

Abstract
Latanoprost was first developed for use in glaucoma therapy as an ocular hypotensive agent targeting the prostaglandin F2α (FP) receptor. Subsequently, latanoprost showed a neuroprotective effect, an additional pharmacological action. However, although it is well‐known that latanoprost exerts an ocular hypotensive effect via the FP receptor, it is not known whether this is also true of its neuroprotective effect. Klotho was firstly identified as the gene linked to the suppression of aging phenotype: the defect of klotho gene in mice results aging phenotype such as hypokinesis, arteriosclerosis, and short lifespan. After that, the function of klotho was also reported to maintain calcium homeostasis and to exert a neuroprotective effect in various models of neurodegenerative disease. However, the function of klotho in eyes including retina is still poorly understood. Here, we show that klotho is a key factor underlying the neuroprotective effect of latanoprost during post‐axotomy retinal ganglion cell (RGC) degeneration. Importantly, a quantitative RT‐PCR gene expression analysis of klotho in sorted rat retinal cells revealed that the highest expression level of klotho in the retina was in the RGCs. Latanoprost acid, the biologically active form of latanoprost, inhibits post‐traumatic calpain activation and concomitantly facilitates the expression and shedding of klotho in axotomized RGCs. This expression profile is a good match with the localization, not of the FP receptor, but of organic anion transporting polypeptide 2B1, known as a prostaglandin transporter, in the ocular tissue. Furthermore, an organic anion transporting polypeptide 2B1 inhibitor suppressed latanoprost acid‐mediated klotho shedding ex vivo, whereas an FP receptor antagonist did not. The klotho fragments shed from the RGCs reduced the intracellular level of reactive oxygen species, and a specific klotho inhibitor accelerated and increased RGC death after axotomy. We conclude that the shed klotho fragments might contribute to the attenuation of axonal injury‐induced calpain activation and oxidative stress, thereby protecting RGCs from post‐traumatic neuronal degeneration.
Keywords: calpain, glaucoma, klotho, latanoprost
Abbreviations used
- DSL
d‐saccharic acid 1,4‐lactone
- ERG
electroretinograms
- FACS
fluorescence‐activated cell sorting
- GCL
ganglion cell layer
- INL
inner nuclear layer
- IOP
intraocular pressure
- IP
immunoprecipitation
- IPL
inner plexiform layer
- LA
latanoprost acid
- MMP
matrix metalloproteinase
- OAG
open‐angle glaucoma
- OATP2B1
organic anion transporting polypeptide 2B1
- ONL
outer nuclear layer
- pSTR
positive scotopic threshold response
- RGC
retinal ganglion cell
- ROS
reactive oxygen species
- RPE
retinal pigment epithelium
- TPA
twelve‐O‐tetradecanoylphorbol‐13‐acetate
Glaucoma is characterized an irreversible optic neuropathy and progressive loss of retinal ganglion cells (RGCs). Elevated intraocular pressure (IOP) is one of the risk factors and ocular hypotensive medication is commonly applied for the treatment of open‐angle glaucoma. Prostaglandin analog are mostly used as first choice for patients with open‐angle glaucoma because of their prominent efficacy at lowering intraocular pressure by increasing the uveoscleral outflow. Latanoprost, a prostaglandin F2α (PGF2α) analog with an ester group, is immediately converted to latanoprost acid (LA) by endogenous esterase as it passes through the corneal tissue into the anterior chamber after ocular instillation. The major pharmacological target of these prostaglandin‐related compounds is the PGF2α (FP) receptor, for which latanoprost has a lower affinity than its free acid hydrolytic product (Sharif et al. 2003). Latanoprost has a potent IOP lowering effect in wild‐type mice, but has no effect on IOP in FP receptor‐deficient mice (Ota et al. 2005). Thus, the FP receptor is considered to be the main mediator of the various pharmacological actions of LA, and to have a key role in the IOP‐reducing effect of latanoprost.
Latanoprost/LA also exert a neuroprotective effect in the retinal ganglion cells under the ischemia and reperfusion stresses (Emre et al. 2009). Several studies have demonstrated that latanoprost/LA protect the RGCs against neuronal degeneration after axonal injury, such as transection or crushing of the optic nerve (Kudo et al. 2006; Kanamori et al. 2009). Nevertheless, FP receptor expression is diffuse in the retina, in contrast to its much higher expression in the ocular anterior segment (Ocklind et al. 1997). The role of the retinal FP receptors in mediating the neuroprotective effect of latanoprost/LA in the RGCs is thus controversial.
It has been hypothesized that in addition to the FP receptor, organic anion transporting polypeptide 2B1 (OATP2B1) is also a pharmacological target of LA. This hypothesis was suggested by the role of OATP2B1 in transporting unoprostone carboxylate, another PGF2α analog (Gao et al. 2005). OATP2B1 is expressed in human retina (Kraft et al. 2010; Gao et al. 2015); however, there is currently little information on the physiological role of OATP2B1 in the retina.
The klotho gene was originally identified as an aging suppressor in an insertion mutagenesis analysis in mice. Klotho encodes a type‐I single‐pass transmembrane protein whose extracellular domain is composed of the KL1 and KL2 domains, homologous to β‐glucuronidase (Kuro‐o et al. 1997). After this extracellular domain is shed, it circulates in the urine, blood, and cerebrospinal fluid and exerts biological effects on target cells (Imura et al. 2004). Treatment with specific β‐glucuronidase inhibitors, such as d‐saccharic acid 1,4‐lactone (DSL), inhibits their effects (Chang et al. 2005). Moreover, defects in klotho disrupt calcium homeostasis (Kuro‐o et al. 1997) and cause the over‐activation of a calcium‐dependent protease, calpain (Manya et al. 2002). Many recent reports have found that calpain activation has a crucial role in post‐axonal injury neuronal degeneration, including in the RGCs (Ryu et al. 2012; Liu et al. 2014). Thus, klotho may be related to axonal injury‐induced calpain activation, but such a relationship has not yet been reported.
Recently, many studies have examined the expression and function of klotho in the posterior segment of the rodent eye. Klotho protein has strongly up‐regulation in degenerating photoreceptor in transgenic mouse and rat models of retinitis pigmentosa (Farinelli et al. 2013), and help preserve retinal pigment epithelium (RPE) function, and to protect the RPE from oxidative stress (Kokkinaki et al. 2013). Klotho is also known to exert a neuroprotective effect in various neurodegenerative disease models (Zeldich et al. 2014), suggesting that it should also have a similar effect in the RGCs. However, the biological function of klotho in the RGCs remains unclear.
The above leads us to hypothesize that LA promotes RGC neuroprotection through the OATP2B1 transporter, not the FP receptor, and that this suppresses excess calpain activation by modulating klotho activation. Thus, the main objectives of this study were to examine the mechanism of LA‐modulated neuroprotection through the OATP2B1 pathway, with a focus on the protein kinase C (PKC) pathway and matrix metalloproteinase (MMP) activation, and to investigate the antioxidant and RGC death‐reducing effects of klotho in rat retinas.
Material and methods
Reagents
Dulbecco's modified Eagle's medium, Neurobasal A medium, Dulbecco's phosphate‐buffered saline, Ca2+/Mg2+‐free Hanks’ buffered saline solution, B‐27 supplement with/without antioxidants and Alamar Blue reagent were purchased from Life Technologies Inc. (Carlsbad, CA, USA). Papain was purchased from Worthington (Lakewood, NJ, USA). AL8810 was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). Bovine serum albumin (BSA), deoxyribonuclease I (DNase I) from bovine pancreas, insulin from bovine pancreas, catalase from bovine liver, l‐cysteine, l‐glutamate, gentamicin, protease inhibitor cocktail, phosphatase inhibitor cocktail 2, LA, rifamycin SV, DSL, and 1,2‐bis (2‐aminophenoxy) ethane N,N,N′,N′‐tetraacetic acid acetoxymethyl ester were purchased from Sigma‐Aldrich (St Louis, MO, USA).
Twelve‐O‐tetradecanoylphorbol‐13‐acetate (TPA) was purchased from Cell Signaling Technology (Beverly, MA, USA). GM 6001, TAPI‐1, GF109203X, and calpain substrate IV were purchased from Calbiochem (San Diego, CA, USA).
Animals
Nine‐ to twelve‐week‐old male Sprague–Dawley rats were purchased from SLC (Shizuoka, Japan). The rats were maintained under a 12 h light (~ 50 lux)/dark cycle. Food and water were freely available. The animals in these experiments were used in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the Guidelines for Animal Experiments of Tohoku University. All animal experiments were conducted with the approval of the ethics committee for animal experiments at the Tohoku University Graduate school of Medicine. The protocol number approved by our Institute is 2016‐245.
Optic nerve transection
Optic nerve transection was performed as previously described (Nakazawa et al. 2002; Kudo et al. 2006). Briefly, after anesthesia was induced with an intramuscular injection of 45 mg/kg sodium pentobarbital (Kyoritsu Pharmaceuticals, Tokyo, Japan), 2 μL of one of the three pharmacological agents under investigation (100 μM LA dissolved in 5% dimethylsulfoxide (DMSO) and 100 μM DSL dissolved in saline) was injected intravitreally with a Hamilton syringe and a 32‐gauge needle. Fifteen minutes after the injection of LA and DSL, the optic nerve was transected at approximately 1 mm posterior to the eyeball, with care taken not to damage the retinal blood supply. Animals were excluded if the lens was injured during the course of intraocular injection.
