

Abstract
A strategy for the preparation of homogeneous antibody–drug conjugates (ADCs) containing multiple payloads has been developed. This approach utilizes sequential unmasking of cysteine residues with orthogonal protection to enable site‐specific conjugation of each drug. In addition, because the approach utilizes conjugation to native antibody cysteine residues, it is widely applicable and enables high drug loading for improved ADC potency. To highlight the benefits of ADC dual drug delivery, this strategy was applied to the preparation of ADCs containing two classes of auristatin drug‐linkers that have differing physiochemical properties and exert complementary anti‐cancer activities. Dual‐auristatin ADCs imparted activity in cell line and xenograft models that are refractory to ADCs comprised of the individual auristatin components. This work presents a facile method for construction of potent dual‐drug ADCs and demonstrates how delivery of multiple cytotoxic warheads can lead to improved ADC activities. Lastly, we anticipate that the conditions utilized herein for orthogonal cysteine unmasking are not restricted to ADCs and can be broadly utilized for site‐specific protein modification.
Keywords: antibodies, bioconjugate, cancer, cysteine, drug delivery
Antibody–drug conjugates (ADCs) combine the tumor targeting specificity of monoclonal antibodies with the potent cell‐killing activity of cytotoxic warheads. There has been a surge of interest in designing new ADC formats due in part to the recent clinical success of ADCs, which includes the approvals of brentuximab vedotin (ADCETRIS) in relapsed Hodgkin lymphoma and anaplastic large‐cell lymphoma, and ado‐trastuzumab mertansine (KADCYLA) in HER2‐positive metastatic breast cancer.1 Most of these new methodologies have focused on addressing some of the shortcomings of existing clinical molecules, such as heterogeneous drug loading, limited drug‐linker stability, and warheads with activities that are restricted to a subset of cancer types. To enable improved ADCs, much notable advancement has been made in the field. These include site‐specific drug‐linker conjugation strategies that enable homogeneous loading, drug‐linker attachment modalities with improved stability, potent new payloads, and linker strategies that utilize alternative release mechanisms.1c, 2
Almost all effective cancer chemotherapy utilizes complementary drug combinations designed to overcome differential drug sensitivities within heterogeneous tumor cell populations.3 This strategy has recently also been applied to ADCs, which are now being tested in combination with unconjugated, clinically approved anticancer drugs.4 In addition, emerging clinical and preclinical data for ADCs has demonstrated that insensitivity to a particular ADC can be overcome through delivery of an alternative warhead using the same antibody.5 For these reasons, complementary drug payloads within an ADC would likely constitute a significant advancement in the field of targeted drug delivery. Here, we describe an accessible dual‐cytotoxic drug conjugate technology for native, non‐engineered IgGs and demonstrate the first use of orthogonal thiol protecting groups on a folded protein. We present the first data demonstrating that dual‐drug ADCs have enhanced in vitro and in vivo activities compared to conventional ADCs.
The conjugation of two different highly potent auristatin molecules with complementary physiochemical properties presents an intriguing route to enhance ADC activity on heterogeneous cell populations. Commonly employed auristatin drug‐linkers include mc‐MMAF (1), mc‐vc‐MMAF (2), and mc‐vc‐MMAE (3). The released drug from a mc‐vc‐MMAE drug linker, monomethyl auristatin E (MMAE), is cell permeable and exhibits bystander activity, or the killing of neighboring antigen‐negative cells.7 However, MMAE is also a substrate for MDR exporters and has diminished activity on cells with high pump expression.8 Conversely, MMAF and cys‐mcMMAF, released from mc‐vc‐MMAF and mc‐MMAF ADCs, respectively, are not susceptible to drug export and retain activity on MDR(+) cells but are minimally cell permeable.7b, 9 Thus, they do not exhibit bystander activity and have little activity on antigen‐negative tumor cells. We reasoned that combining the features of these types of drugs could provide complementary activities on cancers, yielding ADCs with enhanced cytotoxicity profiles.
