

Abstract
Performance traits are economically important and are targets for selection in breeding programs, especially in the poultry industry. To identify regions on the chicken genome associated with performance traits, different genomic approaches have been applied in the last years. The aim of this study was the application of CornellGBS approach (134,528 SNPs generated from a PstI restriction enzyme) on Genome-Wide Association Studies (GWAS) in an outbred F2 chicken population. We have validated 91.7% of these 134,528 SNPs after imputation of missed genotypes. Out of those, 20 SNPs were associated with feed conversion, one was associated with body weight at 35 days of age (P < 7.86E-07) and 93 were suggestively associated with a variety of performance traits (P < 1.57E-05). The majority of these SNPs (86.2%) overlapped with previously mapped QTL for the same performance traits and some of the SNPs also showed novel potential QTL regions. The results obtained in this study suggests future searches for candidate genes and QTL refinements as well as potential use of the SNPs described here in breeding programs.
Production efficiency in the poultry industry is constantly improving as a result of selection for growth rate, feed efficiency and carcass traits for broilers, and egg production and quality traits for layers1,2. The understanding of genomic information of loci controlling those traits are important to improvement of selection efficiencies of breeding programs1.
Several studies have been conducted in chickens using markers randomly distributed in the genome (microsatellites), which have allowed the identification of several QTLs for production traits3,4,5,6,7,8,9,10,11,12,13. Following these, some of the studies have focused their attention on the identification of SNPs in functional and positional candidate genes and to test their association on target QTL regions14,15,16,17,18,19. With the advent of next-generation sequencing (NGS), it was possible to identify a global SNP profile and to perform genome-wide association studies (GWAS) to find novel QTL regions20,21,22,23,24,25,26 and also to refine the previously published regions27,28,29,30,31.
Despite the high-throughput data generation by NGS, which have facilitated the identification of SNPs in several populations, the use of this method for GWAS is still a limitation. This is caused by the high cost involved with the generation of data to be applied in a large number of individuals. To solve this high cost problem, SNP panels were designed to be applied in GWAS32,33. However, some important regions in the genome are inaccessible to sequence capture approaches34 mainly because they are based on predesigned SNP profiles. To overcome those limitations, and to present a unique SNP profile, we used the PstI-derived SNPs dataset from CornellGBS optimized approach. This dataset was originated from a SNP call from the reduced representation of the sequenced genome (~5%) through PstI restriction enzyme35. This SNP dataset is reliable and reproducible, showing a unique profile of SNPs with microchromosome enrichment35 that contains 2-4 times higher gene density than macrochromosomes36,37.
The aim of this study was to identify genetic markers using PstI-derived SNPs dataset, and further use that information to conduct a GWAS with performance traits in chickens. In addition, we have performed a linkage disequilibrium (LD) analyses in the parental, F1 and F2 generations, to better understand the segregation of haplotype blocks, and the population structure, from the associated and suggestively associated SNPs identified. Finally, we have compared the location of these mentioned SNPs with known QTLs, with the objective to validate and to refine the regions of known QTLs.
Results
Animals and Phenotypes
The descriptive statistics for the eight performance traits analyzed can be observed in Table 1. Detailed description of these animals and traits were provided elsewhere7,16. The large variability is expected since the animals are from a broiler x layer F2 population.
Table 1. Means, standard deviations (SD), maximum (max) and minimum (min) values for performance traits of 444 individuals from the F2 population.
| Traits | Average (SD) | Max | Min |
|---|---|---|---|
| Feed conversion from 35-41 days | 2.87±0.71 | 7.55 | 1.52 |
| Feed intake from 35–41 days (g) | 603.43±139.74 | 1176.00 | 258.00 |
| Feed efficiency from 35–41 days | 0.36±0.07 | 0.66 | 0.13 |
| Birth weight (g) | 44.96±4.15 | 55.50 | 34.60 |
| Body weight at 35 days (g) | 789.86±138.60 | 1309.00 | 480.00 |
| Body weight at 41 days (g) | 1006.82±188.34 | 1686.00 | 407.00 |
*g is the weight measured in grams.
Genotypes
In our previously work35, using a minimum taxon call rate of 90%, we have identified 67,096 SNPs originated from 462 chickens using the GBS approach. However, in this study, different filter parameters were applied. We have reduced the loci call rate filtering criteria to 70%. This parameter is the minimum threshold of individuals call rate for each loci to be included in the output. This reduction had minimal impact on sample call rate (proportion of missing genotypes per individual) and large impact on number of SNPs. The sample call rate reduced from 99.96% ± 0.04% to 99.90% ± 0.1% and the number of SNPs increased from 67,096 to 134,528. This allowed us to capture more SNPs, but the number of missing genotypes increased (for details, see M&M section). To overcome this, we have imputed the missing genotypes using Beagle 4.1 software38. This approach resulted in a panel of 134,528 derived PstI-SNPs present in all animals.
SNPs validation
The dataset of 134,528 SNP chromosomal positions obtained with the CornellGBS before and after the imputation analysis was compared with the 600 K Affymetrix® HD genotyping array dataset in order to perform a method validation, since both sets were obtained from the same animals (5 individuals from F2-7810 family). The genotype concordance of the SNPs with concordant chromosomal positions detected between the two methods is shown in Table 2. On average, 91.80%, and 91.66% of the SNPs had concordant genotypes between the CornellGBS and 600 K datasets before and after imputation, respectively. The accuracy of the heterozygous genotypes was slightly lower after the imputation. Reduced representation methods, like CornellGBS, has limitations calling the heterozygous markers39. In our study, we have observed that 82.14 and 82.30% of heterozygous SNPs, while 97.97 and 97.65% of all homozygous SNPs were validated before and after imputation, respectively.