Retrograde labeling of the RGCs and RGC counting
Retrograde labeling of the RGCs and RGC counting were performed as previously described (Kudo et al. 2006; Nakazawa et al. 2007). Briefly, 7 days before axotomy, 3 μL of 2% aqueous Fluorogold (Fluorochrome; Englewood, CO, USA) containing 1% DMSO was injected into the optic nerve sheath. After the animals were killed, the retinas were dissected, fixed in 4% paraformaldehyde (PFA), flat‐mounted onto glass slides and examined under microscopy. The RGC number was then determined by counting retrogradely labeled RGCs in 12 distinct areas (three areas per quadrant, at 1/6, 3/6, and 5/6 of the distance from the optic nerve head to the periphery, all located along retinal radii). Finally, the average number of retrogradely labeled RGCs in each of the 12 fields was determined and used in the analysis. RGC counting was conducted in a blind fashion. Briefly, one investigator captured and labeled images of masked specimens. Another investigator then counted retrogradely labeled RGCs under masked conditions.
Recording ERGs
Three days and fourteen days after surgery, scotopic electroretinograms (ERGs) were recorded from both eyes simultaneously using a Ganzfeld bowl mounted with a stimulator (PS33‐PLUS; Grass Instruments, East Falmouth MA, USA). The rats were dark adapted overnight and handled under dim red light. They were anesthetized by a single intramuscular injection of a mixture of ketamine (87 mg/kg; Daiichi Sankyo Co., Ltd., Tokyo, Japan) and xylazine (13 mg/kg; Bayer Yakuhin Ltd., Osaka, Japan). The pupils were maximally dilated by topical 0.5% tropicamide (Santen Pharmaceutical Co., Ltd., Osaka, Japan) and 0.5% phenylephrine hydrochloride (Sigma‐Aldrich, Tokyo, Japan), and the cornea was anesthetized with topical 0.4% oxybuprocaine hydrochloride (Santen Pharmaceutical Co., Ltd.). Gold contact lens electrodes (Mayo Co., Nagoya, Japan) were placed on the cornea as active electrodes, and reference electrodes were placed in the mouth. An aluminum sheet under the animals served as the ground electrode. Responses were amplified 105 times and bandpass‐filtered from 0.3 to 500 Hz (PC‐100; Mayo Co.). The intensity of the stimuli was increased by increasing the threshold of the scotopic threshold response (STR) to 0.43 log cd‐s/m2 in 0.23, 0.37, or 0.40 log unit steps. The duration of the stimulus was 10 μs, and the maximum luminance was 0.43 log cd‐s/m2. Approximately 20–30 responses were averaged in computer software, with an inter‐stimulus interval of 5 s for recording the STR.
The STR consists of positive and negative components: the positive STR (pSTR) and negative STR, respectively (Sieving et al. 1986; Sieving and Nino 1988; Frishman et al. 1996). Previous research has demonstrated that the RGCs and their axons are the main generators of the pSTR (Bui and Fortune 2004). We measured the amplitude 120 ms after the onset of the stimulus from the baseline or the trough of the a‐wave, following a previously reported method (Bui and Fortune 2004). The amplitude in the axotomized eyes was normalized to that of the contralateral non‐axotomized control eyes, with the normalized amplitude described as the axotomy/control ratio.
Preparation of klotho‐containing supernatant for application in primary retinal cells, and signaling pathway analysis in an ex vivo axotomy model
The rat retinas were immediately dissected in ice‐cold Dulbecco's phosphate‐buffered saline, placed in microtubes, and incubated in a HEPES‐based buffer (composed of 100 mM NaCl, 3 mM KCl, 1 mM MgCl2, 10 mM d‐glucose, 25 mM HEPES, 10 mM mannitol, and 30 mM NaHCO3 pH 7.4) containing various concentrations of LA (0.1, 10, or 1000 nM).
To assess the inhibitory effect of each signaling pathway, 1 μM LA and an additional agent (one of the following: 50 μM AL8810, rifamycin SV, 10 μM TAPI‐1, GM6001 or 100 nM GF109203X) were incubated in the rat retinas at 37°C for 90 min. No LA was used for TPA treatment at 200 nM. To remove the detached cells, the supernatants were centrifuged at 15 000 g for 5 min at 4°C, and then concentrated with an Amicon ultra‐10 centrifugal filter (Merck‐Millipore, Darmstudt, Germany) at 4000 g for 20 min. For immunoprecipitation, a half‐quantity of supernatant (100 μL) was incubated with rat anti‐klotho antibody (1 μg, KO606; TransGenic Inc.) at 4°C for 1 h. Protein G Mag Sepharose (2.5 μL; Piscataway, GE Healthcare, WI, USA) was then added, and the sample was incubated at 4°C for 1 h. The non‐treated and klotho‐depleted supernatants were moved into a Neurobasal A culture medium containing 2% B‐27 supplement without antioxidants (AO−; Life Technologies Inc.), 5 μg/mL insulin, 0.5 mM l‐glutamate and 250 μg/mL gentamicin, using an Amicon Ultra‐10 centrifugal filter (Millipore Corporation). The protein concentration was determined with a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Hudson, NH, MA, USA).
Primary retinal cultures for the measurement of cell viability and ROS generation
Adult rat primary retinal cultures were prepared as previously described, with minor modifications (Yin et al. 2006; Nakazawa et al. 2007). The rat retinas were incubated at 37°C for 15 min in a digestion solution containing 10 units/mL papain, 75 μg/mL BSA, 10 μg/mL DNase I, 5 μg/mL insulin, 1 μg/mL catalase, and 0.3 mg/mL l‐cysteine in Ca2+/Mg2+‐free Hanks’ buffered saline solution. The dissociated retinal cells were passed through a 40 μm nylon mesh and collected by centrifugation at 900 g for 5 min. The cells were re‐suspended in AO− culture medium, and cell density was adjusted to 1.8 × 105 cells per well of a CC2 surface‐treated 96‐well plate (Thermo Fisher Scientific). Fifteen minutes after incubation at 37°C, the adhered retinal cells were incubated in either non‐treated or klotho‐depleted supernatant (300 μg protein/well). B‐27 supplement with an antioxidant (AO+; Life Technologies Inc.) was used as a positive control. Two hours later, the cells were incubated in AO+ or AO− culture medium containing 10% AlamarBlue reagent (Life Technologies Inc.) at 37°C for 24 h in the dark. Fluorescence intensity was measured at 544 nm excitation and 590 nm emission with a fluorescence microplate FluoroskanAscent reader (Thermo Fisher Scientific). For the measurement of intracellular reactive oxygen species (ROS) levels, the cells were incubated with AO− culture medium containing 5 μM CellROX Deep Red reagent (Life Technologies Inc.) at 37°C for 30 min. Cell fluorescence was then measured at 640 nm excitation and 665 nm emission with a Spectra Max Gemini microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Cell sorting
The rat retinas were digested in a solution containing papain and dissociated as described above. In all steps, including suspension, washing and incubation, an AO+ culture medium was used. Mouse IgG1κ antibody (553447; BD Biosciences, San Jose, CA, USA) was used as an isotype control. The cells were re‐suspended and reacted with mouse anti‐Thy‐1.1 antibody (1 : 20; MAB1406; Millipore Corporation) at 25°C for 30 min. After washing twice, the samples were incubated with an Alexa Fluor 647 donkey anti‐mouse immunoglobulin (IgG) secondary antibody (1 : 200; A31571; Life Technologies Inc.) at room temperature for 30 min and again washed twice. For double staining, the cells were incubated with mouse anti‐CD31 antibody (1 : 10, 550300; BD Pharmingen, San Jose, CA, USA) pre‐labeled with Zenon Alexa Fluor 350 (Life Technologies Inc.) at 25°C for 30 min. After washing twice, the cells were re‐suspended in medium containing 0.5% 7‐aminoactinomycin D (Life Technologies Inc.) to exclude dead cells, and were immediately sorted using fluorescence‐activated cell sorting with the Aria II device (BD Biosciences). The cells were sorted, placed into 350 μL of QIAzol Lysis Reagent (Qiagen, Hilden, Germany) and analyzed with qRT‐PCR. The number of sorted cells in each fraction was matched, and the same number of pre‐sorted cells was used as a reference for klotho expression in the naïve rats.
Real‐time quantitative RT‐PCR
To measure klotho gene expression, the RPE/choroid was removed as a sheet by scraping it from the scleral wall. It was collected following removal of the retina from the eyecup on day 3. The retina, RPE/choroid, and the sorted cells were directly lysed in QIAzol Lysis Reagent (Qiagen). Subsequent RNA extraction was performed with the miRNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Total RNA (50 ng) was reverse transcribed using a SuperScript III First Strand Synthesis kit (Life Technologies Inc.) to synthesize cDNA. Real‐time quantitative RT‐PCR was carried out with a 7500 fast real‐time PCR system (Applied Biosystems, Foster city, CA, USA) using TaqMan probes (Life Technologies Inc.). The catalog numbers of the predesigned TaqMan probes were as follows: Klotho (Rn00580123_m1) and Gapdh (Rn01462662_g1). Relative gene expression levels were calculated using the delta‐delta Ct method.
Stimulation of klotho shedding in ARPE19 cells with LA
Human RPE cell line ARPE19 cells were incubated in serum‐free Dulbecco's modified Eagle's medium containing 1 nM LA for 24 h at 37°C. The culture medium was then collected and centrifuged at 15 000 g for 5 min at 4°C to remove the detached cells. The supernatant was collected and concentrated with an Amicon Ultra‐30 centrifugal filter (Millipore Corporation) at 4000 g for 20 min. The samples were mixed with loading buffer (65.8 mM Tris‐HCl pH 6.8, 26.3% glycerol, 2.1% sodium dodecyl sulfate (SDS), 0.01% bromophenol blue) and then used in an immunoblot analysis.
