Greer laboratories) every 15 min until sneezing, rhinorrhea and nasal congestion were observed47. On both days, 5 ml nasal lavage fluids were collected using previously described techniques 30 min and 5 h post nasal challenge. The fluids were centrifuged and cell-free supernatants were analyzed for MMP7 response at each time point using ELISA (R&D).
Serial sections of bronchial biopsies samples were collected under protocols approved by the UCSF Committee on Human Research and with written informed consent for future use as part of the UCSF Airway Tissue Bank from five untreated asthmatic and five control volunteers were stained for the presence of MMP7 and IL-25 using standard immunohistochemical techniques as described below.
Analysis of asthmatic phenotype
All data were collected 24 h following the final allergen challenge. AHR, a pathognomonic feature of asthma, was assessed by estimating the provocative concentration of acetylcholine in milligrams per gram that causes a 200 percent increase in airway resistance over the baseline (PC200) that was calculated by linear interpolation of appropriate dose response curves, as described before15. Bronchoalveolar lavage (BAL) fluid was collected by instilling and withdrawing 1.6 ml of sterile phosphate buffered saline (PBS) through the tracheal cannula in two aliquots of 0.8 ml. BAL fluid total and differential cell count were measured using the standard hemocytometer and hematoxylin and eosin (H&E) stain of cytospin slides as described previously1. In each experiment, mouse lungs were fixed with intra-tracheal instillations of 800 μl of 4% paraformaldehyde solution via a tracheal cannula and were embedded in paraffin and 4–5 μm sections of paraffin embedded lung tissues were either stained with H&E, periodic acid-Schiff (PAS), and/or were prepared for immunohistochemistry.
Immunohistochemistry
Immunohistochemistry was done using the standard protocol described48. Briefly, deparaffinized sections were washed with 3% H2O2 and 2% Triton X-100 in PBS (PBST), followed by antigen retrieval (Target Retrieval Solution; Dako North America, Inc.). Slides were incubated at 4 °C overnight with either rabbit anti-mouse polyclonal antibody against ALDH1A1 (Abcam Inc.), rat anti-mouse monoclonal antibody against MMP7 (R&D, Clone 377314), monoclonal mouse anti-human MMP7 (Calbiochem, Clone ID2), and monoclonal mouse anti-human IL-17E in 1:50 to 1:100 dilution, followed by incubation with corresponding biotinylated secondary antibodies (1:200 dilution) and Vectastain Elite ABC reagent (Vector) at 25 °C. Slides were then incubated with DAB substrate (Vector) and counterstained with hematoxylin. After addition of bluing reagent (0.08% NH4OH), slides were mounted in xylene.
BAL Cytokine and Chemokine Measurement
CCL11, CCL7, and CCL17 proteins in BAL fluid were measured using standard antibody based ELISA and or BioRad multiplex bead-based cytokine detection kit (BioRad) as previously described16. All recombinant protein, capture antibodies, and their corresponding biotin-conjugated detection antibodies were purchased from R&D system. IL-4, IL-5, IL-13, IL-6 and TNF concentrations in BAL fluid were measured by Lincoplex (Millipore). Equal volumes of human nasal washings were used to detect MMP7 using ELISA kit purchased from R&D system.