Table 2. Assessment of genotype concordance between 134,528 PstI-derived filtered SNPs before and after imputation and genotyped SNPs dataset from 600 K Affymetrix® HD genotyping array from five F2 individuals (F2-7810 family); and genotype validation percentages for homozygous and heterozygous SNPs.
| F2-7810 family | SNP type | Before Imputation |
After Imputation |
Total validated genotypes* |
|||
|---|---|---|---|---|---|---|---|
| Concordant physical position† | Validated genotypes* (%) | Concordant physical position† | Validated genotypes *(%) | Before Imputation *(%) | After Imputation *(%) | ||
| ID-1209 | homoz1 | 2,410 | 97.67 | 2,493 | 97.47 | 93.08 | 92.93 |
| heteroz2 | 1,323 | 85.56 | 1,370 | 85.47 | |||
| ID-1786 | homoz1 | 2,379 | 97.73 | 2,427 | 97.65 | 94.04 | 93.96 |
| heteroz2 | 1,422 | 88.60 | 1,440 | 88.47 | |||
| ID-1787 | homoz1 | 2,296 | 97.99 | 2,356 | 97.87 | 93.72 | 93.42 |
| heteroz2 | 1,470 | 87.68 | 1,512 | 87.17 | |||
| ID-2301 | homoz1 | 1,589 | 98.48 | 2,409 | 97.50 | 87.62 | 87.67 |
| heteroz2 | 948 | 70.25 | 1,457 | 72.06 | |||
| ID-786 | homoz1 | 2,383 | 97.98 | 2,438 | 97.74 | 90.57 | 90.33 |
| heteroz2 | 1,395 | 78.63 | 1,422 | 78.34 | |||
1Homozygous genotype; 2heterozygous genotype; †number of concordant physical positions between both datasets used for validation (from CornellGBS vs 600 K Affymetrix® HD genotyping array); *is the percentage of † that had concordant genotypes.
Homozygous and heterozygous SNPs
Out of 62 million possible genotypes (462 samples × 134,528 sites), the average frequency of heterozygous SNPs was 25.32% (±5.6%) before the imputation and after the imputation, it increased to 27.70% (±5.2%). The average heterozygosity observed per chickens before imputation ranged from 8.30–44.69% and after the imputation between 11.38–44.67%. The proportion of heterozygous SNPs remained virtually unchanged before and after imputation among the lines/generations (Table 3).
Table 3. SNP heterozygosity of genotyped populations (parental, F1 and F2 generations) after and before imputation.
| Population | Number of individuals | Proportion of heterozygous (SD) |
|
|---|---|---|---|
| Before Imputation | After Imputation | ||
| Parental CC | 5 | 0.16 ± 0.01 | 0.17 ± 0.01 |
| Parental TT | 5 | 0.21 ± 0.01 | 0.22 ± 0.01 |
| F1 | 8 | 0.28 ± 0.09 | 0.29 ± 0.08 |
| F2-7765 | 72 | 0.29 ± 0.03 | 0.29 ± 0.03 |
| F2-7810 | 82 | 0.28 ± 0.04 | 0.28 ± 0.04 |
| F2-7816 | 94 | 0.30 ± 0.08 | 0.30 ± 0.08 |
| F2-7971 | 100 | 0.25 ± 0.05 | 0.26 ± 0.04 |
| F2-7978 | 96 | 0.26 ± 0.04 | 0.27 ± 0.03 |
Principal component analyses
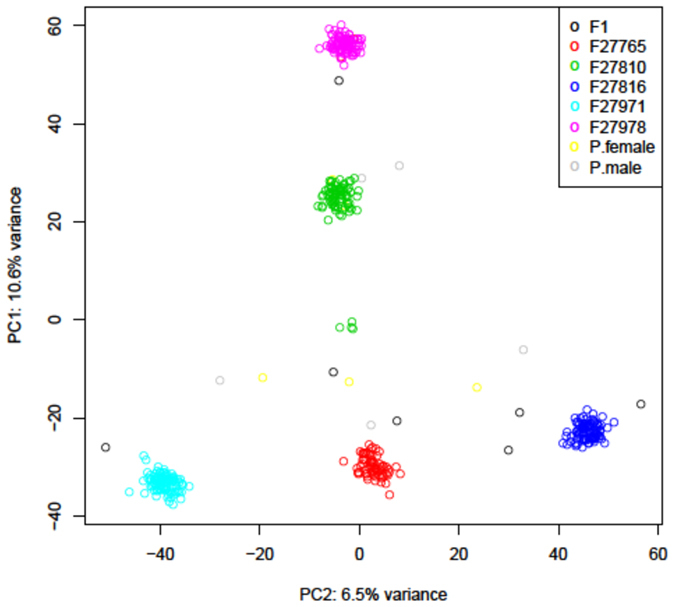
From the list of imputed genotypes we have conducted a principal component analysis (PCA), based on covariance, using Tassel v.5.2.2639 to check the F2 population structure. This plot was useful for visualizing internal structure explained by the variance from PstI-derived SNPs dataset of 134,528 SNPs using eigenvector-based multivariate analyses. Each individual lies in its proper group consistent with our F2 population structure composed by five F2 dame-based families (Fig. 1).
Figure 1. The 444 chickens from five F2 families and18 chickens from the parental lines (P. female and P. male - N = 10) and F1 generation (N = 8) shown in the 2D plane spanned by their first two principal components.
Descriptive Statistics of Heritability
The genetic and residual variance for each trait and their genomic heritability are shown in Table 4. Heritabilities ranging from moderate to high, as is expected40,41, were observed for feed intake and body weights traits, respectively. Low heritabilities were observed for the traits evaluated in short period (between 35 and 41 days) as feed conversion, and feed efficiency, because they are complex traits influenced by several environmental factors42.
Table 4. Genetic and residual variances, and genomic heritability for each trait analyzed in this study.
| Traits | Var_genetic ± SE | Var_error ± SE | Heritability ± SE |
|---|---|---|---|
| Feed conversion 35adj | 0.0045 ± 0.00282 | 0.4363 ± 0.01797 | 0.01 ± 0.006 |
| Feed intake from 35adj | 1492.3 ± 855.94 | 7296.0 ± 830.22 | 0.17 ± 0.094 |
| Feed efficiency 35adj | 0.0005 ± 0.00002 | 0.0036 ± 0.00002 | 0.11 ± 0.005 |
| Birth weight | 2.5719 ± 0.50599 | 3.1009 ± 0.33925 | 0.45 ± 0.073 |
| Body weight at 35 days | 10403 ± 1575.9 | 1879.9 ± 785.7 | 0.85 ± 0.073 |
| Body weight at 41 days | 16790 ± 2936.7 | 5639 ± 1701.1 | 0.75 ± 0.087 |
35adj is adjusted to body weight at 35 days, SE is the standard error, and for Heritability, the SE is the standard deviations obtained from a repeated sampling approach (see M&M section for detailed information).
Genome-wide association study
Twenty significant SNPs (P < 7.86E-07) were associated with feed conversion adjusted to body weight at 35 days (adj35) and one significant SNP associated with body weight at 35 days of age (Fig. 2). In addition to that, 92 suggestive (P < 1.57E-05) SNPs were associated with feed conversion adj35, feed intake adj35, feed efficiency adj35, birth weight, and body weight at 35 and 41 days of age (see Supplementary Spreadsheet S1 for the effects of associated SNPs; Manhattan and QQ plots are available on Supplementary Fig. S1).