Western blotting
Seven days after axonal injury, we performed an immunoblot analysis to evaluate the status of calpain and calpastatin. The retinas were lysed in radioimmunoprecipitation assay buffer (25 mM Tris‐HCl pH 7.6, 150 mM NaCl, 1% NP‐40, 1% sodium deoxycholate, 0.1% SDS) containing 1% protease inhibitor cocktail and 1% phosphatase inhibitor cocktail 2, on ice, and centrifuged at 15 000 g for 10 min at 4°C. To detect klotho in the retinal and ARPE19 cells, HEPES‐buffered saline with Triton X‐100 buffer (10 mM HEPES, 150 mM NaCl, 0.5% Triton X‐100) containing 1% protease and phosphatase inhibitor cocktail was used in the protein extraction. The supernatant of the cell lysate was collected and its protein concentration was determined with a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). After heat treatment in the loading buffer at 98°C for 5 min, the protein samples were separated in a 4–15% SDS‐polyacrylamide gradient gel (Bio‐Rad Laboratories, Hercules, CA, USA) and electroblotted to an Immobilon‐P polyvinylidene difluoride membrane (Millipore Corporation). After blocking the membrane with 4% BlockAce (Yukijirushi, Sapporo, Japan), the membranes were incubated with primary antibodies such as rat anti‐klotho (1 : 1000, KO603; TransGenic Inc.), mouse anti‐α‐fodrin (1 : 1000, ab11755; Abcam, Cambridge, UK), rabbit anti‐calpastatin (1 : 1000, 4146S; Cell Signaling Technology), rabbit anti‐fibroblast growth factor 2 (1 : 200; sc‐79; Santa Cruz Biotechnology, Santa Cruz, Delaware, CA, USA), rabbit anti‐β‐tubulin (1 : 1000, 2146S; Cell Signaling Technology Inc.) and mouse anti‐β‐actin (1 : 5000, A5316; Sigma‐Aldrich) overnight at 4°C. The membranes were then incubated with one of the following secondary antibodies at 25°C for 1 h: horseradish peroxidase‐conjugated goat anti‐mouse IgG (1 : 5000, G21040; Life Technologies Inc.), goat anti‐rat IgG (1 : 5000, A10549; Life Technologies Inc.), or goat anti‐rabbit IgG (1 : 5000, G21234; Life Technologies Inc.). The signals were visualized with the ECL Prime Western Blotting Detection System (GE Healthcare) and measured in Image Lab statistical software (Bio‐Rad).
Immunohistochemistry
Immunostaining was performed 3 days after surgery. Immunohistochemistry was performed as previously described (Nakazawa et al. 2007; Ryu et al. 2012). In brief, the rats were perfused with ice‐cold saline, followed by 2.5% PFA. The eyes of the rats were then collected and fixed in 2.5% PFA for 1 h on ice. Following fixation, the eyes were cryopreserved with increasing concentrations of sucrose (5%, 10%, and 20%) and frozen in Tissue‐Tek OCT compound (Sakura Finetechnical, Tokyo, Japan). After washing with phosphate‐buffered saline (PBS), cryosections were incubated with blocking buffer (10% goat serum, 0.5% gelatin, 3% BSA, and 0.2% Tween 20 in PBS) for 30 min and reacted with primary antibodies, such as rat anti‐klotho (1 : 500, KO606; TransGenic Inc.), rabbit anti‐OATP2B1 (1 : 200, BS‐3913R; Bioss Antibodies Inc., Woburn, MA, USA.), and mouse anti‐C38 (1 : 200; provided by Dr. Jun Kosaka) at 4°C overnight. They were washed three times with PBS containing 0.2% Tween 20 (PBST) and incubated with the following secondary antibodies at 25°C for 1 h: Alexa Fluor 488‐conjugated goat anti‐rat IgG (1 : 200, A11006; Life Technologies Inc.), Alexa Fluor 488‐conjugated goat anti‐rabbit IgG (1 : 200, A11034; Life Technologies Inc.), and Alexa Fluor 546‐conjugated goat anti‐mouse IgG (1 : 200, A11030; Life Technologies Inc.). After washing three times with PBST, the sections were mounted in Vectashield mounting medium with 4′,6‐diamidino‐2‐phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA) and observed using a fluorescence microscope (Axiovert 200; Carl Zeiss, Oberkochen, Germany).
In situ detection of calpain activation
Seven days after optic nerve transection in the presence or absence of LA, the anesthetized rats were injected intravitreally with 2 μL of 100‐μM calpain substrate IV dissolved in saline, with a Hamilton syringe and a 32‐gauge needle. Two hours after injection, the rats were perfused with 10 μM 1,2‐bis (2‐aminophenoxy) ethane N,N,N′,N′‐tetraacetic acid acetoxymethyl ester dissolved in saline, followed by 4% PFA. The eyes of the rats were then collected and fixed in 4% PFA for 1 h on ice. Following fixation, the eyes were cryopreserved with increasing concentrations of sucrose (5%, 10%, and 20%) and frozen in Tissue‐Tek OCT compound (Sakura Finetechnical). Twenty‐micrometer‐thick cryosections were mounted in Vectashield mounting medium (Vector Laboratories) and observed using a fluorescence microscope (Axiovert 200; Carl Zeiss).
Statistical analysis
We used an unpaired t‐test to evaluate statistical differences between pairs of samples. An anova followed by Dunnett's test was used to compare the mean between groups of three. A one‐way repeated‐measures anova with a post hoc Bonferroni test was used to obtain the data shown in Fig. 2. The data in Figure S1 were obtained with an anova followed by Tukey's multiple comparison test. The level of statistical significance was set at p < 0.05.
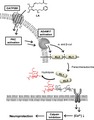
Figure 2.
Functional recovery of the electrophysiological response after latanoprost acid (LA) treatment in axotomized retinas. The figures show representative electroretinogram waveforms elicited by dim stimuli from axotomized eyes that were not treated with LA (a) and were treated with LA (200 pmol/eye) (b). Averaged traces of the scotopic threshold response (STR), measured at dim luminous energies (−6.20 to −4.57 log cd‐s/m2) in axotomized eyes treated without or with LA. Thin line: contralateral eyes; bold line: treated eyes. (c) The amplitude of the axotomized eyes was normalized to that of the contralateral non‐axotomized eyes, and this was designated as the axotomy/control ratio. White circles: control eyes; black circles: axotomized eyes; shaded squares: axotomized eyes treated with LA. Effect of LA on the relative positive STR amplitude on day 3 (d) and on day 14 (e) after axotomy. The data were normalized to the mean of the controls. (**p < 0.01, n = 6 in each group). Error bars denote SD.
Results
The inhibitory effect of LA on the post‐traumatic reduction in RGC in rat retina
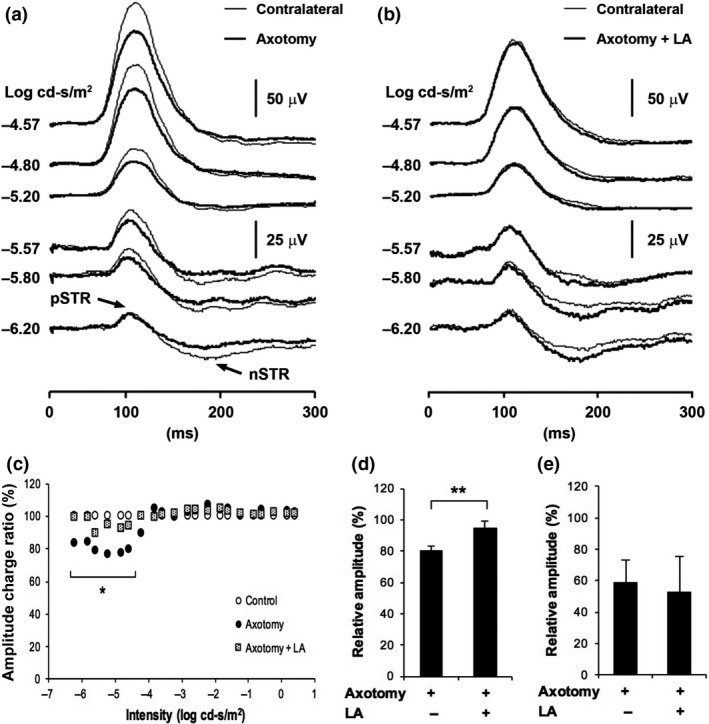
Previously, we demonstrated that latanoprost prevented RGC death following optic nerve transection by counting surviving retrogradely labeled RGCs (Kudo et al. 2006). Our current results reflect this previous finding. Here, we found that when we treated the animals with LA 15 min before axotomy, the number of surviving RGCs 7 days later was 2272 ± 555 RGCs/mm2, whereas when we treated them with 5% DMSO as vehicle, the number was 1478 ± 159 RGCs/mm2. We normalized the number of retrogradely labeled RGCs to the number in a group treated with saline as vehicle in axotomized retina and the ratio of surviving retrogradely labeled RGCs after treatment with 5% DMSO was 70.5 ± 7.6%, and the ratio after treatment with LA was 108.4 ± 26.5% in the axotomized retinas. This was a statistically significant difference (Fig. 1a and b). Our results demonstrated that the RGC survival rate was significantly higher in the LA‐treated group than the vehicle‐treated group.
Figure 1.
