

Abstract
To interpret genetic variants discovered from next-generation sequencing (NGS), integration of heterogeneous information is vital for success. This paper describes a framework named PERCH (Polymorphism Evaluation, Ranking, and Classification for a Heritable trait), available at http://BJFengLab.org/. It can prioritize disease genes by quantitatively unifying a new deleteriousness measure called BayesDel, an improved assessment of the biological relevance of genes to the disease, a modified linkage analysis, a novel rare-variant association test, and a converted variant call quality score. It supports data that contain a various combination of extended pedigrees, trios, and case-controls, and allows for a reduced penetrance, an elevated phenocopy rate, liability classes and covariates. BayesDel is more accurate than PolyPhen2, SIFT, FATHMM, LRT, Mutation Taster, Mutation Assessor, PhyloP, GERP++, SiPhy, CADD, MetaLR, and MetaSVM. The overall approach is faster and more powerful than the existing quantitative method pVAAST, as shown by the simulations of challenging situations in finding the missing heritability of a complex disease. This framework can also classify variants of unknown significance (VUS, or variants of uncertain significance) by quantitatively integrating allele frequencies, deleteriousness, association, and co-segregation. PERCH is a versatile tool for gene prioritization in gene discovery research and variant classification in clinical genetic testing.
Keywords: Variant interpretation, gene prioritization, whole-exome / whole-genome / gene-panel sequencing, functional consequence, co-segregation analysis, rare-variant burden test, gene association network, variants of unknown significance, genetic testing, de novo mutation
Introduction
Next-generation sequencing (NGS) has played a key role in research on human diseases and is now rapidly becoming integrated into clinical practice. A major problem in utilizing such information is in the interpretation of a large number of genetic variants identified that have diverse and unknown clinical significance.
To evaluate whether a variant is a deleterious mutation or a benign polymorphism, many methods have been developed based on sequence conservation, characteristics of amino acid substitution, the location of the variant within protein domains or 3-dimensional protein structure. Others have combined these assessments to produce an overall prediction, yielding a better performance than any predictor alone (González-Pérez and López-Bigas 2011; Kircher et al. 2014; Dong et al. 2015).
Another kind of tool assesses the biological relevance of a gene to the disease in question as a function of association between the gene and a number of other genes known to correlate with the disease, or the seed genes. The association may include sequence homology, co-expression, shared functional annotation, co-occurrence in literature, and physical interaction between gene products (Moreau and Tranchevent 2012). Besides association between genes, others utilize the similarity between phenotypes to infer the correlation of a gene with the disease (Robinson et al. 2014). Additionally, models have been created to integrate deleteriousness predictions with biological relevance assessment, which significantly out-performed the individual deleteriousness scores alone (Sifrim et al. 2013; Dubchak et al. 2014; Wu et al. 2014).
To further narrow down candidate variants from an NGS study, there are scoring or filtering systems to select variants that co-segregate with the disease within a pedigree (Li et al. 2012; Sincan et al. 2012; Aleman et al. 2014; Koboldt et al. 2014; Santoni et al. 2014; Yao et al. 2014; Field et al. 2015). However, due to the assumption of complete co-segregation and/or no allelic heterogeneity, these systems best work for high-penetrance genes for a Mendelian disease, but not for modest-risk (odds ratio ≤2) or intermediate-risk (2<odds ratio<5) genes for a complex disease. Moreover, these systems may impose restrictions on pedigree structure. Alternatively, one can conduct a linkage analysis, which can handle extended pedigrees and an arbitrary number of sequenced individuals, incorporate individual-specific liability classes, and is robust to phenocopies and a reduced penetrance. It is important to note that in linkage analysis the phase of the hypothetical causal variant is normally unknown, whereas in sequencing studies we typically assume that the minor allele is the causal mechanism, thus assuming complete linkage disequilibrium and no recombination between the studied variant and the causal variant can dramatically increase the power of linkage analysis, as described in Thompson et al (Thompson et al. 2003).
Pathogenicity of a variant can also be evaluated by the allele frequency difference between independent cases and controls. For NGS studies, association analysis of each individual variant is not powerful due to the low population frequency and a large number of variants in the genome. Alternatively, aggregate analysis of multiple variants within a genomic region or gene, the rare-variant association test, is more powerful and has less multiple testing penalties (Li and Leal 2008; Morris and Zeggini 2010; Lee et al. 2014). To further increase the power, a program named pVAAST was designed to unify a rare-variant association test with linkage analysis by permutation (Hu et al. 2014).
In addition to pathogenicity, variant call quality should also be considered in the analysis of NGS data. Removing bad variant calls is the most common quality control (QC) procedure, but variants at the borderline of a filtering threshold may contain a relatively high proportion of false calls. Therefore, their quality should be considered in the analyses. For this purpose, VQSLOD calculated by the Genome Analysis Toolkit (DePristo et al. 2011) is a suitable measure.
To increase the power of a study, it is desired to integrate all these lines of evidence for pathogenicity inference. For this purpose, the likelihood-based approach proposed by Goldgar et al (Goldgar et al. 2004) is superior to a filtering-, machine learning- or permutation-based method. Besides the ability to integrate the above-mentioned components in a quantitative fashion and with low computational demands, this approach also has the unique feature of classifying variants of unknown significance (VUS) for genetic testing following the guidelines of the International Agency for Research on Cancer (IARC) (Plon et al. 2008). This study describes the implementation of such an approach, in which each component is developed de novo or improved from existing tools to fit into the scope of the framework.