Proteomics analysis of BAL protein
Proteomics analysis of BAL fluid was performed as described previously17. Briefly, BAL samples from three mice in each group (wild-type and Mmp7−/−) were pooled and concentrated using AMICON filters, and the protein concentration was measured using BCA protein assay (Pierce). Equal amount (50 μg) of protein from each pooled sample was labeled with 400 pmol of charge and molecular weight matched fluorescent dye CyDye (Amersham Biosciences) and brought to a final volume of 250 μl with sample buffer that also contained 0.5% ampholytes (3–11 NL), 0.1% bromophenol blue and 12 μl/ml DeStreak™ reagent (GE Healthcare). Internal standard was made by mixing equal amounts of protein from wild-type and Mmp7−/− samples before CyDye labeling. Wild-type, Mmp7−/− and the internal standard (IS) were labeled and loaded for separation on triplicate gels as follows: gel 1, WT-Cy3, MMP7−/−Cy5, IS- Cy2; gel 2, WT-Cy5, MMP7−/−Cy3, IS-Cy2; gel 3, WT-Cy3, KO-Cy5, IS-Cy2. First dimension separation was performed based on isoelectric focusing using the GE Healthcare IPGPhor II. The second dimension separation was performed using the same IPG strips that were secured in place with agarose on top of a 15% 18 × 16 cm SDS gel and were electrophoresed for 4 h at 20 mA. Electrophoresed gels were scanned using a laser-based image instrument (Typhoon 9400, GE Healthcare) and were digitized using ImageQuant software (GE Healthcare). Gel images were imported into the DeCyder (v. 5.5) Differential In-gel Analysis (GE Healthcare) program and resulting spots were matched among the gels. Using DeCyder Biological Variation Analysis software (GE Healthcare), spot differences were quantified and protein spots that exhibited a significant normalized change in volume, with respect to the internal standard sample (1.5 fold; p < 0.05), were picked for identification. For spot picking, spot map coordinates were transferred to the ETTAN spot handling work-station (GE Healthcare) for automated picking, in-gel trypsin digestion, and MALDI plate spotting.
Quantitative RT-PCR
Total cellular RNA was extracted from RNA Later (Qiagen;) stabilized mouse lung tissues using RNeasy mini kit (Qiagen). One step real-time quantitative RT-PCR was used to determine the relative expression of Ccl11 (eotaxin-1) and Raldh-1 in Real-Time PCR System (7300 Applied Biosystems). The primer and probe mix of target genes, (Ccl11: Mm00441238_m1, Raldh-1: Mm01194995_mH) and 18S rRNA were purchased from Applied Biosystems. Expression of mRNA samples were normalized to 18S rRNA, and relative abundance of the target genes were calculated as described previously16,49. Two-step real time q-RT-PCR was performed to determine relative expression of Il25; expression was normalized with Actb. cDNA was generated using oligo-dT, random hexamers, and SuperScript RT II (Invitrogen). For real time RT reaction, the following primers were used, Il25 primer: 5′-CACACTGCGTCAGCCTACAGA-3′ and 5′-TGTGGTAAAGTGGGACGGAGTT-3, Actb primer 5′-GACGGCCAGGTCATCACTATTG-3′ and 5′AGGAAGGCTGGAAAAGAGCC-3′.
MMP activation and in vitro cleavage assay
Carrier-free recombinant mouse 5 μg IL-25 (rIL-25; R&D), mouse recombinant IL-13 (Peprotech) and mouse recombinant TSLP (R&D) was incubated with 0.5 μg of APMA activated MMP7 in the presence or absence of MMP inhibitor, 10 mM 1,10 phenanthroline (Sigma) at 37 °C for 2 h. Following incubation, equal volumes of each sample (16.5 μl) were reduced and resolved using 16.5% Tricine gel. Cleaved and uncleaved proteins were visualized using Proteosilver plus silver stain kit (Sigma-Aldrich) using manufacturer’s instructions. Alternatively, cleaved IL-25 protein was separated as above, electroblotted in Tris-glycine buffer (25 mM Tris, 192 mM glycine, 10% MeOH (pH 8.3) to a polyvinylidene difluoride (PVDF) membrane and stained with 0.05% Coomassie Blue in 1% acetic acid, 50% methanol. The PVDF membranes were destained in 50% methanol and deionized water. The visible bands were cut out with a clean scalpel blade and air-dried, then N-terminal sequencing of IL-25 was performed using standard protein-sequencing methods (Applied Biosystems Procise 492cLC).
Cell culture
Lymph node cells and splenocytes were isolated from C57BL/6 mice and were stimulated with 2 μg/ml of plate-bound anti-CD3 and 50 U/ml of human IL-2 in the presence of 250 ng/ml of mouse rIL-25 (R&D) or 250 ng/ml of MMP7 cleaved mouse rIL-25 (IL-25′C). After 2 days, culture supernatants were collected and analyzed for IL-4, IL-5, IL-13 and IFN-γ by ELISA as described previously5. On day 3, cells were restimulated with 500 ng/ml ionomycin and 50 ng/ml PMA in the presence of GolgiStop (BD Biosciences) for 5 h. Cells were permeabilized with a Cytofix/Cytoperm kit (BD Biosciences) and analyzed for the intracellular expression of IL-4 (PE conjugated) and IL-5 (APC conjugated) (BD biosciences, Clone 11B11 and TRFK5 respectively).