Latanoprost acid (LA) inhibited axotomy‐induced retinal ganglion cell (RGC) death. (a) Representative images of retrogradely labeled RGCs on day 7 after optic nerve transection. The vehicle was saline containing 5% dimethylsulfoxide. Scale bar = 50 μm. (b) The intravitreal injection of LA (200 pmol/eye) 15 min before axotomy significantly suppressed cell death in axotomized RGCs on day 7 after axotomy. RGC numbers were normalized to the axotomized retina treated with saline as vehicle, expressed as a percentage (*p < 0.05, n = 4 in each group). Error bars denote SD.
Functional recovery of the electrophysiological response after LA treatment in axotomized retinas
Next, we evaluated the physiological function of the retinas with an ERG analysis to determine whether LA treatment could prevent axotomy‐induced degeneration of the RGCs. Scotopic ERGs were recorded from six animals using dim stimuli ranging from −6.20 to 0.43 log cd‐s/m2. The ERGs consist of the pSTR followed by the negative STR. On day 3 after optic nerve transection, the amplitude of the pSTR measured at 120 ms was attenuated at all intensities in the axotomized eyes not treated with LA, in comparison with the contralateral eyes (Fig. 2a). On the other hand, the pSTR amplitude in the axotomized eyes treated with LA was nearly identical to the control eyes (Fig. 2b). The mean axotomy/control ratio in the animals which were treated or not treated with LA was plotted against stimulus intensity, as shown in Fig. 2(c). In the eyes not treated with LA (the black dots) the axotomy/control ratio fell to approximately 80% at stimulus intensities ranging from −6.20 to −4.57 log cd‐s/m2, whereas it remained close to 100% at stimulus intensities over −4.20 log cd‐s/m2. The six lowest stimulus intensities (indicated by the asterisks in Fig. 2c) show the most significant difference between the LA‐treated and untreated eyes, and we therefore averaged the axotomy/control ratio at these intensities to evaluate the function of the RGCs. We defined this average value as the relative amplitude, as shown in Fig. 2(d). The relative amplitude remained at 95% in the axotomized eyes treated with LA, whereas it fell to 80% in the axotomized eyes not treated with LA, a significant difference (relative amplitude in the control group was normalized to 100%) (Fig. 2d). Thus, these results constitute evidence that LA can preserve the function of the RGCs in axotomized eyes. On the other hand, LA treatment did not restore pSTR amplitude on day 14 after axotomy (Fig. 2e). These results suggest that LA‐mediated electrophysiological preservation was effective temporarily.
LA inhibits calpain activation and calpastatin breakdown after optic nerve transection
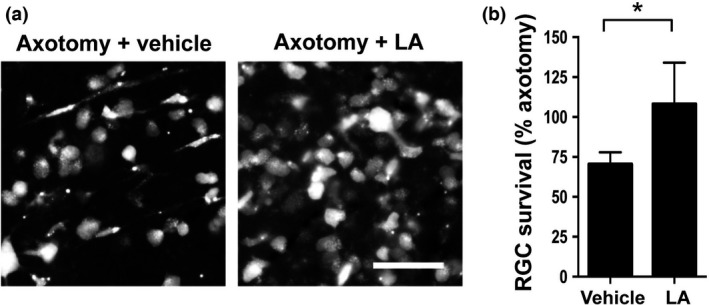
LA is known to have the ability to regulate the intracellular Ca2+ level (Kanamori et al. 2009), prompting us to investigate the potential of LA to suppress post‐injury calpain activation in the retina. As shown in Fig. 3(a), on the seventh day after optic nerve transection, LA had clearly inhibited the axotomy‐induced degradation of α‐fodrin, a prominent substrate for μ‐calpain. The level of cleaved α‐fodrin in the vehicle‐treated group rose significantly, by 10.8‐fold, whereas the level of cleaved α‐fodrin in the LA‐treated group was comparable with the non‐treated group (Fig. 3b). The level of calpastatin, an endogenous μ‐calpain inhibitor, decreased to 12.3% of the normal control level in the vehicle‐treated group. LA treatment significantly ameliorated this decrease; the level of calpastatin decreased to 43.5% of the normal level in the LA‐treated group (Fig. 3c). A fluorescence imaging analysis using a fluorogenic cell‐penetrating calpain substrate revealed calpain activation signals in the ganglion cell layer (GCL). These signals were minimal in both layers after axotomy and LA treatment (Fig. 3d).
Figure 3.
Latanoprost acid (LA) inhibited calpain activation and calpastatin breakdown after optic nerve transection. (a) LA (200 pmol/eye) inhibited the axotomy‐induced degradation of α‐fodrin, a prominent substrate for μ‐calpain, and calpastatin. The full‐length and cleaved forms of α‐fodrin are estimated to comprise 240 and 150 kDa peptides, respectively. Beta‐actin was used as an internal control. (b and c) Quantitative analysis of axotomy‐induced calpain activation and calpastatin breakdown in whole retinas 7 days after axotomy. The signal intensity of each band was normalized to 1 in the non‐treated group. (*p < 0.05, **p < 0.01, n = 5 in each group). Error bars denote SD. (d) Representative fluorescence images of frozen sections showing the results of treatment with a fluorogenic cell‐penetrating calpain substrate (2 μL of 100‐μM) in the eyes. With LA treatment, the signal of the cleaved calpain substrate, which was mainly detected in the GCL, was lower on day 7 after axonal injury. The vehicle was saline containing 5% dimethylsulfoxide. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bar = 50 μm.
LA‐mediated promotion of klotho expression and shedding in the RPE and retina
It has been reported that klotho is a significant upstream regulator affecting μ‐calpain activity. To investigate the expression pattern of klotho in the posterior of the rat eyes, we examined the levels of mRNA expression and protein expression in the RPE/choroid complex or retina during the early stages of axonal injury, before RGC loss. Three days after axotomy, we observed a reduction in klotho expression in the RPE/choroid after vehicle treatment, whereas its expression increased by 3.8‐fold after the topical application of LA (Fig. 4a). In the retina, although the mRNA level of klotho decreased by 5.0‐fold, the expression level of klotho after LA treatment was comparable to the control group. We also found that the transcription level of klotho in the RPE/choroid was approximately fourfold higher in the non‐axotomized LA‐treated eyes than the non‐axotomized, vehicle‐treated eyes. Interestingly, however, the level of klotho mRNA in the overall retinal tissue was lower (less than 10%) in the non‐axotomized LA‐treated eyes than the non‐axotomized vehicle‐treated eyes (Figure S1). This suggests that our original finding of elevated klotho in the RPE/choroid of axotomized, LA‐treated eyes were indeed because of the effect of LA treatment. The extracellular domain of klotho is composed of two homologous domains (KL1 and KL2) and possesses two cleavage sites (Fig. 4b) (Imura et al. 2004). Thus, klotho shedding produces three types of extracellular fragments: KL1, KL2, and soluble full‐length klotho. We found that LA inhibited the post‐injury reduction in KL2 in the retina and increased the protein level of klotho on day 3 (Fig. 4c). The level of KL1 in the axotomized retinas did not significantly differ with the presence or absence of LA. Matching this KL2 expression pattern, klotho elevation was also observed in the GCL of the axotomized rat retinas (Fig. 4d).
Figure 4.
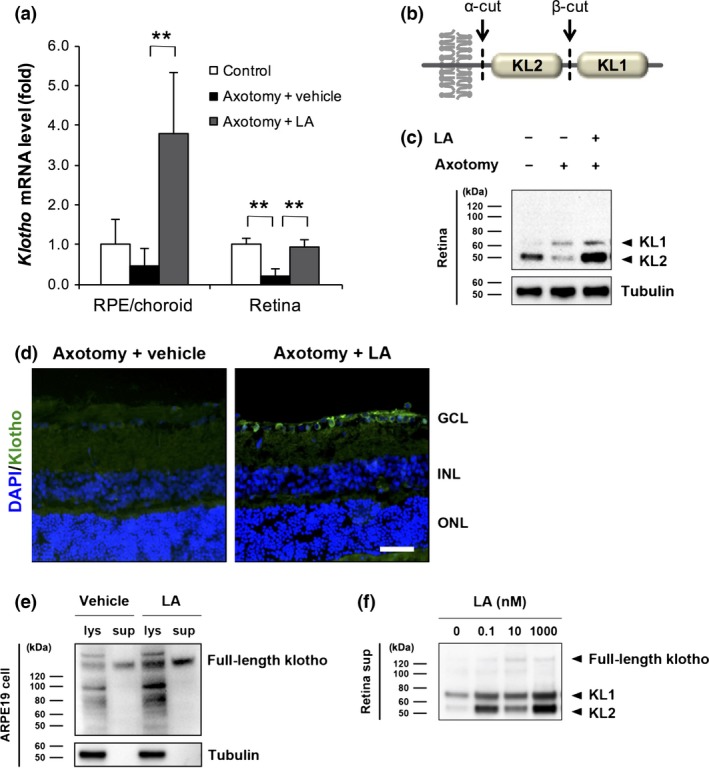
Latanoprost acid (LA)‐mediated promotion of klotho expression and shedding in the retinal pigment epithelium (RPE) and retina. (a) Three days after optic nerve transection, the intravitreal injection of LA (200 pmol/eye) significantly increased the mRNA level of klotho in the RPE/choroid and retina. The vehicle was saline containing 5% dimethylsulfoxide (DMSO). Gapdh was used as an internal control (**p < 0.01, n = 4 in each group). Error bars denote SD. (b) Schematic diagram of the transmembrane klotho structure and klotho shedding. The extracellular domain of klotho protein is composed of two homologous domains, KL1 and KL2. The extracellular domain of klotho possesses two cleavage sites (α‐cut and β‐cut), with three types of fragments (KL1, KL2 and soluble full‐length klotho) being produced extracellularly as a result of klotho shedding. (c) LA inhibited the post‐injury reduction in KL2 in the retina and increased the protein level of klotho on day 3. The level of KL1 in the axotomized rats did not significantly differ with or without LA. Tubulin was used as an internal control. (d) Immunostaining of klotho in retinal sections 3 days after optic nerve transection in LA‐treated rats. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bar = 50 μm. (e) In human ARPE19 cells, the application of LA at 1 nM for 24 h promoted the expression and shedding of full‐length klotho in both cell lysate (lys) and culture supernatant (sup). The vehicle was a culture medium containing 0.1% DMSO. Tubulin was used as an internal control. Neither KL1 nor KL2 were observed in the ARPE19 cells. (f) The promotion of klotho shedding from primary retinal cells at various concentrations of LA (0.1, 10, and 1000 nM).