We prioritized two main criteria for dual‐drug conjugation when initiating this work: the methodology must result in homogeneous and site‐specific loading of both drugs, and it should not require engineered antibodies or enzyme‐mediated conjugations so that drug combinations could be screened on an array of IgGs, including commercial antibodies and hybridoma antibody libraries. To date, only a single example of a multi‐drug conjugate has been reported, but this work was conducted on an antibody Fab fragment and required the genetic introduction of an engineered cysteine residue to enable site‐specific discrimination of conjugation sites.10 A number of other approaches for the site‐specific conjugation of two separate agents to an antibody have been presented (recently reviewed in Ref. 11), but most of these methods require specialized reagents including site‐specific amino acid mutations or custom enzymes, and sometimes require two distinct conjugation handles. All of these factors increase the complexity of reagents required to generate and screen multi‐drug ADCs. One method that fit our criteria utilized pyridazine‐dione re‐bridging of native antibody disulfides followed by dual‐click functionalization to construct a largely homogeneous product, but this method was only used to create a fluorophore–drug antibody conjugate.12
Our solution towards creating a general approach was to utilize a drug carrier that can be conjugated to native antibody interchain disulfides through maleimide chemistry. The multi‐plexing drug carrier (4, Figure 1 B) bears two orthogonally protected cysteine residues that can be sequentially unmasked and conjugated with different drug linkers. Using this approach, an ADC is produced that is homogeneous and bears an average of 16 total drugs, split evenly between the two drug linkers. The carrier utilizes two recent advancements for the construction of ADCs with improved pharmacological activity: a self‐stabilizing maleimide (mDPR) to minimize drug‐linker deconjugation in vivo,2a and a PEG24 stretcher to enable high drug loading without concomitant hydrophobicity‐induced ADC aggregation.6
Figure 1.
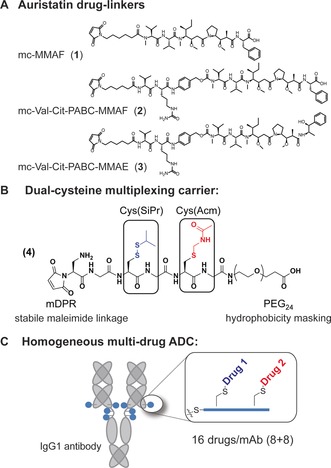
Drug‐linkers and carriers for conjugation to mAbs. A) Maleimide‐auristatin drug‐linkers used in this study. B) A multiplexing drug carrier 4 bearing Cys(SiPr) and Cys(Acm) groups that can be unmasked using orthogonal conditions. The carrier also contains a PEG24 group to mask drug‐linker hydrophicity6 and a self‐stabilizing maleimide (mDPR)2a for antibody attachment. C) Homogeneous dual‐drug ADCs prepared using 4 bear 16 total drugs, split evenly (8+8) between the two component drugs.
To carry out this intended strategy, we needed to identify two distinct cysteine protecting groups that could be unmasked selectively in aqueous conditions without affecting antibody structure or causing ADC aggregation. Cysteine residues bearing reducible disulfide protecting groups, such as S‐(tert‐butyl) or S‐(isopropyl) disulfides can be readily removed with reducing agents and have been used extensively for selective peptide modification.13 We reasoned that these types of protecting groups would be attractive for a 2‐step conjugation on a folded protein because they can be removed using tris‐(2‐carboxylethyl) phosphine (TCEP) under mild aqueous conditions, which are the same conditions used to reduce native antibody interchain disulfides.
To identify a second protecting group for our carrier, we screened for protecting groups that were stable to TCEP reduction but could be removed rapidly in aqueous conditions. Through this work, we identified aqueous conditions for the deprotection of acetamidomethyl (Acm)‐protected cysteines.13b Specifically, maleimidocaproyl‐Cys(Acm) was synthesized and conjugated to the interchain cysteines of the CD30‐directed antibody cAC10 and subjected to potential deprotection conditions. We found that the Acm group could be readily removed with approximately 5–6 molar equivalents (per thiol) of aqueous mercury acetate in 45 min at neutral pH (Figure S1 in the Supporting Information). Subsequent removal of Cys‐bound mercury using an immobilized thiol source (Quadrasil MP resin) was required to liberate the thiol for drug‐linker conjugation (Figure S2). Importantly, the Acm group was stable to TCEP, ensuring that the two protecting groups could be removed orthogonally. In addition, the conditions for Acm removal do not impact antibody disulfide integrity (Figure S3), indicating that these conditions could be applied more generally for bio‐orthogonal protein modification. While the Acm protecting group has been used extensively in peptide synthesis and protein semi‐synthesis,13b, 14 to our knowledge this is the first use of an Acm‐masked Cys on a folded protein.