Methods
Overview of the framework
The proposed framework, PERCH (Polymorphism Evaluation, Ranking and Classification for a Heritable trait), implements a new deleteriousness measure (BayesDel), an improved assessment of biological relevance of genes to the disease of interest (BayesGBA), a modified linkage analysis (BayesSeg), a novel rare-variant association score (BayesHLR), and a converted VQSLOD. All these components produce as output a Bayes factor in log10 scale so that they can be quantitatively integrated. VQSLOD and BayesDel are at the variant level, while BayesGBA, BayesHLR and BayesSeg are at the gene level. To integrate variant level information into a gene level analysis, VQSLOD and BayesDel are combined to calculate a variant weight for BayesHLR and BayesSeg computation. BayesGBA is assessed within a gene network as a function of relatedness between a gene and a set of known disease genes. Finally, PERCH sums the BayesHLR, BayesSeg and BayesGBA scores to calculate an overall score for each gene, which can be used for gene prioritization and candidate gene selection. For variant classification in known disease genes, segregation and association analyses are variant-based. PERCH sums the BayesHLR, BayesSeg and BayesDel to calculate a variant-wise posterior probability of pathogenicity based on a user-defined prior probability. Figure 1 depicts the flowchart of gene prioritization in gene discovery research. The details of each component are described below.
Figure 1.
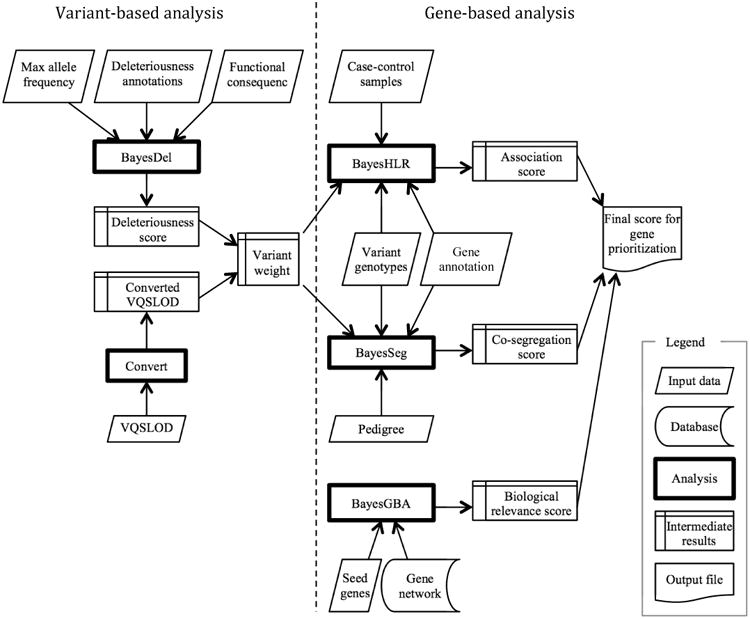
Flowchart of gene prioritization procedures by PERCH.
BayesDel
The goal of this new measure was to achieve an accuracy that is comparable to the state-of-the-art methods and to output a Bayes factor. The proposed measure, BayesDel, combined multiple deleteriousness predictors to create an overall score. Because this tool was designed for large-scale variant analyses, only those predictors that were readily available for exome-wide or genome-wide annotation were considered. These included PolyPhen2 (Adzhubei et al. 2010), SIFT (Kumar et al. 2009), FATHMM (Shihab et al. 2013), LRT (Chun and Fay 2009), Mutation Taster (Schwarz et al. 2010), Mutation Assessor (Reva et al. 2011), PhyloP (Pollard et al. 2010), GERP++ (Davydov et al. 2010), and SiPhy (Garber et al. 2009). These scores were obtained from the dbNSFP version 2.3 (Liu et al. 2011). It is important to note that, similar to sequence conservation measured by GERP++, PhyloP, and SiPhy, population frequency of a variant also correlated with organismal fitness. Therefore, the deleteriousness predictors in this framework included the maximum minor allele frequency across populations (MaxMAF) in the Exome Aggregation Consortium (ExAC) version 0.3 (Lek et al. 2016) and the 1000 Genomes Project (G1K) phase 3 (McVean et al. 2012).
A simple way to combine deleteriousness predictors is the naïve Bayesian approach, which assumes that all predictors are mutually independent. However, this is not true for these predictors because they more or less measure the same characteristics of a variant, such as sequence conservation and physical property of amino acid substitutions. Nevertheless, it has been proven that weighting can alleviate the independence assumption and improves the performance of a naïve Bayesian model (Zhang and Shengli Sheng 2004). The rationale is that correlated predictors are down-weighted so that they jointly make a unit contribution.
Based on this idea, a combined deleteriousness score is defined as a weighted product of likelihood ratios: , where Di is each of the deleteriousness predictors. A likelihood ratio for each score value can be empirically estimated as the probability of observing the score in pathogenic variants divided by the probability in benign variants. However, it is impossible to calculate this for each score for these continuous measures, as the sample size is too small compared to the number of values. Therefore, the range of scores is divided into bins for likelihood ratio calculation. To make the estimation more stable, adjacent bins are merged so that the probability among benign variants, the denominator of the likelihood ratio, is at least 0.02. Using the mean value to represent each bin, a curve of likelihood ratios as a function of score values is generated, which is then smoothed by the least-squares fitting of polynomials to segments of the data. Using this curve, any query score can be translated into a likelihood ratio by linear interpolation. In deriving this curve, pathogenic variants are obtained from the “Pathogenic” or “Likely pathogenic” variants in ClinVar version 2015-08-04 (Landrum et al. 2014) and the “disease” variants in UniProtKB (accessed 2015-08-28) (Yip et al. 2008), non-pathogenic variants are those in the dbSNP version 142, the 1000 Genomes Project, the Exome Aggregation Consortium, and the ALSPAC (Golding et al. 2001) and TWINSUK (Moayyeri et al. 2013) cohorts in the UK10K Project version REL-2012-06-02 (Walter et al. 2015), excluding the above pathogenic variants in ClinVar or UniProtKB.