Figure 2. SNPs associated with feed conversion adj35.

(a) and body weight at 35 days (b) are presented by Manhattan (left side) and QQ (right side) plots. The y-axis is shown as -log10 (p-value). On the left, the red line indicates genome-wise association (P < 7.86E-07) and the blue line, suggestive genome-wise association (P < 1.57E-05) with the respective trait. On the right side, the QQ-plots show the relation of normal theoretical quantiles of the probability distributions between expected (x-axis) and observed (y-axis) p-values from each respective associated trait (feed conversion adj35 and body weight at 35 days).
Linkage disequilibrium analysis
Seventeen haplotype blocks were generated from the associated and suggestively associated SNPs from the F2 population (see Fig. 3, Supplementary Fig. S2 for haplotypes details and Supplementary Table S1 to SNPs’ Mendelian descriptions). We noticed a standard block pattern between the SNPs that matched with the F2 population structure (Fig. 3). Interestingly, we have checked the genotype frequency of blocks formed by LD analysis to determine if the blocks were fixed or not in the parental lines. From the haplotype blocks, we checked the origin of the variation (fixed or variable) and frequency from F2 generation in the parental lines (Supplementary Table S2 and Supplementary Fig. S2 for a more detailed description of frequencies). We also determined the advantageous haplotype for each trait in the F2 generation (Table 5). This information enabled us to identify from which parental line (TT or CC) comes the genotypic variation observed in F2 for each block. All blocks with r2 > 0.56 had the most frequent haplotype agreeing with the advantageous phenotype in the F2 individuals (Fig. 3; Supplementary Fig. S2 and Supplementary Table S2), and this advantageous haplotype (lower feed conversion and higher values of other evaluated traits) was fixed in one of the parental lines, except in blocks 2 and 13. This information is also available for each genome-wide suggestive and/or associated SNPs in Supplementary Spreadsheet S1, as well as the number of genotype observations obtained per SNP.
Figure 3. Haplotype blocks obtained by the solid spine of LD and family structure using Haploview 4.2.

The header represents the physical position of each 94 SNPs selected on chicken genome (Gallus gallus 4.0, NCBI). The family structure is disposed as maternal CC (a) and paternal TT (b) parental pure lines, F1 (c) and F2 (d) generations. The black filled squares indicate r2 = 1 and square numbers represents r2*100 with black color gradient accordingly.
Table 5. TagSNP significance levels from MLM analyses of each 17 blocks obtained by solid spine of LD in the F2 population using Haploview 4.2.
| HaplotypeN° (GGA) | Haplotype | Haplotype Frequency (%) |
Trait | p-value | add p-value | dom p-value | |||
|---|---|---|---|---|---|---|---|---|---|
| Parental CC | Parental TT | F1 | F2 | ||||||
| 1(2) | AC† | 50 | 100 | 75 | 84 | eff.adj35 | 1.36E-05* | 2.46E-03* | 2.11E-05* |
| f.conv.adj35 | 5.11E-09** | 2.04E-07** | 1.05E-09** | ||||||
| 2(4) | CAGAATG† | 0 | 60 | 31 | 42 | bw35 | 6.19E-06* | 3.26E-05* | 1.13E-02* |
| AGTGTCA | 80 | 0 | 0 | 40 | bw41 | 4.70E-06* | 5.31E-06* | 1.25E-02* | |
| 3(4) | TAG† | 0 | 80 | 31 | 44 | bw35 | 3.08E-07** | 1.95E-07** | 8.31E-02* |
| CGA | 90 | 0 | 0 | 38 | bw41 | 1.40E-06* | 1.16E-06* | 6.62E-02* | |
| 4(7) | GC† | 100 | 70 | 94 | 94 | f.conv.adj35 | 1.94E-06* | 3.38E-07** | 7.30E-07** |
| 5(7) | AG | 100 | 80 | 88 | 93 | bw35 | 9.91E-06* | 3.63E-06* | 4.53E-03* |
| f.conv.adj35 | 3.70E-06* | 6.96E-07** | 1.18E-06* | ||||||
| 6(11) | GGA† | 100 | 90 | 88 | 93 | f.conv.adj35 | 1.94E-06* | 3.98E-07** | 7.29E-06* |
| 7(13) | CC† | 80 | 90 | 75 | 78 | f.conv.adj35 | 3.67E-06* | 5.76E-07** | 9.02E-07* |
| 8(13) | TC† | 90 | 100 | 88 | 90 | f.conv.adj35 | 3.63E-06* | 7.39E-07** | 7.51E-07** |
| 9(14) | TGC† | 100 | 60 | 68 | 85 | f.conv.adj35 | 3.31E-07** | 8.88E-08** | 6.11E-08** |
| 10(17) | CGT† | 70 | 100 | 88 | 85 | f.conv.adj35 | 3.15E-06* | 5.18E-07** | 1.48E-06* |
| 11(17) | CC† | 100 | 90 | 94 | 95 | f.conv.adj35 | 2.94E-06* | 7.38E-07** | 6.73E-07** |
| 12(18) | ACC† | 90 | 70 | 94 | 93 | f.conv.adj35 | 3.31E-06* | 5.16E-07** | 3.09E-06* |
| 13(20) | TC† | 70 | 90 | 75 | 78 | f.conv.adj35 | 3.45E-06* | 5.45E-07** | 1.07E-06* |
| 14(26) | CA† | 100 | 60 | 75 | 79 | f.conv.adj35 | 3.12E-07** | 4.54E-08** | 1.75E-06* |
| f.int.adj35 | 1.04E-05* | 1.36E-04* | 1.73E-06* | ||||||
| 15(27) | CAGGGAACCA† | 100 | 90 | 88 | 95 | f.conv.adj35 | 9.00E-07* | 3.26E-07** | 1.77E-05* |
| 16(27) | CG† | 100 | 80 | 63 | 94 | f.conv.adj35 | 2.15E-07** | 1.38E-07** | 4.91E-08** |
| 17(28) | CA† | 70 | 10 | 44 | 35 | bw41 | 2.22E-06* | 4.57E-07** | 1.58E-01* |
| TG | 20 | 50 | 31 | 30 | |||||
| CG | 10 | 40 | 25 | 34 | |||||
(GGA) is the Gallus gallus chromosome number; †Most frequent haplotype in the population; bolded is the advantage haplotype to individuals compared to the assessed trait; additive (add) and dominant (dom) p-values; *suggestive genome-wise significance (P < 1.57E-05) and **genome-wise significance (P < 7.86E-07). Abbreviations: eff.adj35 (feed efficiency adjusted for body weight at 35 days), f.conv.adj35 (feed conversion adjusted for body weight at 35 days), f.int.adj35 (feed intake adjusted for body weight at 35 days), all between 35 and 41 days of age and adjusted for 35 days; bw35, bw41, bw42 is body weight at 35, 41 and 42 days of age, respectively.