Drug carrier 4, bearing Cys(SiPr) and Cys(Acm) residues, was synthesized by solid‐phase peptide synthesis and conjugated to cAC10. The conjugation process and resulting analytics for an ADC bearing drug‐linkers 1 and 3 are shown in Figure 2. First, the antibody interchain disulfides were reduced using 12 equiv of TCEP at pH 7.4 and 37 °C for 90 min. Subsequently, the free sulfhydryls were each reacted with a two‐fold excess of carrier 4, yielding a conjugate with 8 carriers per antibody. The reaction status was checked using a reversed‐phase ultra‐performance liquid chromatography‐mass spectrometry (UPLC‐MS) method that separates light and heavy chain species based on drug (or carrier) loading. This analysis confirmed that the conjugate eluted as a single light and heavy chain, each with the expected mass, indicating full conversion to a homogeneous conjugate with 8 carriers per antibody (Figure 2 A). The cAC10–carrier conjugate was then subjected to reduction with 10 equiv of TCEP for 45 min at 37 °C to remove the S‐(isopropyl) disulfides. Analysis by UPLC‐MS confirmed that the reaction was complete (Figure S4). The un‐masked Cys residues were then conjugated with mc‐MMAF (1) using a 1.5 equiv of drug‐linker per thiol. UPLC‐MS analysis indicated conversion to an 8‐loaded conjugate within 15 min. In order to conjugate the second drug‐linker, the Acm protecting groups were un‐masked using 5–6 equiv of aqueous Hg(OAc)2 per thiol. Complete Acm un‐masking was observed by UPLC‐MS within 45 min at room temperature and neutral pH. Prior to addition of the drug‐linker, excess Hg2+ was captured using Quadrasil MP resin. Addition of 2.0 equiv of mc‐vc‐MMAE drug‐linker (3) per thiol resulted in complete reaction in 15 min, as assessed by UPLC‐MS. As shown in Figure 2, each step of the co‐conjugation process resulted in clean conversion to a single light and heavy chain, demonstrating high levels of homogeneity with an average of 16 drugs/mAb. The conjugate was analyzed by size‐exclusion chromatography (SEC), which demonstrated that the conjugate was 98 % monomeric (Figure S6). Lastly, cell binding analysis demonstrated that the conjugation process did not significantly impact cAC10 binding to CD30 (Figure S7).
Figure 2.
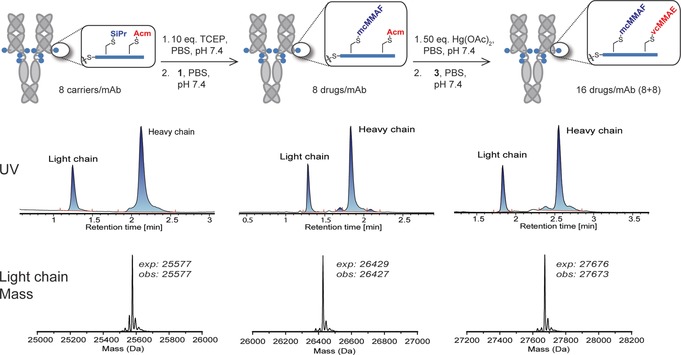
Multi‐drug ADC conjugation process and analytical characterization. Shown is a reaction schematic that includes the conditions for sequential unmasking of Cys(SiPr) and Cys(Acm) residues on carrier 4 and the resulting site‐specific drug‐linker conjugation. Each conjugate was analyzed by reverse‐phase UPLC‐MS. Shown below each intermediate is the UV chromatogram following reverse‐phase separation, and the de‐convoluted light chain mass. Each step proceeded with near quantitative conversion, yielding largely a single light and heavy chain species. The de‐convoluted heavy chain mass for each conjugate is provided in Figure S5.