To obtain the weights, the model is optimized for the area under the receiver operating characteristic curve (AUC) by the controlled random search (CRS) algorithm with the “local mutation” modification (Kaelo and Ali 2006). CRS is a global optimization method that is comparable to a genetic algorithm (Price 1977). The “local mutation” modification makes the CRS more robust in finding the global maximum and more efficient by reducing the number of evaluations (Kaelo and Ali 2006). Variants for training the model are taken from ClinVar and UniProtKB, excluding those in the ENIGMA dataset to avoid overlapping between training and testing. Finally, there are 39395 pathogenic variants and 39978 neutral variants for training. Suppl. Table S1 provides the weight of each component predictor obtained from the training. If some component scores are missing in a real application, the program changes the weights of the missing component to 0, and then normalizes the remaining weights, i.e., the sum of weights is always 1. If too many scores are missing, defined as the sum of weights before normalization is less than 0.5, then BayesDel will be calculated from a genome-wide deleteriousness score such as CADD (Kircher et al. 2014).
BayesGBA
As an example, the proposed framework used the GeneMANIA database (Mostafavi et al. 2008) to infer biological relevance using an improved guilt-by-association (GBA) algorithm, BayesGBA. One of the advantages of GeneMANIA is that it comprehensively incorporates multiple gene-gene association data, such as co-expression, co-localization, shared protein domains, shared pathway annotations, genetic interactions and physical interactions among gene products. Nevertheless, other quantitative approaches could also be applied whenever large-scale genome-wide data were available.
Methods have been proposed to infer biological relevance based on association within a network like GeneMANIA, such as neighbor counting (NC), naïve Bayes label propagation (NB), Gaussian smoothing (GS), InterConnectedness (IC) (Wang and Marcotte 2010; Hsu et al. 2011). However, as has been pointed out previously, these methods favor the genes with higher node degrees (Gillis and Pavlidis 2011, 2012). Thus, they all suffer from the lack of specificity. The current framework uses an improved IC algorithm (described below) to compute GBA, which has unique features that are important for this application: besides its ability to take into account both direct and indirect connection between genes and take into account edge weights, it normalizes node degrees to improve specificity.
The interconnectedness (Iij) between two nodes (i and j) in a weighted undirected network is defined as , where wij is the edge weight between node i and j; si the strength of node i; and u are the set of nodes that are neighbors of both i and j. Edge weights are directly taken from GeneMANIA. The strength of a node in a weighted network is the sum of weights for the edges from the node to its neighbors. The original GBA score of node i by the IC method is calculated as , where Sd is a set of seed genes. It can be seen that this method merely sums up the connections between a node and the seeds, ignoring the fact that different nodes have different connectivity with the entire network, and hence different possibility of connections with the seeds. To address this issue, the IC score in this framework is normalized so that . Next, the scores are standardized (subtracting the mean and then dividing by the standard deviation) so that the final GBA scores are comparable between analyses.
The empirical likelihood ratio of a gene being causal for the disease of interest as a function of a GBA score was estimated by leave-one-out analyses of the DisGeNET gene-disease association database (Bauer-Mehren et al. 2011). In each leave-one-out analysis, seed genes were defined as the known disease genes in DisGeNET excluding the left-out gene, and then GBA scores were calculated for all genes including the left-out one. Because of the normalization and the standardization procedure mentioned above, GBA scores are comparable between leave-one-out analyses and between diseases, so the scores from the whole database can be combined to generate a non-disease specific likelihood ratio curve as a function of GBA, where the nominator was calculated from the left-out genes, and the denominator was from the remaining genes excluding the seeds. As expected, the Bayes factors monotonically and smoothly increased with GBA (Suppl. Figure S1), confirming the validity of the proposed algorithm. In a real study, Bayes factor for a query gene can be calculated by interpolation of the corresponding GBA score against this curve.
BayesHLR
The BayesHLR is written by: , where gi denotes one of the observed haplotype combinations (hereafter referred to as genotype), sj is an indicative variable for affection status (1 for cases and 0 for controls), ni is the number of samples with genotype gi, and ni,j the number of samples with genotype gi and status sj. P(gi) is the population genotype frequency calculated from the data as r(ni1/n*1) + (1 – r)(ni0/n*0), where r is the prevalence of the disease. Expected genotype frequency in cases P(gi|s=1) and in controls P(gi|s=0) based on a specific disease model is calculated by the Bayes theorem: P(gi|sj) = P(sj|gj) P(gi)/Σk P(sj|gk)P(gk), where P(s=1|gi) is the penetrance of genotype gi for cases and P(s=0|gi)=1- P(s=1|gi) for controls. k is summed over the observed genotypes. The penetrance of gi is a function of a genetic model provided by the user and the probability that each of the two haplotypes is pathogenic. The latter is calculated by 1 – ∏ν[1 – wνmν], where mv is the probability of carrying a minor allele for variant v, and wv is the weight for the variant, the probability that the minor allele is pathogenic. It is straightforward to calculate this weight as the posterior probability of pathogenicity using a non-informative prior probability and a Bayes factor calculated from deleteriousness and variant call quality.
BayesSeg
The modified linkage analysis method described in Thompson et al (Thompson et al. 2003) is implemented in this framework. However, this method only provides a likelihood ratio score for each variant. To integrate co-segregation with BayesGBA and BayesHLR, a gene-wise co-segregation LOD score is calculated as the logarithm of the sum of likelihood ratios weighted by deleteriousness and variant call quality. The weights are normalized (sum to one) so that the summation is robust to linkage disequilibrium among rare variants. And because of the normalization, deleteriousness and variant call quality can be reused as weights in a rare-variant association analysis.