Flow cytometry
Total lung eosinophil numbers were quantified using beads enhanced flow cytometry method as described previously50. Briefly, total murine lung cells were stained with MHC-II (FITC conjugated), SiglecF (PE conjugated) (1 μg/106 cells) (BD Biosciences, Clone E50-2440), followed by incubation with Fluorescent beads (Flow-Check™ Fluorospheres, Beckman Coulter). Samples were acquired in flow cytometer (XL2, Beckman Coulter Inc.); cell and bead counts were measured. Absolute numbers of eosinophils were calculated using formulas as described previously50. Abundance of lung T regulatory cells was estimated by staining the cells with Mouse Regulatory T cell Staining Kit (eBioscience); samples were acquired in BD LSRII flow cytometer. Analysis was performed with FlowJo software (version 8.5.3).
High Performance Liquid Chromatography (HPLC)
Pooled BAL samples from WT and Mmp7−/− mice were evaporated under N2 and the dry residue was dissolved in 100 μl of acetonitrile (Fisher). Aliquots of 25 μl were subsequently analyzed by Waters HPLC system consisting of WISP autosampler (model 717 plus), Dual Absorbance detector (model 2487), Pump (model 515) and Nova pak C-18 columns (3.9×150mm). Separation of ATRA was performed using a solvent system composed of 57.5% acetonitrile, 25% of 2% of acetic acid at a flow rate of 1.3 ml/min. Data was analyzed in Waters Millennium software (version 3.2).
IL-25-IL-17RB-Fc binding assay
Recombinant mouse IL-17RB-Fc fusion protein (R&D Systems) or PBS was used to coat 96-microtiter plates. After blocking, recombinant mouse IL-25 or MMP7 cleaved IL-25 (IL-25′C) was added at different concentrations (100 pg/ml to 50,000 pg/ml). After washing, biotinylated anti-mouse IL-25 antibody (R&D Systems) was added and the ability of recombinant IL-25 or IL-25′C to bind to IL-17RB-Fc fusion protein was detected by using standard ELISA protocol.
Statistics
All data are representative of at least 3 independent experiments with 4–5 mice in each in vivo experiment and are expressed as means ± SEM and were analyzed using Prism 4.0a statistical analysis software. We used t-test, one-way analysis of variance (ANOVA) and Bonferroni multiple comparison tests to identify significant differences (P < 0.05) between treatment groups.
Supplementary Material
Acknowledgments
The authors thank D.A. Engler, R.K. Matsunami and K. Gonzalez, for their technical help with proteomic analysis, N. Barrows for editorial support, and all the members of the Kheradmand and Corry laboratory for their comments and criticisms. Supported by grants to F.K (AI070973, HL082487), CD (AI071130 and AR050772) and D.B.C (HL075243, AI057696, AI070973) from the U.S. National Institutes of Health. CD receives a Career Investigator Award from American Lung Association, a Leukemia and Lymphoma Society Scholar Award and a Trust Fellowship of MD Anderson Cancer Center.
Footnotes
Author’s contribution. SG performed animal experiments, in vitro cleavage assay, immuno-histochemistry, Flow, RT-PCR, ELISA and Luminex. PA performed in vitro T cell experiments, WTB and SP performed airway physiology experiments, KJG performed proteomics analysis, AS made liposomal ATRA and helped in HPLC. MS, LS DR, BS, SS performed human Ragweed challenge and ELISA in allergic volunteers. PW performed bronchial biopsies in asthma and control volunteers. SG, FK, DBC and CD designed experiments. SG and FK wrote the manuscript. PA, DBC and CD critically reviewed the manuscript.