To investigate further whether klotho is expressed in RGCs of SD rats, we carried out a flow cytometric analysis using antibodies against Thy1, and used CD31, an endothelial cell specific marker, to determine klotho localization in the rat retina. The mean percentages of Thy1−/CD31−, Thy1+/CD31−, Thy1−/CD31+, and Thy1+/CD31+ cells were 49.9%, 6.7%, 0.0%, and 0.5%, respectively (Figure S2b). In this study, the Thy1+/CD31−, Thy1+/CD31+, and Thy1−/CD31− cells were categorized as either RGCs, vascular endothelial cells, or other retinal cells. Real‐time PCR revealed a significantly lower level of klotho mRNA in the Thy1−/CD31− cells (0.3‐fold), a level that was comparable to that in the vascular endothelial cells (Figure S2c). Sorting significantly increased the mRNA level of klotho in the RGCs, by 2.7‐fold. Furthermore, klotho and the RGC marker C38 were highly co‐localized in the GCL of the naïve rats (Figure S2d).
In turn, we examined klotho expression changes after LA treatment in human RPE cell line ARPE19 cells. We found that LA promoted both full‐length soluble klotho expression and shedding in ARPE19 cells after 24 h (Fig. 4e). No tubulin signals were observed in the supernatant fractions, indicating that they did not include an ARPE19 cell fraction. Unlike the retina, KL1 and KL2 fragments were not observed even in the presence of LA. Thus, we investigated the potential of the retina as a source of shed klotho, as this would provide us with different forms of klotho. We incubated rat retinas in a HEPES‐based buffer containing LA at 0.1, 10, and 1000 nM for 90 min, which we found promoted shedding of KL1 and KL2 (Fig. 4f). The immunoreactive band of the full‐length soluble klotho was faint, whereas the KL1 and KL2 fragments predominated outside the cells. In particular, KL2 fragments were very highly expressed in the supernatant. Nevertheless, we did not observe a dose‐dependent, linear relationship between the strength of KL2 secretion and LA concentration.
Ex vivo analysis of the LA‐mediated klotho shedding pathway
LA is well known as an agonist of the FP receptor. To identify the LA‐mediated klotho shedding pathway, we investigated the involvement of FP receptor signaling using the FP receptor antagonist AL8810. Immunoblot analysis of supernatants revealed that AL8810 had only a modest inhibitory effect on LA‐mediated klotho shedding in the axotomized retinas (Fig. 5a). To confirm that LA activated the FP receptors in the retina, we checked the up‐regulation of fibroblast growth factor 2, a downstream factor of FP receptor activation (Sales et al. 2007) whose up‐regulation in inhibited by AL8810. We also found that LA‐mediated klotho shedding was attenuated when OATP2B1, another pharmacological target of LA, was inhibited by the application of rifamycin SV (Fig. 5b). In addition, the LA transporters OATP2B1 and C38 were highly co‐localized in the GCL of naïve rats (Fig. 5c). An immunoreactive signal for OATP2B1 was barely observed in the inner nuclear layer. Moreover, inhibiting PKC with GF109203X reduced the level of klotho shedding in comparison with the LA‐treated group (lanes 2 and 3; Fig. 5d). Conversely, TPA‐induced PKC activation promoted the shedding of klotho from the retina in comparison with the non‐treated group (lanes 1 and 4). Furthermore, inhibiting MMP with GM6001 suppressed LA‐mediated klotho shedding in comparison with the LA‐treated group (lanes 2 and 6). Inhibiting metalloproteinase 17 (ADAM17) with TAPI‐1 also suppressed LA‐mediated klotho shedding, as GM6001 treatment did, and more than inhibiting MMP (lanes 2, 5, and 6).
Figure 5.
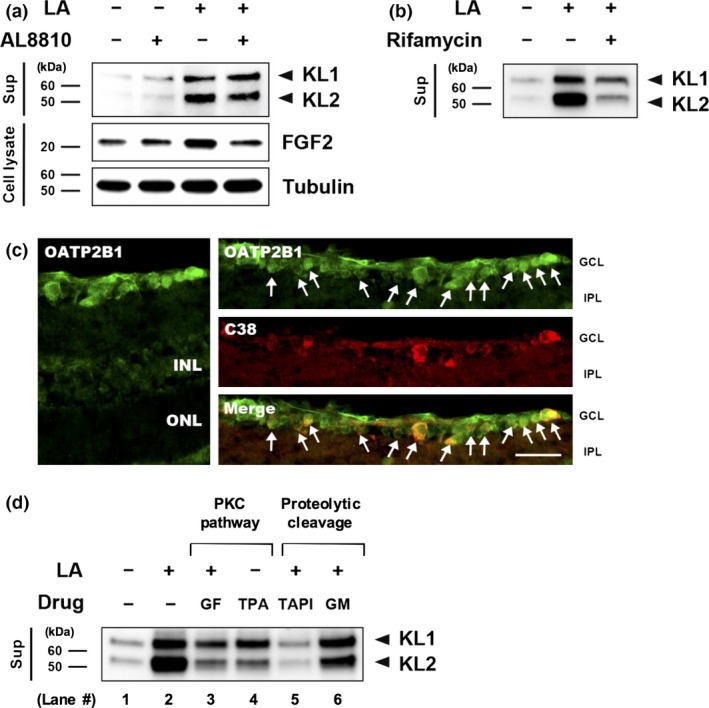
Ex vivo analysis of the latanoprost acid (LA)‐mediated klotho shedding pathway. (a) The FP receptor antagonist AL8810 (50 μM) inhibited LA‐mediated klotho shedding. The non‐treated retinas were incubated in a buffer containing 0.2% dimethylsulfoxide. Tubulin was used as an internal control. (b) Rifamycin SV (50 μM), a potent organic anion transporting polypeptide 2B1 (OATP2B1) inhibitor, attenuated LA‐mediated klotho shedding. (c) The LA transporter OATP2B1 was co‐localized with C38 in the GCL. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bar = 50 μm. (d) protein kinase C (PKC) (GF: GF109203X, 100 nM) and ADAM17 (TAPI: TAPI‐1, 10 μM) inhibitors, as well as a broad spectrum matrix metalloproteinase inhibitor (GM: GM6001, 10 μM), blocked klotho shedding, whereas a PKC activator (twelve‐O‐tetradecanoylphorbol‐13‐acetate, TPA) promoted klotho shedding.
Antioxidative and protective effects of shed klotho on retinal cells
These findings reported above prompted us to investigate the neuroprotective effect of klotho fragments. In addition to being a calcium homeostasis regulator, klotho is known to be an oxidative stress regulator (Yamamoto et al. 2005; Haruna et al. 2007; Zeldich et al. 2014). Thus, we evaluated the antioxidative effects of shed klotho on primary retinal cells, and the contribution of shed klotho to retinal protection in the absence of antioxidants. To deplete klotho protein, we performed immunoprecipitation with an anti‐klotho antibody and collected the supernatant. The supernatant included a detectable amount of KL2, but not KL1 (Fig. 6a). Measuring ROS generation with a CellROX fluorescent probe showed that treatment with a pre‐immunoprecipitation, klotho fragment‐containing supernatant reduced oxidative stress to 89% after 24 h, whereas treatment with a post‐immunoprecipitation supernatant containing a low amount of KL2 did not lead to any changes in oxidative stress compared to a non‐treated group (Fig. 6b). In the absence of antioxidants, the results of an Alamar blue assay revealed that the viability of primary retinal cells decreased to 81% of positive control values after 24 h, whereas cell viability did not significantly change after treatment with a low amount of KL2 containing supernatant, and furthermore, the ROS level decreased (Fig. 6c).
Figure 6.
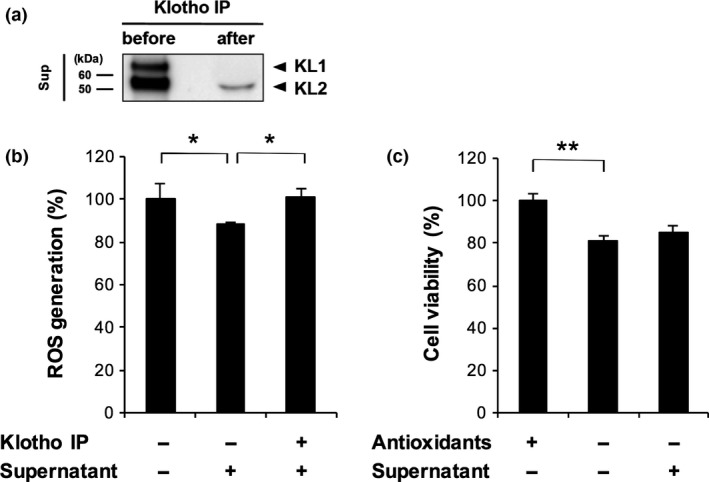
Antioxidative and protective effects of shed klotho on retinal cells. (a) Depletion of klotho in supernatant (sup) by immunoprecipitation (IP). After IP with an anti‐klotho antibody, the supernatant medium did not include a detectable level of KL1, although it did include a minor amount of KL2. (b) Treatment with supernatant had a significant suppressive effect on reactive oxygen species generation in primary retinal cells, whereas treatment with a supernatant depleted of klotho with IP had no effect on oxidative stress (*p < 0.05, n = 4 in each group). The non‐treated retinal cells were incubated in AO−culture medium. (c) The viability of the cultured primary retinal cells did not statistically differ after treatment with normal or klotho‐depleted supernatant media (**p < 0.01, n = 4 in each group). The fluorescence intensity of the cultured primary retinal cells with AO+ treatment was normalized to 100% cell viability. Error bars denote SD.