QTL overlapping SNPs
Through Animal QTLdb, we have selected all the 1,458 known QTLs43 mapped for body weight, feed efficiency, feed conversion and growth, all evaluated in different chicken lines and ages. Out of those, we have observed that 253 QTLs overlapped with 81 of the 94 associated and suggestively associated SNPs with performance traits obtained from the GWAS in this study: 206 QTLs associated with body weight, 39 with postnatal growth, 4 with feed intake, 3 with feed conversion, and 1 with feed efficiency. The complete QTL list that overlapped with these regions can be seen in the Supplementary Table S3 and the graphical representation of the suggestive and significant SNPs distribution in relation to the QTLs can be observed in Fig. 4.
Figure 4. Karyotype of the QTLs (from Animal QTLdb) distribution regions of the chicken genome overlapping suggestive and significant SNPs associated with performance traits (black marks).

These QTLs were mapped for body weight (bw), postnatal growth (grow), feed intake (f.intake), feed conversion (f.conv), and feed efficiency (effic). The colors becoming denser according to the number of QTL previously mapped superposing each other.
Discussion
For better understanding of complex traits control in a segregating F2 population, our research group have focused the attention on genetic association and linkage analyses using different approaches, as: candidate genes14,15,16,17 and QTL mapping3,4,5,6,7, respectively, and more recently, NGS approaches27,28. We have presented here the first study using a higher density of SNPs in this F2 population with GWAS purpose. Therefore, we have optimized a method called CornellGBS in chickens35 to overcome the concept of pre-designed panels, since we planned a method for genotyping efficiently a specific dataset of SNPs in our specific population.
CornellGBS is a widely employed method to genotype large genomes of model and non-model species exploring important regions in the genome34 as microchromosomes36,37, as previously mentioned. This is due to the high coverage of tags (contigs) depending on the number of sequenced individuals of the reduced genome by restriction enzyme cleavage providing a specific SNP profile35. The CornellGBS technique was previously developed for inbreeding population and it is known by its general low sequencing coverage, which can cause significant loss of SNPs, mainly heterozygous39. For our outbreed population, we used a reasonable multiplex of individuals (~48 animals per lane of Illumina flowcell) to maintain a reasonable sequencing coverage per individual (~5X). We also reduced the loci call rate and use imputation to increase the number of SNPs genotyped. The reduction in the loci call rate was also applied in a recent study that used the same PstI restriction enzyme to cleave the cattle genome44. Furthermore, it was already mentioned that the combination of GBS and imputation of missing internal SNPs in haplotype blocks procedures can promote a cost reduction by allowing further reduction of the filtering criteria or sequencing coverage without causing losses in SNP calls34. Using this strategy, we doubled the number of SNPs, successfully imputing all lost genotypes (increasing the individual call rate to 100%), the validation ratio remained > 90%, and the percentage of heterozygous genotypes in our population had an increase of approximately 2% after the imputation.
The use of the GBS SNP panel for GWAS in our outbred F2 crosses resulted in 20 SNPs associated (P < 7.86E-07) with feed conversion adj.35, one SNP associated with body weight at 35 days (BW35) and other 93 SNPs suggestively associated (P < 1.57E-05) with different performance traits (Table 1). Additionally, we noticed that all the evaluated traits, presented an up deviation of the theoretical quantiles (Fig. 2 and Supplementary Fig. S1) of the probability distributions between expected and observed p-values, indicating the existence of QTLs. These results corroborated the Manhattan plot peaks of associated SNPs, indicating that these traits had part of the phenotypic variation significantly explained by the genetic component45. Interestingly, we detected association for several new QTLs located in microchromosomes (GGA11-28). This was only possible because of the distribution of the SNPs used. From our set of SNPs, 38.93% are located in large chromosomes (GGA1-5), 14.15% in intermediate size (GGA6-10), and most, 46.90% are located in microchromosomes (GGA11-28), confirming the microchromossome enrichment mentioned before35. Feed conversion, for exemple, had a high number of significant SNPs (P < 7.86E-07), mainly located in microchromosomes (GGA8, 10, 14, 18, 23, 26, and 27). However, for this trait, the SNP peaks observed by Manhatan plot, in large and intemediate size chromossomes (Fig. 2a), were not well defined, as is usually observed for QTLs peaks46. We believe that this is explained by the SNP profile used in this study, which has a lower density of SNPs on large chromosomes compared to microchromosomes35. Moreover, feed intake is a complex trait subject to a high residual effect and controlled by several genes with a small effect, which require a large sample size to detect associations47,48. This small effect also was previously attributed to the short period used to measure this trait (between 35 and 41 days of age) impairing the animal adaptation to the new enviromental condiction4. On this account, reliable QTLs for this trait were detected in studies that used a larger sampling size (1,534 individuals) for a longer feed intake evaluation period (~4 weeks)49,50 than used in this study.
The GBS strategy can result in clusters of SNPs next to each other35. In order to better define the QTL regions we performed LD analysis. The block pattern between the SNPs matched with our F2 population structure51 (Fig. 3). This allowed us to define possibilities for genetic selection of the lines that did not present the genotype fixed giving attention to the different phenotypic abilities between the CC layer line and the TT broiler line5,16,51. As for example the blocks 2 and 13 (consider Fig. 3(d) for block numbers), which had variable genotypes for both the CC and the TT lines, and the paternal line presented the favorable genotype most frequent in both cases (see Supplementary Fig. S2 and Fig. 3).