Converted VQSLOD score
In this framework, positive VQSLOD is converted to zero, while negative VQSLOD is directly added with BayesDel to calculate a variant weight. The rationale for not using positive VQSLOD scores is, being a true variant doesn't make it more likely to be causal for the disease of interest, but being a false one makes it less likely so. Therefore, variants with positive VQSLOD should not be up-scored, while those with a negative VQSLOD should be down-scored.
Benchmark of gene prioritization
The aim of this simulation was to assess the performance of BayesDel, BayesHLR, BayesSeg, BayesGBA, and their integration for gene prioritization in next-generation sequencing studies of complex human diseases targeting rare variants conferring a modest risk.
The hypothetical disease in this simulation had a total familial relative risk of 2, which was commonly seen in a complex disease such as breast cancer. Assuming a dominant inheritance model that the genotype relative risk was 2.5 to 10, the aggregated risk allele frequency was 0.0004 to 0.006, and the penetrance for non-carrier was 2%, then the hypothetical causal gene explained about 2-6% of the total familial relative risk of the disease. This genetic model was in line with the state-of-the-art complex disease gene research in that the focus was to identify moderate- to high-risk genes explaining a small proportion of the total heritability. In the simulated studies, two cases (cousins) from each of the 20 high-risk pedigrees (with 4 affected members) were sequenced (Suppl. Figure S2). The number of healthy controls for sequencing was four times the number of pedigrees. This is a reasonable number because 1) a large number of public control data will be available for NGS analysis; and 2) the power increase is negligible when the controls-to-cases ratio is beyond 4:1 [59]. Subsequently, the cousins from high-risk pedigrees were used for co-segregation analysis, and the independent cases (one case per pedigree) and controls were used for association analysis. To create realistic sequence data that maintain the linkage disequilibrium among variants and the distribution of allele frequencies, variant types and functional consequences, the 1000 Genomes Project (G1K) Phase 3 data was used as the source of haplotype pools. In each round of simulation, a causal gene for a disease was obtained from DisGeNET, where a proportion (10%, 25% or 50%) of the variants whose minor allele frequency was below a cutoff value (0.0002 or 0.002) were randomly designated as causal variants conferring a relative risk (2.5, 5, 7.5 or 10) to the disease. The relative risk of a haplotype was then multiplicative of the relative risks of the causal variants in the haplotype. High-risk pedigrees were simulated by SLINK (Ott 1989). Causal and non-causal G1K haplotypes were randomly assigned to each sequenced individual in the pedigrees based on the results from SLINK. The advantage of this simulation was that 1) the linkage disequilibrium among variants, the distribution of allele frequencies, variant types, and functional consequences were largely maintained as in a real study; 2) locus and allelic heterogeneity among and within pedigrees were spontaneously simulated as would occur in the nature; 3) the disease model was typical or even more difficult in terms of the proportion of causal variants within a gene, the relative risk and allele frequency of the causal variants, the phenocopy rate, and the proportion of missing heritability explained by the gene and; 4) the research strategy mimic a real study.
The DisGeNET database was used to retrieve the true gene-disease relationships. If a gene was linked with multiple diseases, only one of the diseases was randomly selected for simulation, so that no gene was over-counted. Deleteriousness scores of the causal and non-causal variants were randomly sampled from the BayesDel scores of the “selected” variants and the non-pathogenic non-benign variants from the UK10K Project, respectively. Because these variants were not used in the training of BayesDel, obtaining deleteriousness score distributions from them would not inflate the performance of BayesDel. PERCH and pVAAST analyzed the simulated data. Other methods were not considered for comparison here since they either were unable to integrate co-segregation, association, and biological relevance assessment, or the integration were not quantitative. Penetrance for the PERCH association and segregation analysis was 0.05, 0.5, 0.5 for 0, 1, 2 copies of a causal allele, respectively. Due to the high computational demands, the number of permutations for pVAAST was 1000 times. This number was large enough to separate the top 20 genes given that there was a total of 17377 genes computed by pVAAST. For both methods, analyses were restricted to functional variants whose minor allele frequency was less than 0.005. The accuracy of gene prioritization was evaluated as the probability of finding the disease gene among the top 20, 50, 100, and 200 genes. The McNemar's test was used to test for significant differences between analysis methods.
Deleteriousness of variants other than missense variants
Causal variants for a complex disease may not always reside in a coding region and change a protein sequence, and may not always be single nucleotide variants (SNV). To assess deleteriousness of all variants in the whole genome, this framework calculates deleteriousness scores from different sources for different variants. For coding SNVs, it combines the component scores, while for non-coding variants including SNVs and short insertions/deletions (InDels) in transcription factor binding sites, microRNA binding sites or RNA genes, it calculates deleteriousness from the CADD score. Nevertheless, it is expected that more sophisticated tools, such as the later version of CADD or other genome-wide predictors, will be integrated into this framework to fine-tune the deleteriousness predictions in the near future.
Results
Comparison of BayesDel with other deleteriousness scores
It is of interest to compare the performance of BayesDel with each of its components and with other combinatory methods, such as CADD (Kircher et al. 2014), MetaLR and MetaSVM (Dong et al. 2015). As pointed out by Grimm et al (Grimm et al. 2015), the evaluation of deleteriousness prediction tools is hindered by three types of circularity arising from (1) the same variants in both training and testing; (2) the same genes in both training and testing, where all variants in a gene are unanimously pathogenic or neutral; (3) the same variant classification mechanism in both training and testing. These are called the Type 1, Type2, or Type 3 Circularity hereafter. Considering these, the criteria in choosing test data to evaluate BayesDel were: (1) there should not be any overlapping variants between the training and testing dataset; (2) exclude genes with variants that are almost exclusively pathogenic or neutral; (3) since allele frequency is incorporated in BayesDel, benign variants should not be defined by allele frequency or the presence of homozygous alternative allele genotypes; (4) deleteriousness tools should be removed from the variant classification scheme in the definition of pathogenic variants whenever possible. Among these criteria, the first one controls for the Type 1 Circularity, the second one for the Type 2 Circularity, and the last two for the Type 3 Circularity. Per Grimm et al (Grimm et al. 2015), the fourth criterion is hard to meet due to the lack of documentation of all lines of evidence for pathogenicity in most variant databases. Fortunately, controlling for this kind of bias is not impossible, as demonstrated by the first testing dataset of this study (see below).