References
- 1.Kheradmand F, et al. A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J Immunol. 2002;169:5904–11. doi: 10.4049/jimmunol.169.10.5904. [DOI] [PubMed] [Google Scholar]
- 2.Kiss A, et al. A new mechanism regulating the initiation of allergic airway inflammation. Journal of Allergy & Clinical Immunology. 2007;120:334–42. doi: 10.1016/j.jaci.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 3.Li L, et al. Effects of Th2 cytokines on chemokine expression in the lung: IL-13 potently induces eotaxin expression by airway epithelial cells. J Immunol. 1999;162:2477–87. [PubMed] [Google Scholar]
- 4.Zhu J, et al. Exacerbations of Bronchitis: bronchial eosinophilia and gene expression for interleukin-4, interleukin-5, and eosinophil chemoattractants. Am J Respir Crit Care Med. 2001;164:109–16. doi: 10.1164/ajrccm.164.1.2007050. [DOI] [PubMed] [Google Scholar]
- 5.Angkasekwinai P, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–17. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–74. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 7.Pan G, et al. Forced expression of murine IL-17E induces growth retardation, jaundice, a Th2-biased response, and multiorgan inflammation in mice. J Immunol. 2001;167:6559–67. doi: 10.4049/jimmunol.167.11.6559. [DOI] [PubMed] [Google Scholar]
- 8.Kim MR, et al. Transgenic overexpression of human IL-17E results in eosinophilia, B-lymphocyte hyperplasia, and altered antibody production. Blood. 2002;100:2330–40. doi: 10.1182/blood-2002-01-0012. [DOI] [PubMed] [Google Scholar]
- 9.Ballantyne SJ, et al. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2007;120:1324–31. doi: 10.1016/j.jaci.2007.07.051. [DOI] [PubMed] [Google Scholar]
- 10.Fallon PG, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203:1105–16. doi: 10.1084/jem.20051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. [Review] [322 refs] Physiological Reviews. 2007;87:69–98. doi: 10.1152/physrev.00022.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nature Reviews Immunology. 2008;8:337–48. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 13.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–29. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 14.McMillan SJ, et al. Matrix Metalloproteinase-9 Deficiency Results in Enhanced Allergen-Induced Airway Inflammation. J Immunol. 2004;172:2586–2594. doi: 10.4049/jimmunol.172.4.2586. [DOI] [PubMed] [Google Scholar]
- 15.Corry DB, et al. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. Faseb J. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. Epub 2004 Apr 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corry DB, et al. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat Immunol. 2002;3:347–53. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenlee KJ, et al. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. Journal of Immunology. 2006;177:7312–21. doi: 10.4049/jimmunol.177.10.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–46. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 19.Wilson CL, et al. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–7. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 20.Gearing AJ, et al. Matrix metalloproteinases and processing of pro-TNF-alpha. J Leukoc Biol. 1995;57:774–7. doi: 10.1002/jlb.57.5.774. [DOI] [PubMed] [Google Scholar]
- 21.Corry DB. IL-13 in allergy: home at last. Curr Opin Immunol. 1999;11:610–4. doi: 10.1016/s0952-7915(99)00025-4. [DOI] [PubMed] [Google Scholar]
- 22.Corry DB, et al. Requirements for allergen- induced airway hyperreactivity in T and B cell- deficient mice. Molecular Medicine. 1998;4:344–355. [PMC free article] [PubMed] [Google Scholar]
- 23.Grunig G, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–3. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–94. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 25.Mucida D, et al. Reciprocal Th-17 and Regulatory T Cell Differentiation Mediated by Retinoic Acid. Sciencexpress.org. 2007 doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 26.Connor MJ, Smit MH. Terminal-group oxidation of retinol by mouse epidermis. Inhibition in vitro and in vivo. Biochem J. 1987;244:489–92. doi: 10.1042/bj2440489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung PF, Wong CK, Ip WK, Lam CW. IL-25 regulates the expression of adhesion molecules on eosinophils: mechanism of eosinophilia in allergic inflammation. Allergy. 2006;61:878–85. doi: 10.1111/j.1398-9995.2006.01102.x. [DOI] [PubMed] [Google Scholar]