In order to demonstrate the benefits of the approach described here, we compared the in vitro and in vivo activities of dual‐auristatin ADCs compared to 8‐loaded cAC10‐1, cAC10‐2, or cAC10‐3 ADCs on cells with differential sensitivities to the individual drugs. All ADCs tested, including single‐drug ADCs, employed drug‐linker conjugation to carrier 4, and for the single‐drug ADCs, the second Cys residue on carrier 4 was capped with N‐ethyl maleimide. We first investigated the activity of ADCs in systems with high MDR expression, where MMAE can have impaired activity due to drug transport. Cytotoxicity experiments were conducted in vitro on a cAC10‐vc‐MMAE resistant cell line, DEL‐BVR (Figure 3 A). This cell line was generated after prolonged exposure of the DEL anaplastic large cell lymphoma (ALCL) cell line to cAC10‐3, which renders it refractory to treatment with mc‐vc‐MMAE ADCs due to high MDR levels. On this cell line, cAC10‐3 was completely inactive at the highest concentrations tested (up to 2000 ng mL−1), whereas both cAC10‐2 and cAC10‐(2+3) retained potent activity (IC50<1.0 ng mL−1). In contrast, all three conjugates were highly active on parental DEL, with IC50 values below 1.5 ng mL−1. The dual‐drug conjugate was most potent on this cell line, owing to the increase in drug loading from 8 to 16 drugs per antibody. All three conjugates were inactive on CD30(−) U‐266 cells, demonstrating that activity was not due to insufficient drug‐linker stability. Encouraged by these results, we examined ADC activity in a mouse xenograft model using the DEL‐BVR cell line (Figure 3 B). At a dose of 3 mg kg−1, cAC10‐3 showed no activity. Meanwhile, the dual drug conjugate cAC10‐(2+3) displayed potent antitumor activity at the same dose, resulting in 3/5 cures. This activity was slightly better than cAC10‐2, which provided 1/5 cures. These results demonstrate that dual‐drug ADCs prepared using this approach are active in vivo and that insensitivity to MDR can be overcome through addition of a complementary payload.
Figure 3.
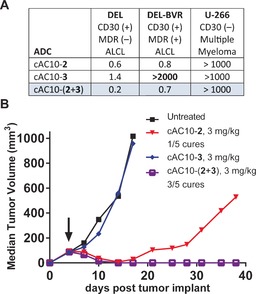
Dual‐drug ADC activity on MDR(+) DEL‐BVR cells in vitro (A) and in vivo (B). In vitro cytotoxicity values are reported as IC50 in ng mL−1 of ADC. ALCL=anaplastic large cell lymphoma.
We next sought to enhance the activity of MMAF‐based ADCs by adding the cell permeability and bystander activity of MMAE. In this case, cAC10‐1, cAC10‐3, and cAC10(1+3) were tested in an in vitro model of bystander activity, where MMAF drugs have little or no activity (Figure 4). In this assay, CD30(−) U‐266 multiple myeloma cells that express luciferase are cultured in a 1:1 mixture with CD30(+) L540cy cells. After treatment with the ADCs, the viability of the U‐266luc cells was assessed by decrease in luciferase activity, which would reflect the diffusion of active drug released within the CD30(+) L540cy cells. In this model, cAC10‐1 had minimal activity on the mixed co‐culture, since the released drug, cys‐mc‐MMAF, is not cell permeable. Meanwhile, cAC10‐3 displayed potent bystander activity with an IC50 of 3.9 ng mL−1, as did cAC10‐(1+3). All three conjugates were inactive on CD30(−) U‐266luc cells, and a non‐binding ADC bearing (1+3) had no activity on L540cy cells, demonstrating immunological specificity (Figure S8). Encouraged with these in vitro results, we tested the same conjugates in an in vivo model that has heterogeneous CD30 expression. The xenograft model consists of an admixed population of CD30(+) and CD30(−) Karpas 299 cells.7b In this model, cAC10‐1 showed only a modest tumor growth delay when dosed at 3 mg kg−1. Meanwhile, treatment with both cAC10‐3 and cAC10‐(1+3) provided a robust antitumor response at the same dose, resulting in 5/5 cures and 4/5 cures, respectively. Importantly, these results combined with those from the DEL‐BVR xenograft (Figure 3 B) demonstrate that dual‐auristatin ADCs are active on tumors that are both high in MDR expression and have heterogeneous antigen levels.