Promotion of axotomy‐induced RGC death via klotho inhibition
To determine whether active klotho had a neuroprotective effect in the rat retina after axotomy, we attempted to inhibit klotho activity using DSL, which has a potent inhibitory effect on the β‐glucuronidase activity of klotho. We found that after the topical application of 2 μL of 100 μM DSL led to a significant decrease in the number of surviving RGCs, to 1659 ± 254 RGCs/mm2, whereas the number of the RGCs in a group of saline‐treatment as vehicle decreased only to 2096 ± 231 RGCs/mm2 on day 7 after axotomy. This retrogradely labeled RGCs was normalized to the number in a group treated with saline as vehicle in axotomized retina and the ratio of surviving RGCs after treatment with saline was 100 ± 11.0%, and the ratio after treatment with DSL was 79.2 ± 12.1% (Fig. 7a and b). This result suggests that klotho fragments act as enzymes contributing to RGC protection in axotomized eyes.
Figure 7.
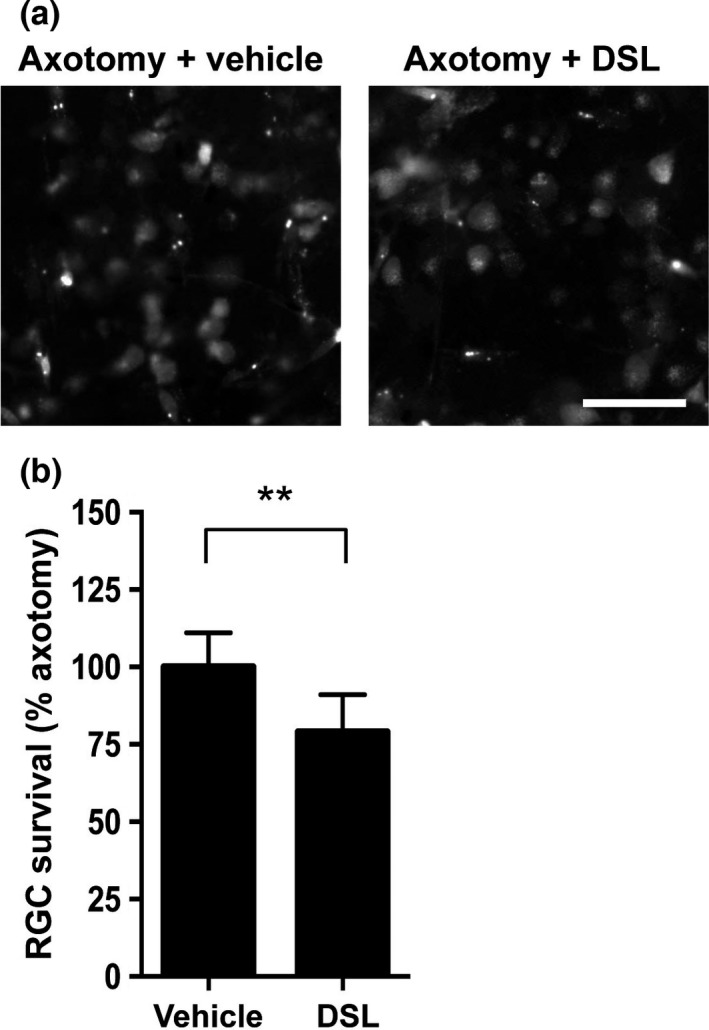
Klotho inhibition promotes retinal ganglion cell (RGC) death after axotomy in rat retinas. (a) Representative images of retrogradely labeled RGCs 7 days after optic nerve transection. The vehicle was saline. Scale bar = 50 μm. (b) The intravitreal injection of 2 μL of 100 μM d‐saccharic acid 1,4‐lactone (DSL), a klotho inhibitor, significantly promoted cell death in axotomized RGCs 7 days after axotomy. RGC numbers were normalized to the axotomized retina treated with saline as vehicle, expressed as a percentage (**p < 0.01, vehicle group: n = 5, DSL group: n = 6). Error bars denote SD.
Discussion
Ocular hypertension is a major associated risk factor and loss of RGCs accounts for visual function deficits for glaucoma. Although it was reported that LA has potential for neuroprotective effect after axonal damage, the molecular mechanisms still remain unclear. In this study, we found that LA treatment induced the expression and shedding of klotho, and that the cleavage of klotho and its release into the outside of the cells were mediated by ADAM17 via mechanisms that depended on OATP2B1 and PKC in rat retinas. These findings suggest that klotho fragments may contribute to the attenuation of axonal injury‐induced calpain activation and oxidative stress, thereby protecting the RGCs against post‐traumatic neuronal degeneration (Fig. 8).
Figure 8.
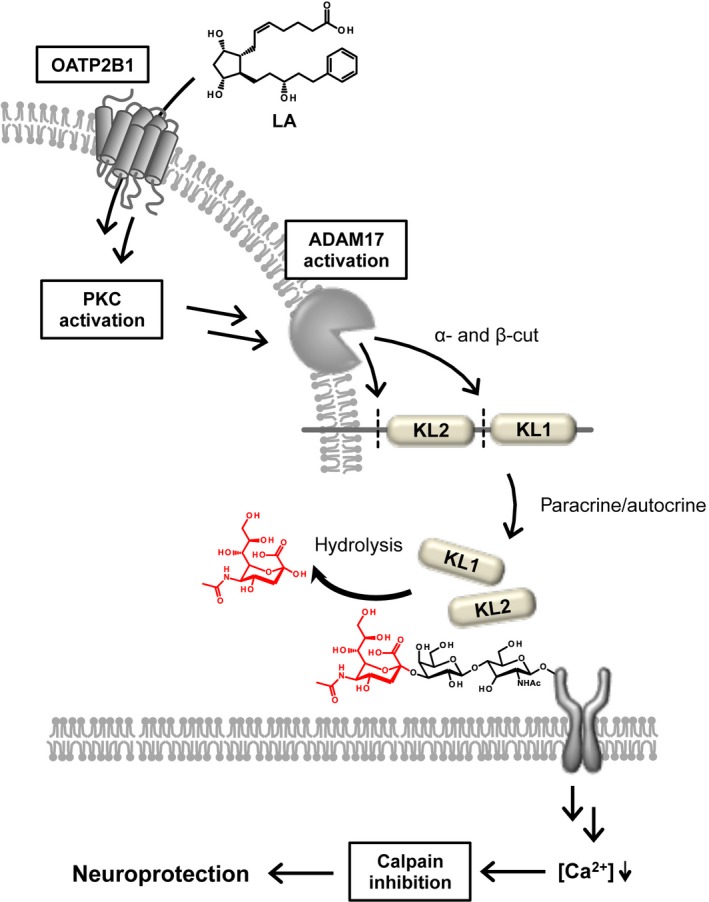
Proposal for the underlying mechanism of the neuroprotective effect of latanoprost acid (LA)‐mediated klotho shedding. LA is transported into the retinal ganglion cells (RGCs) via organic anion transporting polypeptide 2B1 (OATP2B1), where it activates ADAM17 via the protein kinase C (PKC) pathway. Klotho fragments cleaved by ADAM17 might then hydrolyze sugar chains on certain membrane proteins. The klotho fragments also suppress the influx of Ca2+ into the RGCs, and then induce calpain inhibition.
Calpain is a promising therapeutic target for the prevention of neuronal degeneration such as axonal injury‐induced RGC death (Nakazawa et al. 2009; Huang et al. 2010; Ryu et al. 2012; Koch et al. 2014; Liu et al. 2014; Yokoyama et al. 2014; Kobeissy et al. 2015). Indeed, we have previously demonstrated that a selective calpain inhibitor, SNJ‐1945, had a neuroprotective effect in the RGCs after optic nerve crush (Ryu et al. 2012). Klotho has biological function as an upstream regulator affecting μ‐calpain activity and is predominantly expressed in the GCL in naïve CD rats (Manya et al. 2002; Kusaba et al. 2010; Farinelli et al. 2013). Thus, it is likely that LA inhibits axotomy‐induced calpain activation in the GCL, contributing to RGC survival. Calpain activity is closely regulated by calpastatin, an endogenous inhibitor (Melloni et al. 1992; Kawasaki and Kawashima 1996). Several studies have shown that calpastatin is cleaved by caspase‐3, and that this cleavage is essential for the regulation of calpain activity during cell death (Pörn‐Ares et al. 1998; Wang et al. 1998). In this study, we confirmed that LA can attenuate the breakdown of calpastatin after axotomy (Fig. 3a), a finding that is consistent with previous reports that latanoprost inhibited caspase‐3 activation and the elevation of intracellular Ca2+, necessary components of calpain activation (Nakanishi et al. 2006; Kanamori et al. 2009). On the other hand, LA treatment did not restore pSTR amplitude on day 14 after axotomy (Fig. 2e), even though the number of surviving RGCs was higher with treatment (Fig. 1). This finding indicates that even though LA can protect the RGCs, it cannot preserve their physiological function over the long term (i.e., at least 14 days after axotomy).