Also is important to check if these SNPs are within QTL regions previously published. In the past years, many studies identified QTLs associated with performance traits in different chickens populations3,4,5,6,8,9,10,11,12,13,17,52,53 (1,458 QTLs described in the Animal QTLdb) aiming to map loci that control these traits. Recently, to better understand these loci, studies have also applied GWAS with performance traits in chickens13,26,54. The validation of single SNP position obtained by GWAS overlapping with QTL regions can confirm interesting genomic regions to explore. From the 94 genome-wise associated and suggestively associated SNPs with the performance traits analyzed in this study, most of them were fairly distributed in mapped QTL regions in the chicken genome (Fig. 4). Only 13 SNPs did not overlap with QTL regions previously mapped. From these 13 SNPs, one was located on chromosome 1, one in chromosome 8 (GGA1 and 8), one in the Z sex chromosome (GGAZ) and 10 were located in microchromosomes (GGA17, 18, 20, 25, 27 and 28), which confirms the microchromosome enrichment profile obtained by this approach35 and suggests novel QTLs to be explored in these regions. It is also important to mention that most of these 13 SNPs (those located on GGA1, 8, 18, 20, 25 and 27) were associated (P < 7.86E-07) or suggestively associated (P < 1.57E-05) with feed conversion adj35, one with feed efficiency adj35 (GGA17), and two with body weight at 41 days (GGA27) (see Supplementary Spreadsheet S1 for details). The genes where these SNPs are located are mainly related with cell cycle and metabolic pathways (according to the Reactome pathways - http://www.reactome.org/PathwayBrowser) and were within introns, upstream and downstream of these genes (see Supplementary Table S4 for functional annotation).
Despite the importance of the overlap test performed here, previous studies in QTL mapping usually had large confidence intervals (>1 Mbps) and often encompassing several genes, making difficult the selection of candidate genes55. Therefore, we also checked the overlap of these SNPs only with QTLs mapped using specifically the same traits and the same F2 population4,5,14,17 used in this study. From 23 different QTL intervals, we identified 12 SNPs overlapping with seven of them (see the QTLs bolded in Supplementary Tables S3 and S5 to check the QTL list). It is worth mentioning the SNPs located near the QTL regions, or flanking regions (see Supplementary Fig. S3). On GGA1, for exemple one SNP (marker 6, see Supplementary Spreadsheet S1) associated with feed intake (P = 3,83E-07) overlaped with one QTL mapped for the same trait50 in another population, and also with 6 QTLs mapped for body weight at different ages56 and one with feed efficiency57. On the other hand, on GGA4, a well studied chromosome in chickens4,9,11,12,14,16,20,23,24,25,54,58,59,60,61,62,63, we identified three SNPs composing the haplotype 3 (markers 21–23), in which one was associated with BW35 (P < 7.86E-07) and suggestively associated with BW41 (P < 1.57E-05), and two SNPs suggestively associated with BW41 (P < 1.57E-05). These three SNPs overlapped with one QTL region previously mapped in this same population for these same traits4 (QTL_IDs from ChickenQTLdb = 7157; 7162 and 7185). The boundary SNPs from haplotypes 2 and 3 (Fig. 3(d)) are separated by a short distance (less than 4 Mbps), but these QTLs are not linked, beside they have effect on the same traits (BW35 and BW41) (see Supplementary Spreadsheet S1) in our F2 population. It is worth to mention, the haplotype 2 that overllaped with QTLs mapped for different BW in different ages52,53,64,65,66,67 and growth13,52,67 traits in different populations.
To the best of our knowledge, we showed the application of the CornellGBS PstI-derived SNPs to a GWAS for the first time in chickens. We showed a strategy, changing filtering criteria and subsequent genotype imputation, to increase the number of reliable SNPs to be analyzed. We found 13 SNPs indicating new regions associated with performance traits, mainly in microchromosomes, that have not been previously reported. We improved the available information about loci controlling performance traits and we refined these regions to discover novel candidate regions to be explored. Finally, by demonstrating that GBS is a valid strategy for QTL mapping in a species that has genome sequence and SNP panel available, we can argue the validity of GBS in species without genome resources.
Methods
All experimental protocols employed in the present study that relate to animal experimentation were performed in accordance with the resolution number 010/2012 approved by the Embrapa Swine and Poultry Ethics Committee on Animal Utilization to ensure compliance with international guidelines for animal welfare.
Animals and Phenotypes
This study was conducted using 464 chickens from a F2 populations originated from a reciprocal cross-experimental population from Embrapa Swine and Poultry National Research Center, Concórdia, SC, Brazil. We also included in the analysis, 10 chickens from their parental lines TT and CC (5 from each one), and eigth from the F1 generation. This F2 population was developed for QTL mapping studies, and was originated from the crossing of seven males from a broiler line (TT) and seven females from a layer line (CC), resulting in seven full-sib families (F1 generation). Then, twenty-one F1 females were artificially inseminated with seven F1 males (3:1 ratio) to generate the F2. The F2 population comprised seven paternal half-sib families composed of three full-sib families of approximately 100 individuals each, produced in 17 hatches, totaling 2,063 F2 chickens51. For this study, we selected the five most informative families based on the previously QTL studies4,16,63.
The TT broiler line was selected over six generation to improve body weight, feed conversion, carcass and breast yields, viability, fertility, eclodibility, reduction of abdominal fat and metabolic syndromes51. The CC layer line was selected over eight generation to improve egg production, egg weight, feed conversion, viability, sexual maturity, fertility, eclodibility, egg quality and reduction of body weight51.
The F2 chickens were reared as broilers with free access to corn and soybean meal-based diet and water up to 42 days of age. From 35 to 41 days, they were transferred to cages to collect feed intake, and to compute the conversion and efficiency. Body weight was recorded at 1 (birth weight), 35, 41 and 42 (after fasting) days of age. The body weight (BW) at 41 days of age was collected at the end of the conversion measurement and, BW42 days was collected after 6-h fasting period and transportation to the slaughterhouse. More details were previously provided5,16,51. We analysed these six performance traits in this study (feed conversion, feed intake and feed efficiency between 35 to 41 days, birth weight, and body weight at 35 and 41 days of age). A total of 23 missed values from the selected traits were imputed by mean using Tassel v.5.2.26 tool39 among the 446 F2 animals selectect to be evaluated in this study.
DNA, genotypic data and imputation
Genomic DNA was cleaved with the PstI enzyme, ligated to adapters with barcodes identifying individual animals, and then sequenced on Illumina platform. After filtering parameters were applied, 134,528 SNPs were identified from 462 individuals in our experimental population of chickens using minimum minor allele frequency (mnMAF) of 1%; minimum taxon coverage (mntCov) of 20% and minimum site coverage (mnScov) of 70% filter parameters. All procedures to obtain the data were previously described35. After filtering parameters, the number of missing genotypes increased from 0.9 to 5.8 million. This number represents SNPs identified multiplied for the number of individuals genotyped (67,096 derived PstI-SNPs × 462 individuals using 90% of loci call rate and 134,528 derived PstI-SNPs x 462 individuals using 70% of loci call rate). The imputation of missing genotypes was performed using Beagle 4.1 software38 using default parameters, which uses empirical LD model. This model adapts to the local structure in the data using iterative approach to haplotype phasing in which an initial prediction of haplotype phase is made, then the model is fit, and improved estimates of haplotype phase are obtained and the model is refit38.