Figure 4.
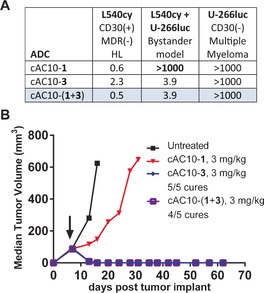
Dual‐drug ADC activity on in vitro (A) and in vivo (B) models that have heterogeneous CD30 expression. In vitro cytotoxicity values are reported as IC50 in ng mL−1 of ADC. The xenograft model consisted of a 1:1 mixture of Karpas 299 (CD30+) and Karpas 35R (CD30−) cells. HL=Hodgkin lymphoma.
In conclusion, we have developed a facile method for the construction of homogenous dual‐drug ADCs. This method is broadly applicable to a variety of antibodies, since recombinant technologies to achieve site‐specificity are not required. The inherent flexibility of this approach enables rapid screening of antibody and drug‐linker libraries to identify dual‐drug ADCs with improved activities. We have provided biological data demonstrating that dual‐drug ADCs are able to kill multiple types of cell populations, including both cells that lack CD30 antigen in mixed cell line models and those that possess MDR activity. Therefore, multi‐drug ADCs are active on cell types that are refractory to either of the individual component drugs. This work highlights the potential for a new class of targeted therapeutics where multiple drugs with complementary or synergistic activities are simultaneously delivered.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
M. R. Levengood, X. Zhang, J. H. Hunter, K. K. Emmerton, J. B. Miyamoto, T. S. Lewis, P. D. Senter, Angew. Chem. Int. Ed. 2017, 56, 733.
References
- 1.
- 1a. Senter P. D., Sievers E. L., Nat. Biotechnol. 2012, 30, 631–637; [DOI] [PubMed] [Google Scholar]
- 1b. Lambert J. M., Chari R. V., J. Med. Chem. 2014, 57, 6949–6964; [DOI] [PubMed] [Google Scholar]
- 1c. Polakis P., Pharmacol. Rev. 2016, 68, 3–19. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Lyon R. P., Setter J. R., Bovee T. D., Doronina S. O., Hunter J. H., Anderson M. E., Balasubramanian C. L., Duniho S. M., Leiske C. I., Li F., Senter P. D., Nat. Biotechnol. 2014, 32, 1059–1062; [DOI] [PubMed] [Google Scholar]
- 2b. Agarwal P., Bertozzi C. R., Bioconjugate Chem. 2015, 26, 176–192; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Chudasama V., Maruani A., Caddick S., Nat. Chem. 2016, 8, 114–119; [DOI] [PubMed] [Google Scholar]
- 2d. de Goeij B. E., Lambert J. M., Curr. Opin. Immunol. 2016, 40, 14–23; [DOI] [PubMed] [Google Scholar]
- 2e. Chari R. V., Miller M. L., Widdison W. C., Angew. Chem. Int. Ed. 2014, 53, 3796–3827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3872–3904. [Google Scholar]
- 3.