On the basis of the robust ability of LA to inhibit post‐injury calpain activation, we speculated that the neuroprotective effect of LA might be related to klotho. We therefore confirmed that klotho was localized in the retina, a finding that was compatible with previous work showing that α‐klotho is predominantly expressed in the GCL (Farinelli et al. 2013). Our data strongly suggest that klotho is predominantly expressed in the RGCs of naïve rats (Figure S2). In axotomized rats, LA inhibited the cleavage of α‐fodrin (Fig. 3a), and an inverse correlation was observed in the GCL between calpain activity and the expression level of klotho (Figs 3d and 4d). This implies that LA‐induced klotho contributes to calpain inhibition and the subsequent increase in RGC survival. Moreover, LA treatment facilitated both α‐ and β‐cleavages of klotho, and induced more KL2 fragments than KL1 fragments in both the retina and its supernatant. Thus, KL2 appeared to be more involved in the neuroprotective effect of LA. However, klotho fragments did not increase in a dose‐dependent manner (Fig. 4f). Previous work showed that an LA concentration of 1000 nM had cytotoxic properties, but that concentrations of 1 nM and 10 nM LA had a neuroprotective effect (Drago et al. 2001). Therefore, even though 1000 nM of LA was very efficient at promoting the shedding of KL1 and 2, it may not be the best choice for maintaining primary cultures of retinal cells.
Since LA is a prostaglandin‐related compound primarily targeting the FP receptor, we attempted to determine whether the FP receptor mediated LA‐induced klotho cleavage and the release of klotho to the outside of the cells. Unexpectedly, we found that klotho shedding in LA‐treated axotomized retinas was mediated not via the FP receptor, but rather via the OATP2B1 transporter. In addition, the expression pattern of OATP2B1 in naïve retinas matched that of klotho after axotomy with LA treatment (Figs 4d and 5c), indicating that OATP2B1 might be an upstream regulator of LA‐mediated klotho shedding. We also confirmed that OATP2B1 was expressed in the RGCs even after axotomy (data not shown). Moreover, it has been reported that the extracellular domain of klotho undergoes proteolytic cleavage by ADAM17, which is up‐regulated and activated in the RGCs after axonal injury (Ahmed et al. 2006; Chen et al. 2007). Reinforcing this result, we found that the signal intensity of klotho fragments in a group treated with TAPI‐1, an ADAM17 inhibitor, remained at a level comparable to a non‐treated group, whereas the LA‐induced klotho shedding was only slightly affected by GM6001, a broad spectrum MMP inhibitor (Fig. 5d). The PKC pathway is known as one of the regulatory mechanisms underlying ADAM17 activation (Nagano et al. 2004), and in fact, interfering with the PKC pathway with inhibitors (such as GF109203X) or activators (such as TPA) had only a modest effect on LA‐induced klotho shedding. This result indicates that in addition to the PKC pathway, another mechanism may regulate ADAM17. One possibility for this alternative mechanism is the phosphatidylinositol 3‐kinase (PI3K)‐Akt pathway, because PI3K inhibitors inhibit insulin‐mediated klotho shedding via ADAM17. Furthermore, latanoprost can activate the PI3K‐Akt pathway (Chen et al. 2007; Zheng et al. 2011). However, further investigation is needed to identify new intracellular pharmacological targets of LA after it is transported into the RGCs, and to better understand the mechanism of LA‐mediated ADAM17 activation.
Klotho functions not only as an enzyme that modifies the sugar chain in membrane proteins, but also as a cofactor that modulates the affinity of various receptors after binding, which in turn regulates calcium homeostasis and oxidative stress (Kurosu et al. 2005; Yamamoto et al. 2005; Urakawa et al. 2006; Cha et al. 2008; Kusaba et al. 2010). It has been reported that oxidative stress occurs in the RGCs immediately after axonal injury and leads to RGC degeneration (Kanamori et al. 2010; Yamamoto et al. 2014). Our results showed that KL1 and KL2, both found in the supernatant did not change retinal cell viability in vitro (Fig. 6c). On the other hand, KL1 and KL2 were responsible for the attenuation of oxidative stress (Fig. 6b), suggesting that KL1 and KL2 have antioxidant function based on each cell. Meanwhile, the DSL‐induced inhibition of klotho enzymatic activity accelerated RGC death after axotomy, suggesting that klotho fragments contribute to RGC protection by acting as enzymes, rather than as cofactors.
In conclusion, the neuroprotective effect of latanoprost may arise because of a mechanism by which KL2 is produced via the OATP2B1 transporter, rather than via the FP receptor, whereupon it attenuates axonal injury‐induced calpain activation and oxidative stress in the RGCs (Fig. 8). Furthermore, our findings show that klotho may have potential as a new therapeutic tool to prevent degeneration of the RGCs in retinal diseases such as glaucoma.
Supporting information
Figure S1. LA‐mediated klotho expression in non‐axotomized rat eyes.
Figure S2. The localization of klotho in naïve rat retinas.
Acknowledgments and conflict of interest disclosure
This study was supported by grants from Pfizer, Inc. (WS2252097) and by a Grant‐in‐Aid from the Ministry of Education, Science and Technology of Japan (K.S. 26893019 and 15K20247, K.O. 15H06041, M. R. 25462749, T.N. 22689045 and 26670751). There is a conflict of interest. We thank Mr. Tim Hilts for reviewing and editing the language of the manuscript, Ms. Natsumi Konno, Ms. Junko Sato, and Ms. Kanako Sakai for the technical assistance. We also thank the Biomedical Research Core of Tohoku University Graduate School of Medicine for technical support. Authors thank Dr. Jun Kosaka (Okayama University) for gift of C38 antibody.
All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Ahmed Z., Suggate E. L., Brown E. R., Dent R. G., Armstrong S. J., Barrett L. B., Berry M. and Logan A. (2006) Schwann cell‐derived factor‐induced modulation of the NgR/p75NTR/EGFR axis disinhibits axon growth through CNS myelin in vivo and in vitro. Brain 129, 1517–1533. [DOI] [PubMed] [Google Scholar]
- Bui B. V. and Fortune B. (2004) Ganglion cell contributions to the rat full‐field electroretinogram. J. Physiol. 555, 153–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha S. K., Ortega B., Kurosu H., Rosenblatt K. P., Kuro‐O M. and Huang C. L. (2008) Removal of sialic acid involving Klotho causes cell‐surface retention of TRPV5 channel via binding to galectin‐1. Proc. Natl Acad. Sci. USA 105, 9805–9810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q., Hoefs S., van der Kemp A. W., Topala C. N., Bindels R. J. and Hoenderop J. G. (2005) The beta‐glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310, 490–493. [DOI] [PubMed] [Google Scholar]
- Chen C. D., Podvin S., Gillespie E., Leeman S. E. and Abraham C. R. (2007) Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc. Natl Acad. Sci. USA 104, 19796–19801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago F., Valzelli S., Emmi I., Marino A., Scalia C. C. and Marino V. (2001) Latanoprost exerts neuroprotective activity in vitro and in vivo. Exp. Eye Res. 72, 479–486. [DOI] [PubMed] [Google Scholar]
- Emre S., Gul M., Ates B., Esrefoglu M., Koc B., Erdogan A. and Yesilada E. (2009) Comparison of the protective effects of prostaglandin analogues in the ischemia and reperfusion model of rabbit eyes. Exp. Anim. 58, 505–513. [DOI] [PubMed] [Google Scholar]
- Farinelli P., Arango‐Gonzalez B., Völkl J., Alesutan I., Lang F., Zrenner E., Paquet‐Durand F. and Ekström P. A. (2013) Retinitis Pigmentosa: over‐expression of anti‐ageing protein Klotho in degenerating photoreceptors. J. Neurochem. 127, 868–879. [DOI] [PubMed] [Google Scholar]
- Frishman L. J., Shen F. F., Du L., Robson J. G., Harwerth R. S., Smith E. L., Carter‐Dawson L. and Crawford M. L. (1996) The scotopic electroretinogram of macaque after retinal ganglion cell loss from experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 37, 125–141. [PubMed] [Google Scholar]
- Gao B., Huber R. D., Wenzel A., Vavricka S. R., Ismair M. G., Remé C. and Meier P. J. (2005) Localization of organic anion transporting polypeptides in the rat and human ciliary body epithelium. Exp. Eye Res. 80, 61–72. [DOI] [PubMed] [Google Scholar]
- Gao B., Vavricka S. R., Meier P. J. and Stieger B. (2015) Differential cellular expression of organic anion transporting peptides OATP1A2 and OATP2B1 in the human retina and brain: implications for carrier‐mediated transport of neuropeptides and neurosteriods in the CNS. Pflugers Arch. 467, 1481–1493. [DOI] [PubMed] [Google Scholar]