SNPs validation
The validation was performed by the comparison of the filtered 134,528 PstI-derived SNPs dataset with the 600 K Affymetrix® HD genotyping array SNP dataset using five individuals from F2-7810 family. The SNPs in both datasets were located on GGA1-28, 32, W and Z chromosomes on the Gallus-gallus-4.0 reference genome. The validation standards used in this study were based on a methodology previously proposed68 considering chromosomal positions and genotype concordances. With these concordances, we estimate the accuracy for homozygous and heterozygous SNPs.
Principal component analysis
The principal component analyses (PCA) of imputed genotype data were performed using Tassel v.5.2.26 39 considering five principal components. This tool transforms a set of correlated variables into successive orthogonal PCs accounted for the maximum variance providing a way to highlight groups of individuals differing at the level of minor allele frequency39. The PCA graph was produced using the plot function in R 2.13.2 software (http://www.r-project.org/).
Genome-Wide Association Study
The compressed mixed linear model (MLM) implemented in TASSEL v.5.2.26 software45 was used for GWAS. The following form represents the statistical model:
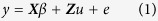 |
where y is the vector of the dependent variables, β is the vector containing fixed effects, including the sex (male/female), hatch (1–17) and SNP (−1, 0, 1) effects, and the covariate body weight at 35 days for traits measured from 35 to 41 days of age (feed intake, feed efficiency and feed conversion); u is the vector of random additive genetic effects from background QTL for individuals, X and Z are design matrices, and e is the vector of random residuals. We assumed that u and e vectors are normally distributed with null mean and variance of
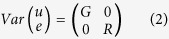 |
where 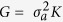 ;
; 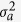 is an unknown additive genetic variance and K is the kinship (co-ancestry) matrix calculated from SNPs and provided by the same software using centered identity by state (IBS) method. For the residual effect, homogeneous variance was assumed, with
is an unknown additive genetic variance and K is the kinship (co-ancestry) matrix calculated from SNPs and provided by the same software using centered identity by state (IBS) method. For the residual effect, homogeneous variance was assumed, with 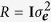 , where I is an identity matrix and
, where I is an identity matrix and 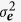 is the unknown residual variance. The Restricted Maximum Likelihood (REML) estimates of
is the unknown residual variance. The Restricted Maximum Likelihood (REML) estimates of 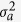 and
and 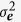 were obtained by the Efficient Mixed-Model Association (EMMA) algorithm69. Heritability (h2) was calculated as the ratio of the additive genetic variance (
were obtained by the Efficient Mixed-Model Association (EMMA) algorithm69. Heritability (h2) was calculated as the ratio of the additive genetic variance (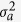 ) to the phenotypic variance (
) to the phenotypic variance (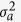 +
+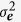 ). Tassel program does not provide the standard errors of the estimates. Thus, standard errors were estimated using the REML method with an average information (AI) algorithm by AIREMLF90 software70. Standard errors for additive genetic and residual variance were computed as square roots of diagonal elements of the inverse of the average information matrix. For Heritability, standard deviations obtained from the repeated sampling approach were considered as their standard error71. Each SNP allele was fit as a separate class with heterozygotes fit as additional SNP classes. The total SNP effect was not decomposed in additive and dominance effects but tested for overall significance45
). Tassel program does not provide the standard errors of the estimates. Thus, standard errors were estimated using the REML method with an average information (AI) algorithm by AIREMLF90 software70. Standard errors for additive genetic and residual variance were computed as square roots of diagonal elements of the inverse of the average information matrix. For Heritability, standard deviations obtained from the repeated sampling approach were considered as their standard error71. Each SNP allele was fit as a separate class with heterozygotes fit as additional SNP classes. The total SNP effect was not decomposed in additive and dominance effects but tested for overall significance45
Quantile-quantile (Q-Q) plots for each trait and Manhattan plots of genome-wide association analyses were performed in R using ggd.qqplot and Manhattan functions. The thresholds were corrected for multiple testing (Bonferroni) established by the estimated number of independent SNPs and LD blocks (pairwise r2 values > 0.40)72 that was 63,640 SNPs. We set two thresholds from our data: P < 1.57E-05 (1/63,640) for suggestive significance and P < 7.86E-07 (0.05/63,640) for 5% genome-wise significance level54,60.
Linkage disequilibrium analysis
The linkage disequilibrium (LD) analysis was performed with Haploview 4.273 as well as the LD graphs. To perform the pairwise comparison of ours SNPs considering 1 Mb apart, we selected 94 genome-wise associated and suggestively associated SNPs with the traits analyzed in this study (feed conversion, feed intake, feed efficiency, and weight gain between 35–41 days of age, birth weight and body weight at 35 and 41 days). We have defined the haplotype blocks by the solid spine of LD and the family structure of parental lines (CC and TT pure lines), F1 and F2 generations (Fig. 3) using Haploview 4.2.
QTL overlapping with SNPs
The QTL data was obtained from QTLdb43 and the overlapping test was performed using the GenomicRanges package from R 3.3.1 software (http://www.r-project.org/). For the data presentation, we designed a figure using ggplot2 package karyogram layout from R 3.3.1 software.
Additional Information
How to cite this article: Pértille, F. et al. Genome-wide association study for performance traits in chickens using genotype by sequencing approach. Sci. Rep. 7, 41748; doi: 10.1038/srep41748 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The authors are grateful for the support from the University of São Paulo, thematic project (2014/08704-0) from São Paulo Research Foundation (FAPESP) and the Embrapa Swine and Poultry National Research Center. The F2 population genotyping was funded by the PRODETAB project (no. 038-01/01). Fábio Pértille received a fellowship from CAPES. Luiz L. Coutinho is recipient of productivity fellowship from CNPq.
Footnotes
The authors declare no competing financial interests.
Author Contributions F.P. analyzed the data and wrote the manuscript with the valuable help of R.Z and C.B. G.C.M.M. and R.Z. helped with bioinformatics analysis. J.R.N. helped with the CornellGBS method related. C.B. helped with the CornellGBS dataset interpretation. M.C.L. was responsible for the design of the EMBRAPA F2 Chicken Resource Population, provided the biological material and phenotypes. G.A.R., G.B.M., R.Z. and M.C.L. provided guidance to the GWAS analyses. L.L.C. provided overall supervision of the research. All authors reviewed and approved the last version of the manuscript.