The first dataset for benchmarking BayesDel was the BRCA1 (MIM *113705) and BRCA2 (MIM *600185) variant classification data from the ENIGMA consortium (Spurdle et al. 2012). Variants in the ENIGMA data were classified by a method integrating association with the disease of interest in cases and controls, co-segregation with the disease in families, family history analysis, the co-occurrence of the variant in trans with a pathogenic variant, population frequency, functional assay, and deleteriousness predictions (Goldgar et al. 2004). This classification method was well established, comprehensive, and had been adapted for clinical practice for 10 years (Eggington et al. 2014). These facts, together with the large number of pedigrees, cases, and controls collected by the ENIGMA consortium, allowed for a high proportion (99.8%) of variants to be confidently classified. Therefore, these data were among the most accurate databases. Instead of defining benign variants by high allele frequency in controls, the ENIGMA classification used here was based strictly on family-history, co-occurrence, and co-segregation. Therefore, it was suitable for benchmarking a predictor that uses allele frequency in its assessment. More importantly, the deleteriousness component was removed from the ENIGMA classification scheme in this study, so there was no overlapping tool for variant classification between the gold standard and the methods in question. As stated in Methods, ENIGMA variants were removed from the training of BayesDel. Although some component predictors were trained on datasets that may contain some of the ENIGMA variants, its effect was negligible because the ENIGMA variants constitute only a tiny proportion of the training datasets for those predictors. Furthermore, both the training and testing data had a mixture of pathogenic and neutral variants for each gene, so this dataset fulfills all the four criteria for test data selection. Finally, there were 26 Likely Pathogenic or Pathogenic variants and 147 Likely Not Pathogenic or Not Pathogenic variants for testing. The results of this benchmarking were in line with previously reported findings, where the ensemble scores MetaSVM, MetaLR and CADD had a better performance than most individual methods. Nevertheless, BayesDel yielded a significantly higher area under the receiver operating characteristic curve (AUC) than MetaSVM (0.94 vs. 0.90, p=0.024), MetaLR (0.94 vs. 0.88, p=0.008), and CADD (0.94 vs. 0.88, p=0.022) (Figure 2).
Figure 2.

Receiver operating characteristic (ROC) curve for the prediction of pathogenic variants in test dataset 1. Numbers in parentheses are areas under the curves (AUC).
Although the ENIGMA dataset was accurate and unbiased for benchmarking, it contained pathogenic and benign variants in only two genes, making it unsuitable to generalize the conclusions to other diseases or genes. Therefore, a second dataset was used to assess deleteriousness predictions. The pathogenic variants in this dataset were the “selected” variants created by Grimm et al (accessed 2015-08-28) (Grimm et al. 2015), excluding variants in ClinVar or UniProtKB that have been used in the training of BayesDel. In Grimm et al, the variants were “selected” to avoid overlapping with variants in the training of some deleteriousness scores including PolyPhen2 and Mutation Assessor, a feature that was also important for the benchmarking of BayesDel. Control variants were those from the ALSPAC and TWINSUK datasets within the UK10K Project, excluding pathogenic or benign variants from ClinVar, UniProtKB, or the above-mentioned pathogenic variants for testing. To fulfill the second criterion for test data selection, genes with a pathogenic variant rate of less than 20% or more than 80% were excluded. To this point, this dataset fulfilled the first three but not the fourth criteria, and thus the Type 3 Circularity was not fully controlled for. After filtering, there were 7323 pathogenic and 8164 benign variants for testing. In agreement with the previous findings, BayesDel outperformed all other methods, including the individual or combinatory scores (Figure 3). Compared to the second best method, MetaSVM, BayesDel had a significantly higher AUC (0.80 vs. 0.74, p=2×10-138). Changing the within-gene pathogenic variant rate threshold doesn't affect this conclusion.
Figure 3.

Receiver operating characteristic (ROC) curve for the prediction of pathogenic variants in test dataset 2. Numbers in parentheses are areas under the curves (AUC).
Performance of PERCH in gene prioritization
To demonstrate the power of integrating BayesDel (d), BayesSeg (S), BayesHLR (H) and BayesGBA (G) for gene prioritization, whole exome sequencing studies of high-risk pedigrees for complex diseases were simulated imitating realistic but challenging situations in such studies. In these studies, each of the high-risk pedigrees had 4 affected cases, but only 2 were sequenced to reduce the cost while maintaining the ability to perform co-segregation analysis. Along with the familial cases, a number of independent control samples were also sequenced for the rare-variant association test. The performance of each analysis method was measured by the probability of finding the simulated causal genes among the top 20, 50, 100 and 200 genes. Different disease models were simulated based on the minor allele frequency cutoff for low-frequency variants, the proportion of low-frequency variants that were causal to the disease, and the relative risk of the causal variants. Results (Figure 4) showed that, even without the integration of biological relevance and deleteriousness assessment, PERCH was more accurate than pVAAST 2.1.6 (pVAAST vs. H+S) by all measures for all simulated models. The difference was significant (p<0.001) when the simulated relative risk was greater than or equal to 5. For intermediate-risk genes (RR=2.5), the difference was significant for the “top 20” but not the other comparisons probably due to the small sample size. Figure 4 also indicated that BayesGBA alone was not a strong classifier for causal genes, but it consistently and significantly (p<0.001) improved the overall accuracy of gene prioritization (H+S+G vs. H+S). Lastly, this benchmarking also showed that using BayesDel as a variant weight in BayesSeg and BayesHLR significantly (p<0.001) improved the overall ranking accuracy (H[d]+S[d] vs. H+S).