- 28.Fort MM, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–95. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 29.Zheng T, et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–93. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swee M, Wilson CL, Wang Y, McGuire JK, Parks WC. Matrix metalloproteinase-7 (matrilysin) controls neutrophil egress by generating chemokine gradients. Journal of Leukocyte Biology. 2008;83:1404–12. doi: 10.1189/jlb.0108016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu D, et al. Overexpression of matrix metalloproteinase-7 (MMP-7) correlates with tumor proliferation, and a poor prognosis in non-small cell lung cancer. Lung Cancer. 2007;58:384–91. doi: 10.1016/j.lungcan.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 32.Ito TK, Ishii G, Chiba H, Ochiai A. The VEGF angiogenic switch of fibroblasts is regulated by MMP-7 from cancer cells. Oncogene. 2007;26:7194–203. doi: 10.1038/sj.onc.1210535. [DOI] [PubMed] [Google Scholar]
- 33.Upham JW, et al. Retinoic acid modulates IL-5 receptor expression and selectively inhibits eosinophil-basophil differentiation of hemopoietic progenitor cells. J Allergy Clin Immunol. 2002;109:307–13. doi: 10.1067/mai.2002.121527. [DOI] [PubMed] [Google Scholar]
- 34.Takamura K, et al. Retinoic acid inhibits interleukin-4-induced eotaxin production in a human bronchial epithelial cell line. Am J Physiol Lung Cell Mol Physiol. 2004;286:L777–85. doi: 10.1152/ajplung.00289.2003. [DOI] [PubMed] [Google Scholar]
- 35.Takaki H, et al. STAT6 Inhibits TGF-beta1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. Journal of Biological Chemistry. 2008;283:14955–62. doi: 10.1074/jbc.M801123200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGowan SE, Holmes AJ, Smith J. Retinoic acid reverses the airway hyperresponsiveness but not the parenchymal defect that is associated with vitamin A deficiency. American Journal of Physiology – Lung Cellular & Molecular Physiology. 2004;286:L437–44. doi: 10.1152/ajplung.00158.2003. [DOI] [PubMed] [Google Scholar]
- 37.Maret M, et al. Liposomal retinoic acids modulate asthma manifestations in mice. J Nutr. 2007;137:2730–6. doi: 10.1093/jn/137.12.2730. [DOI] [PubMed] [Google Scholar]
- 38.Schuster GU, Kenyon NJ, Stephensen CB. Vitamin A deficiency decreases and high dietary vitamin A increases disease severity in the mouse model of asthma. J Immunol. 2008;180:1834–42. doi: 10.4049/jimmunol.180.3.1834. [DOI] [PubMed] [Google Scholar]
- 39.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take center stage. Nature Reviews Immunology. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nature Reviews Immunology. 2008;8:435–46. doi: 10.1038/nri2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun CM, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid.[see comment] Journal of Experimental Medicine. 2007;204:1775–85. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation.[see comment] Journal of Experimental Medicine. 2007;204:1765–74. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elias KM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–20. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peters-Golden M. The alveolar macrophage: the forgotten cell in asthma. Am J Respir Cell Mol Biol. 2004;31:3–7. doi: 10.1165/rcmb.f279. [DOI] [PubMed] [Google Scholar]
- 45.Careau E, et al. Antigen sensitization modulates alveolar macrophage functions in an asthma model. Am J Physiol Lung Cell Mol Physiol. 2006;290:L871–9. doi: 10.1152/ajplung.00219.2005. [DOI] [PubMed] [Google Scholar]
- 46.Koshkina NV, Gilbert BE, Waldrep JC, Seryshev A, Knight V. Distribution of camptothecin after delivery as a liposome aerosol or following intramuscular injection in mice. Cancer Chemotherapy & Pharmacology. 1999;44:187–92. doi: 10.1007/s002800050966. [DOI] [PubMed] [Google Scholar]
- 47.Diaz-Sanchez D, Tsien A, Fleming J, Saxon A. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J Immunol. 1997;158:2406–13. [PubMed] [Google Scholar]
- 48.Grumelli S, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1:e8. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SH, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nature Medicine. 2007;13:567–9. doi: 10.1038/nm1583. [DOI] [PubMed] [Google Scholar]
- 50.Montes M, Jaensson EA, Orozco AF, Lewis DE, Corry DB. A general method for bead-enhanced quantitation by flow cytometry. Journal of Immunological Methods. 2006;317:45–55. doi: 10.1016/j.jim.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.