References
- Fulton J. E. Genomic selection for poultry breeding. Anim. Front. 2, 30–36 (2012). [Google Scholar]
- Blackburn H. D. The National Animal Germplasm Program: challenges and opportunities for poultry genetic resources. Poult. Sci. 85, 210–215 (2006). [DOI] [PubMed] [Google Scholar]
- Ambo M. et al. Genetic linkage maps of chicken chromosomes 6, 7, 8, 11 and 13 from a Brazilian resource population. Sci. Agric. 65, 447–452 (2008). [Google Scholar]
- Ambo M. et al. Quantitative trait loci for performance traits in a broiler x layer cross. Anim. Genet. 40, 200–8 (2009). [DOI] [PubMed] [Google Scholar]
- Nones K. et al. Mapping QTLs on chicken chromosome 1 for performance and carcass traits in a broiler x layer cross. Anim. Genet. 37, 95–100 (2006). [DOI] [PubMed] [Google Scholar]
- Nones K. et al. Quantitative trait loci associated with chemical composition of the chicken carcass. Anim. Genet. 43, 570–6 (2012). [DOI] [PubMed] [Google Scholar]
- Campos R. L. R. et al. Quantitative trait loci associated with fatness in a broiler-layer cross. Anim. Genet. 40, 729–36 (2009). [DOI] [PubMed] [Google Scholar]
- Tatsuda K. & Fujinaka K. Genetic mapping of the QTL affecting body weight in chickens using a F 2 family. Br. Poult. Sci. 42, 333–337 (2001). [DOI] [PubMed] [Google Scholar]
- Koning D. J. de et al. Quantitative trait locus detection in commercial broiler lines using candidate regions. J Anim Sci 81, 1158–1165 (2003). [DOI] [PubMed] [Google Scholar]
- Ikeobi C. O. et al. Quantitative trait loci for meat yield and muscle distribution in a broiler layer cross. Livest. Prod. Sci. 87, 143–151 (2004). [Google Scholar]
- Nassar M. K., Goraga Z. S. & Brockmann G. a. Quantitative trait loci segregating in crosses between New Hampshire and White Leghorn chicken lines: II. Muscle weight and carcass composition. Anim. Genet. 43, 739–45 (2012). [DOI] [PubMed] [Google Scholar]
- Nassar M. K., Goraga Z. S. & Brockmann G. a. Quantitative trait loci segregating in crosses between New Hampshire and White Leghorn chicken lines: III. Fat deposition and intramuscular fat content. Anim. Genet. 44, 62–8 (2013). [DOI] [PubMed] [Google Scholar]
- Zhou H., Deeb N., Evock-Clover C. M., Ashwell C. M. & Lamont S. J. Genome-wide linkage analysis to identify chromosomal regions affecting phenotypic traits in the chicken. I. Growth and average daily gain. Poult. Sci. 85, 1700–11 (2006). [DOI] [PubMed] [Google Scholar]
- Felício A. M. et al. Polymorphisms in FGFBP1 and FGFBP2 genes associated with carcass and meat quality traits in chickens. Genet. Mol. Res. 12, 208–22 (2013). [DOI] [PubMed] [Google Scholar]
- Felício a. M. et al. Identification and association of polymorphisms in CAPN1 and CAPN3 candidate genes related to performance and meat quality traits in chickens. Genet. Mol. Res. 12, 472–82 (2013). [DOI] [PubMed] [Google Scholar]
- Pértille F. et al. Identification of polymorphisms associated with production traits on chicken (Gallus gallus) chromosome 4. Genet. Mol. Res. 14, 10717–10728 (2015). [DOI] [PubMed] [Google Scholar]
- Boschiero C. et al. Association of IGF1 and KDM5A polymorphisms with performance, fatness and carcass traits in chickens. J. Appl. Genet. 54, 103–12 (2013). [DOI] [PubMed] [Google Scholar]
- Shen X. et al. The GTPase activating Rap/RanGAP domain-like 1 gene is associated with chicken reproductive traits. PLoS One 7, e33851 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Q.-H., Zhang X.-Q. & Lei M.-M. Single nucleotide polymorphism and its use in chicken QTL mapping. Yi Chuan 25, 729–34 (2003). [PubMed] [Google Scholar]
- Sun Y. et al. The identification of 14 new genes for meat quality traits in chicken using a genome-wide association study. BMC Genomics 14, 458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. P. et al. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. USA 110, 453–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M. N. et al. Genome-wide Association Study of Chicken Plumage Pigmentation. Asian-Australasian J. Anim. Sci. 26, 1523–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C. et al. Genome-wide association study of antibody response to Newcastle disease virus in chicken. BMC Genet. 14, 42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C. et al. Genetic parameters and genome-wide association study of hyperpigmentation of the visceral peritoneum in chickens. BMC Genomics 14, 334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. et al. Genome-wide linkage analysis and association study identifies loci for polydactyly in chickens. G3 (Bethesda). 4, 1167–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L. et al. Genome-wide association study identified a narrow chromosome 1 region associated with chicken growth traits. PLoS One 7, e30910 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira G. C. M. et al. Variant discovery in a QTL region on chromosome 3 associated with fatness in chickens. Anim. Genet. 46, 141–147 (2015). [DOI] [PubMed] [Google Scholar]
- Godoy T. F. et al. SNP and INDEL detection in a QTL region on chicken chromosome 2 associated with muscle deposition. Anim. Genet. 46, 158–163 (2015). [DOI] [PubMed] [Google Scholar]
- Ahsan M. et al. Identification of candidate genes and mutations in QTL regions for chicken growth using bioinformatic analysis of NGS and SNP-chip data. Front. Genet. 4, 1–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux P.-F. et al. Re-Sequencing Data for Refining Candidate Genes and Polymorphisms in QTL Regions Affecting Adiposity in Chicken. PLoS One 9, e111299 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. Using targeted re-sequencing for identification of candidate genes and SNPs for a QTL affecting the pH value of chicken muscle. bioRxiv 5, 17186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen M. a. M. et al. The development and characterization of a 60K SNP chip for chicken. BMC Genomics 12, 274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranis A. et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genomics 14, 59 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J. et al. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 5, 484 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pértille F. et al. High-throughput and Cost-effective Chicken Genotyping Using Next-Generation Sequencing. Sci. Rep. 6, 26929 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermann F. a. et al. Arrangements of macro- and microchromosomes in chicken cells. Chromosome Res. 9, 569–84 (2001). [DOI] [PubMed] [Google Scholar]
- Smith J. et al. Differences in gene density on chicken macrochromosomes and microchromosomes. Anim. Genet. 31, 96–103 (2000). [DOI] [PubMed] [Google Scholar]