Figure 4.
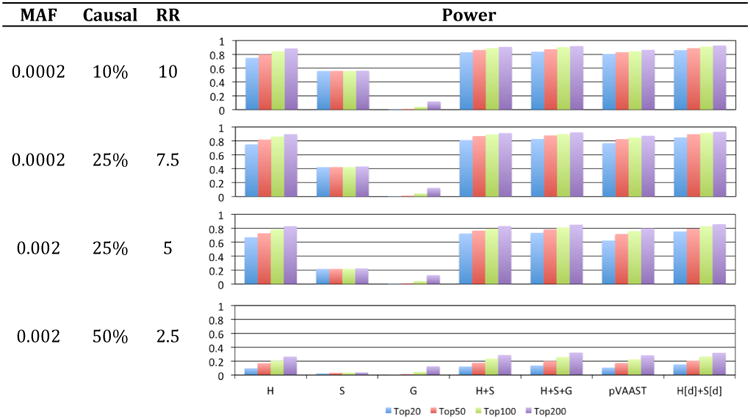
Performance of PERCH and pVAAST in gene prioritization for complex diseases. H: BayesHLR, S: BayesSeg, G: BayesGBA, d: BayesDel; H[d]+S[d]: H+S weighted by BayesDel. MAF: minor allele frequency cutoff for low-frequency variants; Causal: the percentage of low-frequency variants that are causal; RR: relative risk of causal variants; Power: the probability of finding the causal genes among the top 20, 50, 100 and 200.
Software implementation
The software was designed to be flexible. A new component can be directly added whenever it is independent of the others in the framework, without the need to re-train any model or perform a permutation test. For example, it is straightforward to integrate LOD scores from a previous linkage study if it does not double count the same information. It is also easy to incorporate expression-profiling results. Likewise, removing a component from the model is also straightforward. Besides employing methods provided by the framework, it is also a trivial task to replace some of the components with external software packages, such as SEQlinkage for linkage analysis (Wang et al. 2015). In addition, the programs can filter variants by call quality, chromosomal region, allele frequency, deleteriousness, loss-of-function consequence, and whether the variant is a de novo mutation.
This software suite includes a number of command-line programs and uses the Unix pipe to organize a sequence of analyses. Without the need to permute or load all data before computation, PERCH is fast with low demands on memory. In an analysis of the whole-exome sequencing of 1156 independent cases, 1176 healthy controls, and 50 high-risk pedigrees, it took less than 8 minutes to run on a computer cluster with dual-socket 8-core Intel Xeon processors and 64G memory. All programs were written in C++. The current version is 1.0. This framework is available at http://BJFengLab.org/.
Discussion
This paper describes a novel framework for gene prioritization in disease gene discovery research, which quantitatively unifies deleteriousness, allele frequency, call quality, segregation, association, and the biological relevance of genes to the disease of interest. This framework can also be used for the classification of VUS (variants of unknown significance) in clinical genetic testing.
The combined deleteriousness score proposed in this paper has many advantages. First, it is more accurate than other tested methods, including the combined scores. Secondly, the naïve Bayesian approach treats missing values naturally, so it is not necessary to do imputation, which may introduce bias and hinders its usage. As a Bayes factor, it can be directly applied to the classification of variants of unknown significance (VUS), as shown in this framework. One of the caveats in the evaluation of deleteriousness scores in this study was that Type 3 Circularity was not addressed by the second test dataset, while the first test data included only two genes. However, as stated by Grimm et al (Grimm et al. 2015), Type 3 Circularity was an issue “to beware of in the future” because its solution requires a variant database to document each line of evidence for pathogenicity. Nevertheless, the conclusion that BayesDel is more accurate than other deleteriousness scores should not be affected by circularity, as demonstrated by the first test dataset. Another caveat in BayesDel is the potential over-fitting to known variants, so that the performance may be different between known and novel variants. This is a pervasive problem in ensemble classifiers. It is expected that larger and more accurate variant databases will be generated by functional assays and comprehensive variant classification schemes. It will be interesting to see how these methods perform in the new data in the near future.
Additionally, users can choose the data source for allele frequency, or even not to include allele frequency in BayesDel. This is an important feature since it is particularly beneficial to calculate allele frequency in the studied population and/or obtain them from a sequence data that will be freely available in the future. It also may be desirable to customize a public data set to remove related phenotypes. For example, one may use the subset of ExAC (Exome Aggregation Consortium) without TCGA (The Cancer Genome Atlas) samples in a study of cancer. In some research where only extremely rare variants were selected for analysis, the contribution of allele frequency is negligible. PERCH has an option to use a model without allele frequency, which was separately trained and thus has different weights. In summary, users have full control over the usage of allele frequency based on their study designs.
This framework implements a novel rare variant association analysis called BayesHLR. It allows for a disease model with a reduced penetrance (dominant, recessive or additive). By stratified analysis, it can adjust for cryptic population structures, confounding loci, and environmental risk factors. However, because BayesHLR aggregates the minor alleles of rare variants within a gene, it has the same limitations as other burden tests. Namely, it is not robust to protective rare variants and is sensitive to the proportion of causal rare variants in a gene. Still, this method is useful for most situations, since protective alleles for a severe disease are unlikely to be rare due to organismal fitness, and the problem of the small proportion of causal variants can be alleviated by allele frequency filtering, deleteriousness weighting, and domain-based analysis. Nevertheless, the output scores of the proposed framework can also be used as variant weights in other rare-variant association tests such as SSU (Basu and Pan 2011) and SKAT-O (Lee et al. 2012), which are more robust with respect to the percentage of causal variants and the presence of protective rare variants.