- Haruna Y., Kashihara N., Satoh M., Tomita N., Namikoshi T., Sasaki T., Fujimori T., Xie P. and Kanwar Y. S. (2007) Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc. Natl Acad. Sci. USA 104, 2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Fileta J., Rawe I., Qu J. and Grosskreutz C. L. (2010) Calpain activation in experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 51, 3049–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura A., Iwano A., Tohyama O., Tsuji Y., Nozaki K., Hashimoto N., Fujimori T. and Nabeshima Y. (2004) Secreted Klotho protein in sera and CSF: implication for post‐translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 565, 143–147. [DOI] [PubMed] [Google Scholar]
- Kanamori A., Naka M., Fukuda M., Nakamura M. and Negi A. (2009) Latanoprost protects rat retinal ganglion cells from apoptosis in vitro and in vivo. Exp. Eye Res. 88, 535–541. [DOI] [PubMed] [Google Scholar]
- Kanamori A., Catrinescu M. M., Kanamori N., Mears K. A., Beaubien R. and Levin L. A. (2010) Superoxide is an associated signal for apoptosis in axonal injury. Brain 133, 2612–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H. and Kawashima S. (1996) Regulation of the calpain‐calpastatin system by membranes (review). Mol. Membr. Biol. 13, 217–224. [DOI] [PubMed] [Google Scholar]
- Kobeissy F. H., Liu M. C., Yang Z. et al (2015) Degradation of βII‐spectrin protein by calpain‐2 and caspase‐3 under neurotoxic and traumatic brain injury conditions. Mol. Neurobiol. 52, 696–709. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Koch J. C., Tönges L., Barski E., Michel U., Bähr M. and Lingor P. (2014) ROCK2 is a major regulator of axonal degeneration, neuronal death and axonal regeneration in the CNS. Cell Death Dis. 5, e1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkinaki M., Abu‐Asab M., Gunawardena N., Ahern G., Javidnia M., Young J. and Golestaneh N. (2013) Klotho regulates retinal pigment epithelial functions and protects against oxidative stress. J. Neurosci. 33, 16346–16359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft M. E., Glaeser H., Mandery K. et al (2010) The prostaglandin transporter OATP2A1 is expressed in human ocular tissues and transports the antiglaucoma prostanoid latanoprost. Invest. Ophthalmol. Vis. Sci. 51, 2504–2511. [DOI] [PubMed] [Google Scholar]
- Kudo H., Nakazawa T., Shimura M., Takahashi H., Fuse N., Kashiwagi K. and Tamai M. (2006) Neuroprotective effect of latanoprost on rat retinal ganglion cells. Graefes Arch. Clin. Exp. Ophthalmol. 244, 1003–1009. [DOI] [PubMed] [Google Scholar]
- Kuro‐o M., Matsumura Y., Aizawa H. et al (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51. [DOI] [PubMed] [Google Scholar]
- Kurosu H., Yamamoto M., Clark J. D. et al (2005) Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusaba T., Okigaki M., Matui A. et al (2010) Klotho is associated with VEGF receptor‐2 and the transient receptor potential canonical‐1 Ca2+ channel to maintain endothelial integrity. Proc. Natl Acad. Sci. USA 107, 19308–19313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Yin F., Zhang J. and Qian Y. (2014) The role of calpains in traumatic brain injury. Brain Inj. 28, 133–137. [DOI] [PubMed] [Google Scholar]
- Manya H., Inomata M., Fujimori T., Dohmae N., Sato Y., Takio K., Nabeshima Y. and Endo T. (2002) Klotho protein deficiency leads to overactivation of mu‐calpain. J. Biol. Chem. 277, 35503–35508. [DOI] [PubMed] [Google Scholar]
- Melloni E., Salamino F. and Sparatore B. (1992) The calpain‐calpastatin system in mammalian cells: properties and possible functions. Biochimie 74, 217–223. [DOI] [PubMed] [Google Scholar]
- Nagano O., Murakami D., Hartmann D., De Strooper B., Saftig P., Iwatsubo T., Nakajima M., Shinohara M. and Saya H. (2004) Cell‐matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca(2+) influx and PKC activation. J. Cell Biol. 165, 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y., Nakamura M., Mukuno H., Kanamori A., Seigel G. M. and Negi A. (2006) Latanoprost rescues retinal neuro‐glial cells from apoptosis by inhibiting caspase‐3, which is mediated by p44/p42 mitogen‐activated protein kinase. Exp. Eye Res. 83, 1108–1117. [DOI] [PubMed] [Google Scholar]
- Nakazawa T., Nakano I., Sato M., Nakamura T., Tamai M. and Mori N. (2002) Comparative expression profiles of Trk receptors and Shc‐related phosphotyrosine adapters during retinal development: potential roles of N‐Shc/ShcC in brain‐derived neurotrophic factor signal transduction and modulation. J. Neurosci. Res. 68, 668–680. [DOI] [PubMed] [Google Scholar]
- Nakazawa T., Hisatomi T., Nakazawa C. et al (2007) Monocyte chemoattractant protein 1 mediates retinal detachment‐induced photoreceptor apoptosis. Proc. Natl Acad. Sci. USA 104, 2425–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T., Shimura M., Mourin R., Kondo M., Yokokura S., Saido T. C., Nishida K. and Endo S. (2009) Calpain‐mediated degradation of G‐substrate plays a critical role in retinal excitotoxicity for amacrine cells. J. Neurosci. Res. 87, 1412–1423. [DOI] [PubMed] [Google Scholar]
- Ocklind A., Lake S., Krook K., Hallin I., Nistér M. and Westermark B. (1997) Localization of the prostaglandin F2 alpha receptor in rat tissues. Prostaglandins Leukot. Essent. Fatty Acids 57, 527–532. [DOI] [PubMed] [Google Scholar]
- Ota T., Aihara M., Narumiya S. and Araie M. (2005) The effects of prostaglandin analogues on IOP in prostanoid FP‐receptor‐deficient mice. Invest. Ophthalmol. Vis. Sci. 46, 4159–4163. [DOI] [PubMed] [Google Scholar]
- Pörn‐Ares M. I., Samali A. and Orrenius S. (1998) Cleavage of the calpain inhibitor, calpastatin, during apoptosis. Cell Death Differ. 5, 1028–1033. [DOI] [PubMed] [Google Scholar]
- Ryu M., Yasuda M., Shi D. et al (2012) Critical role of calpain in axonal damage‐induced retinal ganglion cell death. J. Neurosci. Res. 90, 802–815. [DOI] [PubMed] [Google Scholar]
- Sales K. J., Boddy S. C., Williams A. R., Anderson R. A. and Jabbour H. N. (2007) F‐prostanoid receptor regulation of fibroblast growth factor 2 signaling in endometrial adenocarcinoma cells. Endocrinology 148, 3635–3644. [DOI] [PubMed] [Google Scholar]
- Sharif N. A., Kelly C. R., Crider J. Y., Williams G. W. and Xu S. X. (2003) Ocular hypotensive FP prostaglandin (PG) analogs: PG receptor subtype binding affinities and selectivities, and agonist potencies at FP and other PG receptors in cultured cells. J. Ocul. Pharmacol. Ther. 19, 501–515. [DOI] [PubMed] [Google Scholar]
- Sieving P. A. and Nino C. (1988) Scotopic threshold response (STR) of the human electroretinogram. Invest. Ophthalmol. Vis. Sci. 29, 1608–1614. [PubMed] [Google Scholar]
- Sieving P. A., Frishman L. J. and Steinberg R. H. (1986) Scotopic threshold response of proximal retina in cat. J. Neurophysiol. 56, 1049–1061. [DOI] [PubMed] [Google Scholar]
- Urakawa I., Yamazaki Y., Shimada T., Iijima K., Hasegawa H., Okawa K., Fujita T., Fukumoto S. and Yamashita T. (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774.17086194 [Google Scholar]
- Wang K. K., Posmantur R., Nadimpalli R. et al (1998) Caspase‐mediated fragmentation of calpain inhibitor protein calpastatin during apoptosis. Arch. Biochem. Biophys. 356, 187–196. [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Clark J. D., Pastor J. V. et al (2005) Regulation of oxidative stress by the anti‐aging hormone klotho. J. Biol. Chem. 280, 38029–38034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K., Maruyama K., Himori N., Omodaka K., Yokoyama Y., Shiga Y., Morin R. and Nakazawa T. (2014) The novel Rho kinase (ROCK) inhibitor K‐115: a new candidate drug for neuroprotective treatment in glaucoma. Invest. Ophthalmol. Vis. Sci. 55, 7126–7136. [DOI] [PubMed] [Google Scholar]
- Yin Y., Henzl M. T., Lorber B., Nakazawa T., Thomas T. T., Jiang F., Langer R. and Benowitz L. I. (2006) Oncomodulin is a macrophage‐derived signal for axon regeneration in retinal ganglion cells. Nat. Neurosci. 9, 843–852. [DOI] [PubMed] [Google Scholar]
- Yokoyama Y., Maruyama K., Yamamoto K., Omodaka K., Yasuda M., Himori N., Ryu M., Nishiguchi K. M. and Nakazawa T. (2014) The role of calpain in an in vivo model of oxidative stress‐induced retinal ganglion cell damage. Biochem. Biophys. Res. Commun. 451, 510–515. [DOI] [PubMed] [Google Scholar]
- Zeldich E., Chen C. D., Colvin T. A., Bove‐Fenderson E. A., Liang J., Tucker Zhou T. B., Harris D. A. and Abraham C. R. (2014) The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J. Biol. Chem. 289, 24700–24715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J., Feng X., Hou L., Cui Y., Zhu L., Ma J., Xia Z., Zhou W. and Chen H. (2011) Latanoprost promotes neurite outgrowth in differentiated RGC‐5 cells via the PI3K‐Akt‐mTOR signaling pathway. Cell. Mol. Neurobiol. 31, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. LA‐mediated klotho expression in non‐axotomized rat eyes.
Figure S2. The localization of klotho in naïve rat retinas.