- Browning S. R. & Browning B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–97 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaubitz J. C. et al. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9, e90346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaya L. G. et al. Heritability and Genetic Correlation Estimates for Performance and Carcass and Body Composition Traits in a Male Broiler Line. Poult. Sci. 85, 837–843 (2006). [DOI] [PubMed] [Google Scholar]
- Rovadoscki G. A. et al. Genetic parameters for growth characteristics of free-range chickens under univariate random regression models. Poult. Sci. 95, 1989–1998 (2016). [DOI] [PubMed] [Google Scholar]
- Reyer H., Hawken R., Murani E., Ponsuksili S. & Wimmers K. The genetics of feed conversion efficiency traits in a commercial broiler line. Sci. Rep. 5, 16387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z.-L., Park C. A., Wu X.-L. & Reecy J. M. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 41, D871–D879 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Donato M., Peters S. O., Mitchell S. E., Hussain T. & Imumorin I. G. Genotyping-by-sequencing (GBS): a novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS One 8, e62137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007). [DOI] [PubMed] [Google Scholar]
- Fragomeni B. D. O. et al. Changes in variance explained by top SNP windows over generations for three traits in broiler chicken. Front. Genet. 5, 1–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J. et al. Genome-wide association studies for feed intake and efficiency in two laying periods of chickens. Genet. Sel. Evol. 47, 82 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. et al. Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 19, 807–812 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuiskula-Haavisto M. et al. Mapping of quantitative trait loci affecting quality and production traits in egg layers. Poult. Sci. 81, 919–27 (2002). [DOI] [PubMed] [Google Scholar]
- Tuiskula-Haavisto M. et al. Quantitative trait loci with parent-of-origin effects in chicken. Genet. Res. 84, 57–66 (2004). [DOI] [PubMed] [Google Scholar]
- Rosário M. F. do, Ledur M. C., Moura A. S. A. M. T., Coutinho L. L. & Garcia A. A. F. Genotypic characterization of microsatellite markers in broiler and layer selected chicken lines and their reciprocal F1s. Sci. Agric. 66, 150–158 (2009). [Google Scholar]
- Nassar M. K., Goraga Z. S. & Brockmann G. A. Quantitative trait loci segregating in crosses between New Hampshire and White Leghorn chicken lines: IV. Growth performance. Anim. Genet. 46, 441–446 (2015). [DOI] [PubMed] [Google Scholar]
- Goraga Z. S., Nassar M. K. & Brockmann G. A. Quantitative trait loci segregating in crosses between New Hampshire and White Leghorn chicken lines: I. egg production traits. Anim. Genet. 43, 183–189 (2012). [DOI] [PubMed] [Google Scholar]
- Gu X. et al. Genome-Wide Association Study of Body Weight in Chicken F2 Resource Population. PLoS One 6, e21872 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledur M. C., Navarro N. & Pérez-Enciso M. Large-scale SNP genotyping in crosses between outbred lines: how useful is it? Heredity (Edinb). 105, 173–182 (2010). [DOI] [PubMed] [Google Scholar]
- Liu X. et al. Mapping quantitative trait loci affecting body weight and abdominal fat weight on chicken chromosome one. Poult. Sci. 86, 1084–9 (2007). [DOI] [PubMed] [Google Scholar]
- Hansen C. et al. Identification of QTL for production traits in chickens. Anim. Biotechnol. 16, 67–79 (2005). [DOI] [PubMed] [Google Scholar]
- Rabie T. S. K. M. et al. Genetic mapping of quantitative trait loci affecting susceptibility in chicken to develop pulmonary hypertension syndrome. Anim. Genet. 36, 468–76 (2005). [DOI] [PubMed] [Google Scholar]
- Ankra-Badu G. a. et al. Mapping QTL for growth and shank traits in chickens divergently selected for high or low body weight. Anim. Genet. 41, 400–5 (2010). [DOI] [PubMed] [Google Scholar]
- Liu R. et al. Genome-Wide Association Study Identifies Loci and Candidate Genes for Body Composition and Meat Quality Traits in Beijing-You Chickens. PLoS One 8, e61172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abasht B. & Lamont S. J. Genome-wide association analysis reveals cryptic alleles as an important factor in heterosis for fatness in chicken F2 population. Anim. Genet. 38, 491–8 (2007). [DOI] [PubMed] [Google Scholar]
- Liu W. et al. A genome-wide SNP scan reveals novel loci for egg production and quality traits in white leghorn and brown-egg dwarf layers. PLoS One 6, e28600 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron E. E. et al. QTL for percentage of carcass and carcass parts in a broiler x layer cross. Anim. Genet. 42, 117–124 (2011). [DOI] [PubMed] [Google Scholar]
- Podisi B. K., Knott S. A., Burt D. W. & Hocking P. M. Comparative analysis of quantitative trait loci for body weight, growth rate and growth curve parameters from 3 to 72 weeks of age in female chickens of a broiler–layer cross. BMC Genet. 14, 22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podisi B. K. et al. Overlap of quantitative trait loci for early growth rate, and for body weight and age at onset of sexual maturity in chickens. Reproduction 141, 381–389 (2011). [DOI] [PubMed] [Google Scholar]
- Sewalem A. et al. Mapping of quantitative trait loci for body weight at three, six, and nine weeks of age in a broiler layer cross. Poult. Sci. 81, 1775–81 (2002). [DOI] [PubMed] [Google Scholar]
- Carlborg O., Hocking P. M., Burt D. W. & Haley C. S. Simultaneous mapping of epistatic QTL in chickens reveals clusters of QTL pairs with similar genetic effects on growth. Genet. Res. 83, 197–209 (2004). [DOI] [PubMed] [Google Scholar]
- Eck S. H. et al. Whole genome sequencing of a single Bos taurus animal for single nucleotide polymorphism discovery. Genome Biol. 10, R82 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H. M. et al. Efficient Control of Population Structure in Model Organism Association Mapping. Genetics 178, 1709–1723 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misztal I. et al. BLUPF90 and related programs (BGF90). Proc. 7th World Congr. Genet. Appl. to Livest. Prod. 28, 21–22 (2002). [Google Scholar]
- Meyer K. & Houle D. Sampling based approximation of confidence intervals for functions of genetic covariance matrices. Proc. Assoc. Advmt. Anim. Breed. Genet. 20, 523–526 (2013). [Google Scholar]
- Nicodemus K. K., Liu W., Chase G. a., Tsai Y.-Y. & Fallin M. D. Comparison of type I error for multiple test corrections in large single-nucleotide polymorphism studies using principal components versus haplotype blocking algorithms. BMC Genet. 6, S78 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett J. C., Fry B., Maller J. & Daly M. J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.