A caveat of BayesHLR and BayesSeg is that they require the user to specify a genetic model for the disease. For most complex diseases high-penetrance rare mutations should have been found by linkage studies, and common variants with low effect sizes should have been discovered by GWAS. Thus, the aims of most next-generation sequencing studies are variants with in-between effects. Within this range, BayesHLR and BayesSeg are robust toward misspecification with regard to disease models, as indicated in the simulation where the disease model for testing was different from the disease model for data generation.
Supplementary Material
Acknowledgments
The author gratefully acknowledges the supports of the NIH (grants CA116167, CA192393, CA155767, CA164944), the National Psoriasis Foundation, the Utah Psoriasis Initiative, the Utah Genome Project, and the PERSPECTIVE project (PErsonalised Risk Stratification for Prevention and Early deteCTIon of breast cancer) that is funded from the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the Ministère de l'Économie, de la Science et de l'Innovation du Québec through Genome Québec, and the Quebec Breast Cancer Foundation. The author would like to thank the ENIGMA Consortium for providing the BRCA1 and BRCA2 data for comparison. This study makes use of data generated by the UK10K Consortium, derived from samples from ALSPAC and TWINSUK. A full list of the investigators who contributed to the generation of the data is available from www.UK10K.org. Funding for UK10K was provided by the Wellcome Trust under award WT091310. The author would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about. The support and resources from the Center for High-Performance Computing at the University of Utah are gratefully acknowledged. Bryony A. Thompson and Hai-De Qin greatly helped to improve the earlier version of this manuscript. The author is immensely grateful to his mentor, David E. Goldgar, for the tremendous support and the critical review of this manuscript.
Grant Sponsor: The author gratefully acknowledges the supports of the NIH (grants CA116167, CA192393, CA155767, CA164944), the National Psoriasis Foundation, the Utah Psoriasis Initiative, the Utah Genome Project, and the PERSPECTIVE project that is funded from the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the Ministère de l'Économie, de la Science et de l'Innovation du Québec through Genome Québec, and the Quebec Breast Cancer Foundation.
Footnotes
The Author declares that there is no conflict of interest.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman A, Garcia-Garcia F, Salavert F, Medina I, Dopazo J. A web-based interactive framework to assist in the prioritization of disease candidate genes in whole-exome sequencing studies. Nucleic Acids Res. 2014;42:W88–W93. doi: 10.1093/nar/gku407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Pan W. Comparison of statistical tests for disease association with rare variants. Genet Epidemiol. 2011;35:606–619. doi: 10.1002/gepi.20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer-Mehren A, Bundschus M, Rautschka M, Mayer MA, Sanz F, Furlong LI. Gene-Disease Network Analysis Reveals Functional Modules in Mendelian, Complex and Environmental Diseases. PLoS ONE. 2011;6:e20284. doi: 10.1371/journal.pone.0020284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Angel G del, Rivas MA, Hanna M, McKenna A, Fennell TJ, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–2137. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubchak I, Balasubramanian S, Wang S, Meyden C, Sulakhe D, Poliakov A, Börnigen D, Xie B, Taylor A, Ma J, Paciorkowski AR, Mirzaa GM, et al. An Integrative Computational Approach for Prioritization of Genomic Variants. PLoS ONE. 2014;9:e114903. doi: 10.1371/journal.pone.0114903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Bowles KR, Moyes K, Manley S, Esterling L, Sizemore S, Rosenthal E, Theisen A, Saam J, Arnell C, Pruss D, Bennett J, et al. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet. 2014;86:229–237. doi: 10.1111/cge.12315. [DOI] [PubMed] [Google Scholar]
- Field MA, Cho V, Cook MC, Enders A, Vinuesa CG, Whittle B, Andrews TD, Goodnow CC. Reducing the search space for causal genetic variants with VASP. Bioinformatics. 2015;31:2377–2379. doi: 10.1093/bioinformatics/btv135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Guttman M, Clamp M, Zody MC, Friedman N, Xie X. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics. 2009;25:i54–i62. doi: 10.1093/bioinformatics/btp190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis J, Pavlidis P. The Impact of Multifunctional Genes on “Guilt by Association” Analysis. PLoS ONE. 2011;6:e17258. doi: 10.1371/journal.pone.0017258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis J, Pavlidis P. “Guilt by Association” Is the Exception Rather Than the Rule in Gene Networks. PLoS Comput Biol. 2012;8:e1002444. doi: 10.1371/journal.pcbi.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro ANA, Tavtigian SV, Couch FJ Breast Cancer Information Core (BIC) Steering Committee. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–544. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding J, Pembrey M, Jones R ALSPAC Study Team. ALSPAC--the Avon Longitudinal Study of Parents and Children. I. Study methodology. Paediatr Perinat Epidemiol. 2001;15:74–87. doi: 10.1046/j.1365-3016.2001.00325.x. [DOI] [PubMed] [Google Scholar]
- González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm DG, Azencott CA, Aicheler F, Gieraths U, MacArthur DG, Samocha KE, Cooper DN, Stenson PD, Daly MJ, Smoller JW, Duncan LE, Borgwardt KM. The Evaluation of Tools Used to Predict the Impact of Missense Variants Is Hindered by Two Types of Circularity. Hum Mutat. 2015;36:513–523. doi: 10.1002/humu.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CL, Huang YH, Hsu CT, Yang UC. Prioritizing disease candidate genes by a gene interconnectedness-based approach. BMC Genomics. 2011;12(3):S25. doi: 10.1186/1471-2164-12-S3-S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Roach JC, Coon H, Guthery SL, Voelkerding KV, Margraf RL, Durtschi JD, Tavtigian SV, Shankaracharya, Wu W, Scheet P, Wang S, et al. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol. 2014;32:663–669. doi: 10.1038/nbt.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelo P, Ali MM. Some Variants of the Controlled Random Search Algorithm for Global Optimization. J Optim Theory Appl. 2006;130:253–264. [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Larson DE, Sullivan LS, Bowne SJ, Steinberg KM, Churchill JD, Buhr AC, Nutter N, Pierce EA, Blanton SH, Weinstock GM, Wilson RK, et al. Exome-based mapping and variant prioritization for inherited Mendelian disorders. Am J Hum Genet. 2014;94:373–384. doi: 10.1016/j.ajhg.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Abecasis GR, Boehnke M, Lin X. Rare-Variant Association Analysis: Study Designs and Statistical Tests. Am J Hum Genet. 2014;95:5–23. doi: 10.1016/j.ajhg.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostat Oxf Engl. 2012;13:762–775. doi: 10.1093/biostatistics/kxs014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, Gui HS, Kwan JSH, Bao SY, Sham PC. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012;40:e53–e53. doi: 10.1093/nar/gkr1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA, Altshule DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, Gabriel SB, Gibbs RA, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayyeri A, Hammond CJ, Valdes AM, Spector TD. Cohort Profile: TwinsUK and healthy ageing twin study. Int J Epidemiol. 2013;42:76–85. doi: 10.1093/ije/dyr207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau Y, Tranchevent LC. Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nat Rev Genet. 2012;13:523–536. doi: 10.1038/nrg3253. [DOI] [PubMed] [Google Scholar]
- Morris AP, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol. 2010;34:188–193. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott J. Computer-simulation methods in human linkage analysis. Proc Natl Acad Sci U S A. 1989;86:4175–4178. doi: 10.1073/pnas.86.11.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FBL, Hoogerbrugge N, Spurdle AB, Tavtigian SV IARC Unclassified Genetic Variants Working Group. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price WL. A controlled random search procedure for global optimisation. Comput J. 1977;20:367–370. [Google Scholar]
- Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Köhler S, Oellrich A, Wang K, Mungall CJ, Lewis SE, Washington N, Bauer S, Seelow D, Krawitz P, Gilissen C, et al. Sanger Mouse Genetics Project. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 2014;24:340–348. doi: 10.1101/gr.160325.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoni FA, Makrythanasis P, Nikolaev S, Guipponi M, Robyr D, Bottani A, Antonarakis SE. Simultaneous identification and prioritization of variants in familial, de novo, and somatic genetic disorders with VariantMaster. Genome Res. 2014;24:349–355. doi: 10.1101/gr.163832.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, Day INM, Gaunt TR. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifrim A, Popovic D, Tranchevent LC, Ardeshirdavani A, Sakai R, Konings P, Vermeesch JR, Aerts J, Moor B, De Moreau Y. eXtasy: variant prioritization by genomic data fusion. Nat Methods. 2013;10:1083–1084. doi: 10.1038/nmeth.2656. [DOI] [PubMed] [Google Scholar]
- Sincan M, Simeonov DR, Adams D, Markello TC, Pierson TM, Toro C, Gahl WA, Boerkoel CF. VAR-MD: a tool to analyze whole exome-genome variants in small human pedigrees with mendelian inheritance. Hum Mutat. 2012;33:593–598. doi: 10.1002/humu.22034. [DOI] [PubMed] [Google Scholar]
- Spurdle AB, Healey S, Devereau A, Hogervorst FBL, Monteiro ANA, Nathanson KL, Radice P, Stoppa-Lyonnet D, Tavtigian S, Wappenschmidt B, Couch FJ, Goldgar DE, et al. ENIGMA--evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat. 2012;33:2–7. doi: 10.1002/humu.21628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D, Easton DF, Goldgar DE. A full-likelihood method for the evaluation of causality of sequence variants from family data. Am J Hum Genet. 2003;73:652–655. doi: 10.1086/378100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JRB, Xu C, Futema M, Lawson D, Iotchkova V, Schiffels S, et al. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GT, Zhang D, Li B, Dai H, Leal SM. Collapsed haplotype pattern method for linkage analysis of next-generation sequence data. Eur J Hum Genet. 2015;23:1739–1743. doi: 10.1038/ejhg.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PI, Marcotte EM. It's the machine that matters: Predicting gene function and phenotype from protein networks. J Proteomics. 2010;73:2277–2289. doi: 10.1016/j.jprot.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Li Y, Jiang R. Integrating Multiple Genomic Data to Predict Disease-Causing Nonsynonymous Single Nucleotide Variants in Exome Sequencing Studies. PLoS Genet. 2014;10:e1004237. doi: 10.1371/journal.pgen.1004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Zhang KX, Kramer M, Pellegrini M, McCombie WR. FamAnn: an automated variant annotation pipeline to facilitate target discovery for family-based sequencing studies. Bioinformatics. 2014;30:1175–1176. doi: 10.1093/bioinformatics/btt749. [DOI] [PubMed] [Google Scholar]
- Yip YL, Famiglietti M, Gos A, Duek PD, David FPA, Gateau A, Bairoch A. Annotating single amino acid polymorphisms in the UniProt/Swiss-Prot knowledgebase. Hum Mutat. 2008;29:361–366. doi: 10.1002/humu.20671. [DOI] [PubMed] [Google Scholar]
- Zhang H, Sheng Shengli. Proceedings of the Fourth IEEE International Conference on Data Mining (ICDM'04) Brighton, United Kingdom: 2004. Learning Weighted Naive Bayes with Accurate Ranking. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.