- 3a. V. T. DeVita, Jr. , Chu E., Cancer Res. 2008, 68, 8643–8653; [DOI] [PubMed] [Google Scholar]
- 3b. McGranahan N., Swanton C., Cancer Cell 2015, 27, 15–26; [DOI] [PubMed] [Google Scholar]
- 3c. Meacham C. E., Morrison S. J., Nature 2013, 501, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Fanale M. A., Horwitz S. M., Forero-Torres A., Bartlett N. L., Advani R. H., Pro B., Chen R. W., Davies A., Illidge T., Huebner D., Kennedy D. A., Shustov A. R., J. Clin. Oncol. 2014, 32, 3137–3143; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Younes A., Connors J. M., Park S. I., Fanale M., O'Meara M. M., Hunder N. N., Huebner D., Ansell S. M., Lancet Oncol. 2013, 14, 1348–1356. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Loganzo F., Tan X., Sung M., Jin G., Myers J. S., Melamud E., Wang F., Diesl V., Follettie M. T., Musto S., Lam M. H., Hu W., Charati M. B., Khandke K., Kim K. S., Cinque M., Lucas J., Graziani E., Maderna A., O'Donnell C. J., Arndt K. T., Gerber H. P., Mol. Cancer Ther. 2015, 14, 952–963; [DOI] [PubMed] [Google Scholar]
- 5b. Yu S. F., Zheng B., Go M., Lau J., Spencer S., Raab H., Soriano R., Jhunjhunwala S., Cohen R., Caruso M., Polakis P., Flygare J., Polson A. G., Clin. Cancer Res. 2015, 21, 3298–3306. [DOI] [PubMed] [Google Scholar]
- 6. Lyon R. P., Bovee T. D., Doronina S. O., Burke P. J., Hunter J. H., Neff-LaFord H. D., Jonas M., Anderson M. E., Setter J. R., Senter P. D., Nat. Biotechnol. 2015, 33, 733–735. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Kovtun Y. V., Audette C. A., Mayo M. F., Jones G. E., Doherty H., Maloney E. K., Erickson H. K., Sun X., Wilhelm S., Ab O., Lai K. C., Widdison W. C., Kellogg B., Johnson H., Pinkas J., Lutz R. J., Singh R., Goldmacher V. S., Chari R. V., Cancer Res. 2010, 70, 2528–2537; [DOI] [PubMed] [Google Scholar]
- 7b. Li F., Emmerton K. K., Jonas M., Zhang X., Miyamoto J. B., Setter J. R., Nicholas N. D., Okeley N. M., Lyon R. P., Benjamin D. R., Law C. L., Cancer Res. 2016, 76, 2710–2719; [DOI] [PubMed] [Google Scholar]
- 7c. Okeley N. M., Miyamoto J. B., Zhang X., Sanderson R. J., Benjamin D. R., Sievers E. L., Senter P. D., Alley S. C., Clin. Cancer Res. 2010, 16, 888–897. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Chen R., Hou J., Newman E., Kim Y., Donohue C., Liu X., Thomas S. H., Forman S. J., Kane S. E., Mol. Cancer Ther. 2015, 14, 1376–1384; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. O'Brien C., Cavet G., Pandita A., Hu X., Haydu L., Mohan S., Toy K., Rivers C. S., Modrusan Z., Amler L. C., Lackner M. R., Cancer Res. 2008, 68, 5380–5389. [DOI] [PubMed] [Google Scholar]
- 9. Doronina S. O., Mendelsohn B. A., Bovee T. D., Cerveny C. G., Alley S. C., Meyer D. L., Oflazoglu E., Toki B. E., Sanderson R. J., Zabinski R. F., Wahl A. F., Senter P. D., Bioconjugate Chem. 2006, 17, 114–124. [DOI] [PubMed] [Google Scholar]
- 10. Puthenveetil S., Musto S., Loganzo F., Tumey L. N., O'Donnell C. J., Graziani E., Bioconjugate Chem. 2016, 27, 1030–1039. [DOI] [PubMed] [Google Scholar]
- 11. Maruani A., Richards D. A., Chudasama V., Org. Biomol. Chem. 2016, 14, 6165–6178. [DOI] [PubMed] [Google Scholar]
- 12. Maruani A., Smith M. E., Miranda E., Chester K. A., Chudasama V., Caddick S., Nat. Commun. 2015, 6, 6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Yuan Y., Wang X., Mei B., Zhang D., Tang A., An L., He X., Jiang J., Liang G., Sci. Rep. 2013, 3, 3523; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Andreu D., Albericio F., Sole N. A., Munson M. C., Ferrer M., Barany G., Methods Mol. Biol. 1994, 35, 91–169. [DOI] [PubMed] [Google Scholar]
- 14. Maity S. K., Jbara M., Laps S., Brik A., Angew. Chem. Int. Ed. 2016, 55, 8108–8112; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8240–8244. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary