

Abstract
Objective
To assess the efficacy and safety of anifrolumab, a type I interferon (IFN) receptor antagonist, in a phase IIb, randomized, double‐blind, placebo‐controlled study of adults with moderate‐to‐severe systemic lupus erythematosus (SLE).
Methods
Patients (n = 305) were randomized to receive intravenous anifrolumab (300 mg or 1,000 mg) or placebo, in addition to standard therapy, every 4 weeks for 48 weeks. Randomization was stratified by SLE Disease Activity Index 2000 score (<10 or ≥10), oral corticosteroid dosage (<10 or ≥10 mg/day), and type I IFN gene signature test status (high or low) based on a 4‐gene expression assay. The primary end point was the percentage of patients achieving an SLE Responder Index (SRI[4]) response at week 24 with sustained reduction of oral corticosteroids (<10 mg/day and less than or equal to the dose at week 1 from week 12 through 24). Other end points (including SRI[4], British Isles Lupus Assessment Group [BILAG]–based Composite Lupus Assessment [BICLA], modified SRI[6], and major clinical response) were assessed at week 52. The primary end point was analyzed in the modified intent‐to‐treat (ITT) population and type I IFN–high subpopulation. The study result was considered positive if the primary end point was met in either of the 2 study populations. The Type I error rate was controlled at 0.10 (2‐sided), within each of the 2 study populations for the primary end point analysis.
Results
The primary end point was met by more patients treated with anifrolumab (34.3% of 99 for 300 mg and 28.8% of 104 for 1,000 mg) than placebo (17.6% of 102) (P = 0.014 for 300 mg and P = 0.063 for 1,000 mg, versus placebo), with greater effect size in patients with a high IFN signature at baseline (13.2% in placebo‐treated patients versus 36.0% [P = 0.004] and 28.2% [P = 0.029]) in patients treated with anifrolumab 300 mg and 1,000 mg, respectively. At week 52, patients treated with anifrolumab achieved greater responses in SRI(4) (40.2% versus 62.6% [P < 0.001] and 53.8% [P = 0.043] with placebo, anifrolumab 300 mg, and anifrolumab 1,000 mg, respectively), BICLA (25.7% versus 53.5% [P < 0.001] and 41.2% [P = 0.018], respectively), modified SRI(6) (28.4% versus 49.5% [P = 0.002] and 44.7% [P = 0.015], respectively), major clinical response (BILAG 2004 C or better in all organ domains from week 24 through week 52) (6.9% versus 19.2% [P = 0.012] and 17.3% [P = 0.025], respectively), and several other global and organ‐specific end points. Herpes zoster was more frequent in the anifrolumab‐treated patients (2.0% with placebo treatment versus 5.1% and 9.5% with anifrolumab 300 mg and 1,000 mg, respectively), as were cases reported as influenza (2.0% versus 6.1% and 7.6%, respectively), in the anifrolumab treatment groups. Incidence of serious adverse events was similar between groups (18.8% versus 16.2% and 17.1%, respectively).
Conclusion
Anifrolumab substantially reduced disease activity compared with placebo across multiple clinical end points in the patients with moderate‐to‐severe SLE.
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease 1 leading to significant morbidity and shortened lifespan 2, 3, 4. Treatment of SLE is challenging because of the limited efficacy and poor tolerability of standard therapy 5, 6, 7.
Evidence supports activation of the type I interferon (IFN) system as a central pathogenic mediator in SLE 8, 9, 10, 11. Cell signaling by all type I IFNs, including IFNα, IFNβ, IFNɛ, IFNκ, and IFNω, is mediated by the type I IFN‐α/β/ω receptor (IFNAR), resulting in IFN‐stimulated gene transcription, otherwise known as the IFN gene signature 12. Consequently, blockade of IFNAR may reverse some of the immune dysregulation that occurs in SLE 13, 14.
Results of recent phase II clinical studies of anti‐IFNα antibodies have been mixed. Rontalizumab did not meet the primary or secondary end points in a recent study 15. However, a post hoc analysis suggested potential benefit in a small subset of patients with a low baseline IFN signature. In contrast, sifalimumab met primary and some secondary end points 16, but the treatment effects were modest. Both of these molecules have specificity only for IFNα, leaving other type I IFNs unaffected and able to bind IFNAR.
Anifrolumab is a fully human, IgG1κ monoclonal antibody that binds to IFNAR and prevents signaling by all type I IFNs 17. Anifrolumab exhibited a favorable safety profile and sustained inhibition of the type I IFN gene signature in a phase I study of patients with scleroderma 18. Given the similarities in type I IFN activity in SLE and scleroderma 19 the present phase IIb study was conducted to evaluate the efficacy and safety of 2 fixed intravenous dosages of anifrolumab in adults with moderately to severely active SLE with inadequate responses to standard therapy.
PATIENTS AND METHODS
Patients
This study included patients ages 18–65 years with SLE. Patients had to weigh ≥40 kg and fulfill ≥4 of the 11 American College of Rheumatology 1997 classification criteria for SLE 20, 21. At screening, all patients were required to have antinuclear antibodies and/or anti–double‐stranded DNA (anti‐dsDNA) antibodies, and/or anti‐Sm antibodies, and to be receiving treatment with at least one of the following: oral prednisone (≤40 mg/day or equivalent), azathioprine (≤200 mg/day), an antimalarial, mycophenolate mofetil/mycophenolic acid (≤2.0 gm/day), or methotrexate (≤25 mg/week).
Treatments for SLE had to be administered for at least 24 weeks prior to study entry and at stable dosages for ≥2 weeks (for prednisone and nonsteroidal antiinflammatory drugs) or ≥8 weeks (for other therapies) before screening. Biologic agents and protocol‐prohibited immunosuppressants had to be discontinued before the study.
Patients had to meet all of the following disease activity criteria at screening: a score of ≥6 on the SLE Disease Activity Index 2000 (SLEDAI‐2K) 22, excluding points attributable to lupus headache or organic brain syndrome; a British Isles Lupus Assessment Group (BILAG) 2004 23 organ domain score of ≥1A or ≥2B 24; and a physician's global assessment of disease activity of ≥1 on a visual analog scale from 0 (none) to 3 (severe disease). In addition, patients were required to have a score of ≥4 in clinical components of the SLEDAI‐2K (clinical SLEDAI‐2K; points attributed to laboratory components were excluded) at week 1 (prior to receiving the study drug). Patients with active and severe lupus nephritis or neuropsychiatric SLE were excluded from the study. Additional study exclusion criteria are provided in the Supplementary Methods, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39962/abstract.
Study design
This randomized, double‐blind, placebo‐controlled, parallel‐group phase IIb study (A Phase II, Randomized Study to Evaluate the Efficacy and Safety of MEDI‐546 in Subjects with Systemic Lupus Erythematosus [MUSE]; ClinicalTrials.gov identifier: NCT01438489) was conducted at 101 sites in 15 countries (see Supplementary Methods, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39962/abstract). Patients were randomized 1:1:1 to receive intravenous infusions of placebo, anifrolumab 300 mg, or anifrolumab 1,000 mg. Treatment was administered every 4 weeks with the final dose administered at week 48. The primary and secondary efficacy measures were assessed at week 24 and week 52, respectively. All patients were required to complete a 12‐week follow‐up period after administration of the final dose of the study drug. During the treatment period, there were 13 scheduled monthly visits. The follow‐up period consisted of 3 visits, conducted 4, 8, and 12 weeks after the last dose. Randomization was stratified by type I IFN gene signature (IFN high or IFN low), dosage of oral corticosteroids (<10 mg/day or ≥10 mg/day of prednisone or equivalent), and SLEDAI‐2K score (<10 or ≥10) at screening. IFN gene signature was determined at a central laboratory using an analytically validated 4‐gene (IFI27, IFI44, IFI44L, and RSAD2) quantitative polymerase chain reaction (PCR)–based test from patients’ whole blood 25. A predetermined, ΔCt‐based cutoff point, in the trough of the bimodal distribution, was used to segregate patients with a high IFN gene signature from patients with a low IFN gene signature at baseline.
Tapering of oral corticosteroids was encouraged but was at the discretion of the investigators. Tapering was allowed after randomization except within 8 weeks of the primary (week 24) and secondary (week 52) end point assessments. A maximum of 2 oral corticosteroid bursts for increased SLE disease activity were allowed during the study. Patients with increased SLE disease activity could receive an oral corticosteroid burst between week 1 and week 10 (increase of ≤40 mg/day prednisone or equivalent), which had to be tapered to the week 1 dosage within 2 weeks of initiation of the burst; alternatively, a single intramuscular dose of methylprednisone (80 or 160 mg or equivalent) was permitted. The course of oral corticosteroid burst could not extend beyond week 10. Patients could receive an additional oral corticosteroid burst for increased SLE disease activity between week 24 and week 40 (increase of ≤20 mg/day prednisone or equivalent), which had to be tapered to the week 24 dosage within 2 weeks of initiation; alternatively, a single intramuscular dose of methylprednisone (80 mg or equivalent) was permitted. The course of oral corticosteroids could not extend beyond week 40.
This study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Guidelines for Good Clinical Practice. Independent ethics committee or independent institutional review board approvals were obtained, and all patients provided written informed consent in accordance with local requirements.
An independent data safety and monitoring board was appointed for the study. Screening assessments of SLE organ system involvement and disease activity requirements were confirmed by an independent external adjudication group that also approved patient randomization and monitored study assessments.
Efficacy and safety evaluations
The primary efficacy end point was a composite of the SLE Responder Index (SRI[4]) 26 at week 24 with a sustained reduction in oral corticosteroids from week 12 through week 24 (<10 mg/day and less than or equal to the dose received at week 1). SRI(4) response is defined as a ≥4‐point reduction in SLEDAI‐2K score; no new BILAG 2004 A or >1 new BILAG 2004 B domain scores; and <0.3‐point deterioration in physician's global assessment.
Secondary efficacy measures included the SRI(4) response rates at week 52 with a sustained oral corticosteroid reduction from week 40 through week 52 and reduction of oral corticosteroid dosage at week 52 to ≤7.5 mg/day in patients who were receiving ≥10 mg/day at baseline. Other efficacy measures included percentages of patients with ≥50% improvement in Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI) 27, 28 for patients with at least moderate skin involvement (CLASI ≥10); ≥50% improvement in swollen and tender joint count (28 joints assessed) for patients with ≥8 swollen and ≥8 tender joints at baseline; response in BILAG‐based Composite Lupus Assessment (BICLA) 29; modified SRIs requiring SLEDAI‐2K reductions of 5–8 points to be considered a responder; physician's global assessment; proportion of subjects with a SLEDAI‐2K score of ≤2; proportion of subjects with a SLEDAI‐2K score of 0; clinical SLEDAI; major clinical response defined as BILAG 2004 score of C or better in all organ domains at week 24 with maintenance of this response through week 52; >3‐point improvement in Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT‐F) 30; Short Form 36 (SF‐36) health survey; anti‐dsDNA; and C3 and C4 complement concentrations.
To ensure that evaluation of SLE disease activity was consistent across study sites, training and certification was provided to investigators and designated site personnel, as appropriate, who were responsible for completing the following disease evaluation assessments: SLEDAI‐2K, BILAG 2004, physician's global assessment, CLASI, and swollen and tender joint count evaluation. Where possible, evaluation of each patient was performed by a single assessor. Independent expert adjudication was undertaken throughout the study. Safety end points included adverse events, adverse events of special interest, laboratory assessments, and vital signs.
Statistical analysis
Analysis of the primary end point compared response rates at week 24 between each anifrolumab group and placebo using a logistic regression model adjusted for randomization stratification factors. The secondary end points and other binary end points were analyzed using the same approach as for the primary end point. Continuous end points were analyzed using an analysis of covariance model adjusted for randomization stratification factors, with the relevant baseline value as the covariate. For responder analyses, patients who were withdrawn from treatment, had increased use of oral corticosteroids beyond the protocol‐permitted dosage, or had initiation of or an increase in dosage of immunosuppressant treatment any time after baseline were defined as nonresponders.
The primary end point was analyzed in the modified intent‐to‐treat (ITT) population (all randomized patients who received any dose of study drug and had baseline primary efficacy measurements) and a modified ITT subpopulation of patients with a high IFN gene signature at screening (IFN‐high subpopulation). The study result was considered positive if the primary end point was met in either of the 2 study populations.
The Type I error rate was controlled at 0.10 (2‐sided), within each of the 2 study populations (modified ITT population and IFN‐high subpopulation) for the primary end point analysis, by performing a Cochran‐Armitage trend test of all treatment groups prior to performing pairwise comparisons between each anifrolumab group and placebo. No multiplicity adjustment for the 2 study populations or other end points was applied.
The target sample size of 100 patients per group was based on providing 88% power at the 0.10 alpha level to detect at least 20% absolute improvement in SRI(4) response rate at week 24 for anifrolumab relative to placebo, assuming a 40% placebo response rate. All data analyses were conducted using the SAS system (SAS Institute).
RESULTS
Study population
Between January 2012 and January 2014, 626 patients were screened, with 307 randomized to receive treatment (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39962/abstract). One patient in the anifrolumab 300‐mg group had poor peripheral venous access, and 1 patient in the placebo group had an entry criteria violation and did not receive study treatment. The modified ITT population therefore consisted of 305 patients (102 receiving placebo, 99 receiving anifrolumab 300 mg, and 104 receiving anifrolumab 1,000 mg). One patient randomized to the placebo group mistakenly received a single dose of anifrolumab (1,000‐mg). This was classified as a protocol deviation. The patient was included in the placebo group of the modified ITT population for the efficacy analyses, but in the anifrolumab 1,000‐mg group of the safety population for the safety analyses. Baseline characteristics (Table 1) were similar between groups, with the following exceptions: disease duration was shorter for the placebo group than for the anifrolumab groups (mean 90.6 months versus 95.9 months and 100.1 months), and fewer patients in the placebo group were receiving methotrexate (15.7% versus 19.2% and 24.0%). The anifrolumab 1,000‐mg group had a greater percentage of white patients than the anifrolumab 300‐mg and placebo groups (49.0% versus 35.4% and 40.2%, respectively).
Table 1.
Demographic and baseline clinical characteristics of the patients with SLE (modified ITT population)a
| Placebo (n = 102) | Anifrolumab 300 mg (n = 99) | Anifrolumab 1,000 mg (n = 104) | |
|---|---|---|---|
| Age, years | 39.3 ± 12.9 | 39.1 ± 11.9 | 40.8 ± 11.6 |
| Sex, no. (%) female | 93 (91.2) | 93 (93.9) | 99 (95.2) |
| Weight, kg | 68.1 ± 19.1 | 69.5 ± 17.2 | 70.7 ± 17.3 |
| Height, cm | 161.2 ± 8.1 | 161.6 ± 8.5 | 161.9 ± 6.7 |
| Race, no. (%) | |||
| White | 41 (40.2) | 35 (35.4) | 51 (49.0) |
| African American | 12 (11.8) | 19 (19.2) | 10 (9.6) |
| Asian | 13 (12.7) | 3 (3.0) | 6 (5.8) |
| American Indian/Alaska Native | 0 (0.0) | 4 (4.0) | 1 (1.0) |
| Other | 36 (35.3) | 38 (38.4) | 36 (34.6) |
| Ethnicity, no. (%) non‐Hispanic | 60 (58.8) | 53 (53.5) | 64 (61.5) |
| Disease duration, monthsb | 90.6 ± 86.3 | 95.9 ± 76.8 | 100.1 ± 90.3 |
| High IFN gene signature, no. (%) | 76 (74.5) | 75 (75.8) | 78 (75.0) |
| SLEDAI‐2K global score | 11.1 ± 4.4 | 10.7 ± 3.7 | 10.9 ± 4.1 |
| BILAG 2004 global score | 19.8 ± 5.8 | 19.6 ± 5.8 | 18.6 ± 5.7 |
| Physician's global assessment | 1.77 ± 0.44 | 1.86 ± 0.39 | 1.86 ± 0.39 |
| CLASI activity score | 6.7 ± 5.1 | 7.5 ± 6.3 | 7.1 ± 6.2 |
| Swollen joint countc | 8.3 ± 6.4 | 8.6 ± 6.0 | 8.3 ± 6.4 |
| Tender joint countc | 10.5 ± 7.4 | 12.2 ± 7.1 | 11.6 ± 7.8 |
| Low complement concentrations, no. (%) | |||
| C3 | 43 (42.2) | 28 (28.3) | 48 (46.2) |
| C4 | 25 (24.5) | 21 (21.2) | 28 (26.9) |
| Elevated anti‐dsDNA, no. (%)d | |||
| Multiplex assay | 27 (26.5) | 24 (24.2) | 28 (26.9) |
| Farr assaye | 66 (80.5) | 56 (72.7) | 63 (76.8) |
| Concomitant immunomodulatory medications | |||
| Corticosteroids | |||
| No. (%) | 88 (86.3) | 79 (79.8) | 91 (87.5) |
| Dosage, mg/day | 12.8 ± 8.1 | 11.3 ± 6.4 | 12.5 ± 7.8 |
| Antimalarials, no. (%) | 75 (73.5) | 76 (76.8) | 68 (65.4) |
| Corticosteroids and antimalarials, no. (%) | 63 (61.8) | 59 (59.6) | 56 (53.8) |
| Azathioprine, no. (%) | 19 (18.6) | 23 (23.2) | 21 (20.2) |
| Methotrexate, no. (%) | 16 (15.7) | 19 (19.2) | 25 (24.0) |
| Mycophenolate, no. (%) | 11 (10.8) | 11 (11.1) | 11 (10.6) |
Except where indicated otherwise, values are the mean ± SD. Treatment was given every 4 weeks from week 1 to week 48. SLE = systemic lupus erythematosus; ITT = intent‐to‐treat; IFN = interferon; BILAG 2004 = British Isles Lupus Assessment Group 2004; CLASI = Cutaneous Lupus Erythematosus Disease Area and Severity Index; anti‐dsDNA = anti–double‐stranded DNA.
From diagnosis to study entry.
Based on the assessment of 28 joints.
The multiplex assay, an AtheNA Multi‐Lyte ANA‐II Plus test system, was used for screening and to calculate Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI‐2K) scores throughout the study. The differences between the multiplex and Farr assays were due to a low sensitivity cutoff point of the multiplex assay (Farr assay ≥5 IU/ml; multiplex assay ≥100 IU/ml) 32.
Data were available for 82 patients receiving placebo, 77 patients receiving anifrolumab 300 mg, and 82 patients receiving anifrolumab 1,000 mg.
Efficacy
Based on the mechanism of action of anifrolumab, the primary end point (SRI[4] response with sustained reduction of oral corticosteroids at week 24) was analyzed in the overall modified ITT population and the IFN‐high subpopulation. The predefined statistical significance level was 0.1; therefore, P values less than 0.1 were considered significant for the analyses of the primary end point. Multiplicity of comparing 2 active dosages with placebo within each study population was adjusted by performing a Cochran‐Armitage trend test of all treatment groups prior to performing pairwise comparisons between each anifrolumab group and placebo. No multiplicity adjustment for the 2 study populations was applied. The Cochran‐Armitage trend test of all treatment groups showed that the number of patients in whom the primary end point was achieved was greater for anifrolumab versus placebo in the modified ITT population (P = 0.072) and in the IFN‐high subpopulation, which included 75% of the patients (P = 0.034). In pairwise comparisons, both anifrolumab groups had greater response rates compared with placebo (17.6%). A response was achieved in 34.3% of the patients treated with anifrolumab 300 mg (P = 0.014 versus placebo) and in 28.8% of the patients treated with anifrolumab 1,000 mg (P = 0.063 versus placebo). The response rates in the IFN‐high subpopulation were 13.2% for the placebo group, 36.0% for the anifrolumab 300‐mg group (P = 0.004 versus placebo), and 28.2% for the anifrolumab 1,000‐mg group (P = 0.029 versus placebo) (see Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39962/abstract).
At week 52, anifrolumab treatment groups demonstrated improvements over placebo across a broad range of global disease activity measures. In the whole modified ITT population, 25.5% of the placebo group achieved SRI(4) response with sustained reduction of oral corticosteroids, compared with 51.5% in the anifrolumab 300‐mg group (P < 0.001) and 38.5% in the anifrolumab 1,000‐mg group (P = 0.048). Response rates in the IFN‐high population were 19.7% for placebo, 52.0% for the anifrolumab 300‐mg group (P < 0.001), and 38.5% for the anifrolumab 1,000‐mg group (P = 0.013). No differences from placebo were seen in the patients with a low IFN gene signature at screening (IFN‐low subpopulation) at week 24 or week 52 (Table 2 and Figure 1).
Table 2.
Summary of efficacy results for patients with SLE treated with placebo or anifrolumaba
| Placebo (n = 102) | Anifrolumab 300 mg (n = 99) | Anifrolumab 300 mg versus placebo | Anifrolumab 1,000 mg (n = 104) | Anifrolumab 1,000 mg versus placebo | |||
|---|---|---|---|---|---|---|---|
| OR (90% CI)b | P b | OR (90% CI)b | P b | ||||
| Week 24 | |||||||
| SRI(4) (including oral corticosteroid taper) | 18/102 (17.6) | 34 (34.3) | 2.38 (1.33–4.26) | 0.014 | 30/104 (28.8) | 1.94 (1.08–3.49) | 0.063 |
| High IFN gene signature | 10/76 (13.2) | 27/75 (36.0) | 3.55 (1.72–7.32) | 0.004 | 22/78 (28.2) | 2.65 (1.27–5.53) | 0.029 |
| Low IFN gene signature | 8/26 (30.8) | 7/24 (29.2) | 0.96 (0.34–2.74) | 0.946 | 8/26 (30.8) | 1.04 (0.37–2.88) | 0.953 |
| SRI(4) (excluding oral corticosteroid taper) | 41/102 (40.2) | 53/99 (53.5) | 1.77 (1.10–2.84) | 0.047 | 59/104 (56.7) | 1.98 (1.24–3.16) | 0.016 |
| High IFN gene signature | 29/76 (38.2) | 41/75 (54.7) | 2.03 (1.17–3.52) | 0.034 | 47/78 (60.3) | 2.50 (1.45–4.32) | 0.006 |
| Low IFN gene signature | 12/26 (46.2) | 12/24 (50.0) | 1.13 (0.44–2.93) | 0.832 | 12/26 (46.2) | 0.96 (0.38–2.44) | 0.943 |
| Week 52 | |||||||
| SRI(4) (including oral corticosteroid taper) | 26/102 (25.5) | 51/99 (51.5) | 3.08 (1.86–5.09) | <0.001 | 40/104 (38.5) | 1.84 (1.11–3.04) | 0.048 |
| High IFN gene signature | 15/76 (19.7) | 39/75 (52.0) | 4.30 (2.34–7.91) | <0.001 | 30/78 (38.5) | 2.52 (1.37–4.64) | 0.013 |
| Low IFN gene signature | 11/26 (42.3) | 12/24 (50.0) | 1.47 (0.55–3.93) | 0.514 | 10/26 (38.5) | 0.89 (0.34–2.35) | 0.849 |
| SRI4 (excluding oral corticosteroid taper) | 41/102 (40.2) | 62/99 (62.6) | 2.66 (1.64–4.31) | <0.001 | 56/104 (53.8) | 1.78 (1.11–2.85) | 0.043 |
| High IFN gene signature | 27/76 (35.5) | 45/75 (60.0) | 2.98 (1.69–5.24) | 0.001 | 43/78 (55.1) | 2.33 (1.34–4.04) | 0.012 |
| Low IFN gene signature | 14/26 (53.8) | 17/24 (70.8) | 2.07 (0.77–5.53) | 0.225 | 13/26 (50.0) | 0.85 (0.34–2.12) | 0.765 |
| Meeting oral corticosteroid taper criteriac | 17/64 (26.6) | 31/55 (56.4) | 3.59 (1.87–6.89) | 0.001 | 20/63 (31.7) | 1.23 (0.64–2.37) | 0.595 |
| ≥50% improvement in CLASId | 8/26 (30.8) | 17/27 (63.0) | 4.49 (1.67–12.12) | 0.013 | 14/24 (58.3) | 2.97 (1.08–8.19) | 0.077 |
| ≥50% improvement in joint countse | 18/37 (48.6) | 32/46 (69.6) | 2.67 (1.23–5.82) | 0.038 | 31/48 (64.6) | 1.92 (0.90–4.09) | 0.156 |
| BICLA responderf | 26/101 (25.7) | 53/99 (53.5) | 3.42 (2.06–5.68) | <0.001 | 42/102 (41.2) | 2.06 (1.25–3.42) | 0.018 |
| Modified SRI(5) | 30/102 (29.4) | 49/99 (49.5) | 2.47 (1.51–4.06) | 0.003 | 48/103 (46.6) | 2.14 (1.31–3.49) | 0.010 |
| Modified SRI(6) | 29/102 (28.4) | 49/99 (49.5) | 2.58 (1.57–4.23) | 0.002 | 46/103 (44.7) | 2.07 (1.27–3.37) | 0.015 |
| Modified SRI(7) | 16/93 (17.2) | 33/90 (36.7) | 2.83 (1.58–5.07) | 0.003 | 26/95 (27.4) | 1.83 (1.01–3.32) | 0.094 |
| Modified SRI(8) | 16/92 (17.4) | 32/90 (35.6) | 2.67 (1.49–4.80) | 0.006 | 26/95 (27.4) | 1.82 (1.00–3.29) | 0.099 |
| Physician's global assessment decreaseg | 55/102 (53.9) | 74/99 (74.7) | 2.81 (1.67–4.71) | 0.001 | 63/104 (60.6) | 1.37 (0.85–2.20) | 0.281 |
| Clinical SLEDAIh | 44/102 (43.1) | 62/99 (62.6) | 2.35 (1.45–3.81) | 0.004 | 54/104 (51.9) | 1.46 (0.91–2.32) | 0.185 |
| SLEDAI ≤2 | 18/102 (17.6) | 35/99 (35.4) | 2.68 (1.53–4.70) | 0.004 | 34/104 (32.7) | 2.35 (1.34–4.11) | 0.012 |
| SLEDAI = 0 | 8/102 (7.8) | 18/99 (18.2) | 2.66 (1.25–5.64) | 0.033 | 20/104 (19.2) | 2.90 (1.38–6.08) | 0.018 |
| Major clinical responsei | 7/102 (6.9) | 19/99 (19.2) | 3.24 (1.49–7.04) | 0.012 | 18/104 (17.3) | 2.88 (1.32–6.26) | 0.025 |
| Disease flares | |||||||
| BILAG A or 2B | 17/102 (16.7) | 12/99 (12.1) | 0.71 (0.36–1.42) | 0.421 | 12/104 (11.5) | 0.68 (0.34–1.36) | 0.359 |
| BILAG A | 17/102 (16.7) | 9/99 (9.1) | 0.51 (0.24–1.07) | 0.134 | 11/104 (10.6) | 0.61 (0.30–1.24) | 0.253 |
| Fatiguej | 34/98 (34.7) | 41/96 (42.7) | 1.46 (0.89–2.38) | 0.207 | 43/104 (41.3) | 1.35 (0.83–2.18) | 0.308 |
| SF‐36 | |||||||
| MCSk | 27/102 (26.5) | 36/99 (36.4) | 1.68 (1.00–2.79) | 0.097 | 35/104 (33.7) | 1.44 (0.86–2.39) | 0.240 |
| PCSl | 40/102 (39.2) | 48/99 (48.5) | 1.51 (0.94–2.44) | 0.154 | 43/104 (41.3) | 1.11 (0.69–1.78) | 0.726 |
Values are the number of patients/number assessed (%). Treatment was given every 4 weeks from week 1 to week 48. The observed response rates of each treatment group are shown. SLE = systemic lupus erythematosus; SRI = SLE Responder Index; IFN = interferon; BICLA = British Isles Lupus Assessment Group (BILAG)–based Combined Lupus Assessment; SRI(5) = SRI requiring SLE Disease Activity Index 2000 (SLEDAI‐2K) reductions of 5 points; SF‐36 = Short Form 36; MCS = mental component summary; PCS = physical component summary.
Odds ratio (ORs), 90% confidence intervals (90% CIs), and nominal P values are from a logistic regression model for comparisons of each anifrolumab group versus placebo adjusted for randomization stratification factors.
Reduction of oral corticosteroid dosage to ≤7.5 mg/day in patients who were receiving ≥10 mg/day at baseline.
≥50% decrease in the Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI) activity score in patients who had a score of ≥10 at baseline.
≥50% decrease in the swollen and tender joint count from baseline in patients with ≥8 swollen and ≥8 tender joints at baseline.
Reduction of baseline BILAG 2004 index A to B/C/D and B to C/D, no BILAG 2004 index worsening in other organ domains (no new BILAG score of A or B), increase in total SLEDAI‐2K of <1, and increase in the physician's global assessment of <0.3.
≥0.3‐point improvement from baseline.
≥4‐point reduction in clinical components (no laboratory components) of the SLEDAI.
BILAG 2004 score of C or better in all organ domains at week 24 with maintenance of this response through week 52.
>3‐point improvement from baseline in Functional Assessment of Chronic Illness Therapy–Fatigue score.
≥3.8‐point improvement from baseline.
≥3.1‐point improvement from baseline.
Figure 1.
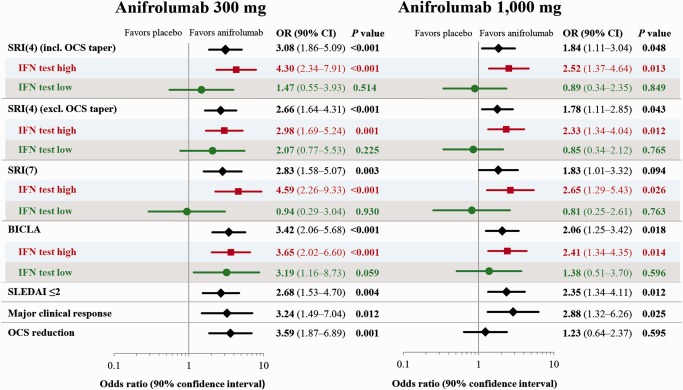
Efficacy results at week 52 in patients with systemic lupus erythematosus (SLE) receiving anifrolumab 300 mg, anifrolumab 1,000 mg, or placebo. Anifrolumab treatment led to a greater rate of response across multiple end points. The benefit observed in the overall modified intent‐to‐treat population was driven by the patients with a high interferon (IFN) gene signature (IFN test high subpopulation), which represents ∼75% of the entire cohort. The odds ratios (ORs), 90% confidence intervals (90% CIs), and P values are from a logistic regression model adjusted for stratification factors. SRI(4) = SLE Responder Index requiring a ≥4‐point reduction in SLE Disease Activity Index 2000 (SLEDAI‐2K) score; OCS = oral corticosteroid; BICLA = British Isles Lupus Assessment Group 2004–based Combined Lupus Assessment.
Consistent with the primary end point results, a greater percentage of patients receiving anifrolumab met criteria for SRI(4) response without the oral corticosteroid taper requirement (Table 2 and Figure 1), and the modified SRI responses, requiring 5‐, 6‐, 7‐, or 8‐point reductions in SLEDAI‐2K scores. Both anifrolumab dosages were associated with higher rates of response in the BICLA compared with placebo (Table 2, Figure 2, and Supplementary Figure 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39962/abstract). The differentiation between placebo and anifrolumab occurred within 6 months and was maintained or increased through week 52 (Figure 2). Improvements in physician's global assessment at week 52 were also greater for the anifrolumab‐treated groups than for the placebo‐treated group (Table 2), with a greater effect observed in the 300‐mg treatment group compared with the 1,000‐mg treatment group. For all outcomes, the treatment effects observed in the overall population were driven by the IFN‐high subset (Table 2 and Figure 2).
Figure 2.

Efficacy results over time in patients with systemic lupus erythematosus (SLE) receiving anifrolumab 300 mg, anifrolumab 1,000 mg, or placebo. Treatment was given every 4 weeks from week 1 to week 48. A, Proportion of patients achieving an SLE Responder Index response over time in the modified intent‐to‐treat (ITT) population and in the interferon (IFN)–high and IFN‐low subsets. B, Proportion of patients achieving a British Isles Lupus Assessment Group 2004–based Combined Lupus Assessment response over time in the modified ITT population and in the IFN‐high and IFN‐low subsets.
The study also assessed various measures of low disease activity. A SLEDAI‐2K score of ≤2 at week 52 was achieved in 17.6% in the placebo group, 35.4% in the anifrolumab 300‐mg group (P = 0.004 versus placebo), and 32.7% in the anifrolumab 1,000‐mg group (P = 0.012 versus placebo) (see Supplementary Figure 4, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39962/abstract), and a score of 0 was achieved in 7.8% in the placebo group, 18.2% in the anifrolumab 300‐mg group (P = 0.033 versus placebo), and 19.2% in the anifrolumab 1,000‐mg group (P = 0.018 versus placebo). Major clinical response, defined as a BILAG 2004 score of C or better in all organ domains at week 24 with maintenance of this response through week 52, was achieved in 6.9%, 19.2% (P = 0.012), and 17.3% (P = 0.025) in the placebo, anifrolumab 300‐mg, and anifrolumab 1,000‐mg groups, respectively (Table 2 and Supplementary Figure 4).
The numbers of patients who developed new BILAG 1A or 2B flares at any time during the study were reduced by 28% and 29%, and the numbers who developed new BILAG A flares were reduced by 46% and 35%, in the anifrolumab 300‐mg and anifrolumab 1,000‐mg groups, respectively (Table 2).
A reduction in background oral corticosteroid dosage to ≤7.5 mg/day at week 52 in those taking ≥10 mg/day at baseline was achieved in 56.4% of the patients in the anifrolumab 300‐mg group (P = 0.001) and 31.7% of the patients in the anifrolumab 1,000‐mg group (P = 0.595), compared with 26.6% of the patients receiving placebo.
Anifrolumab‐treated patients demonstrated greater improvements than those receiving placebo across a range of organ‐specific disease measures and patient‐reported outcomes. The percentage of patients with a baseline CLASI activity score of ≥10 who had a ≥50% reduction in this score by week 52 was greater for both anifrolumab dosages (63.0% for 300 mg [P = 0.013] and 58.3% for 1,000 mg [P = 0.077]) compared with placebo (30.8%) (Table 2 and Figure 3). Improvement in arthritis was also greater in patients receiving anifrolumab (Figure 3 and Supplementary Figure 4). In patients with ≥8 swollen and ≥8 tender joints at baseline, a ≥50% decrease in both the swollen and tender joint count was achieved in a greater percentage of anifrolumab‐treated patients than placebo‐treated patients (48.6% for placebo, 69.6% for anifrolumab 300 mg [P = 0.038], and 64.6% for anifrolumab 1,000 mg [P = 0.156]). Compared with placebo, greater proportions of patients in the anifrolumab 300‐mg group achieved a >3‐point improvement in the FACIT‐F scale, a ≥3.1‐point improvement in the physical component summary score of the SF‐36 health survey, and a ≥3.8‐point improvement in the mental component summary score of the SF‐36 health survey. However, none of these comparisons achieved statistical significance (Table 2).
Figure 3.
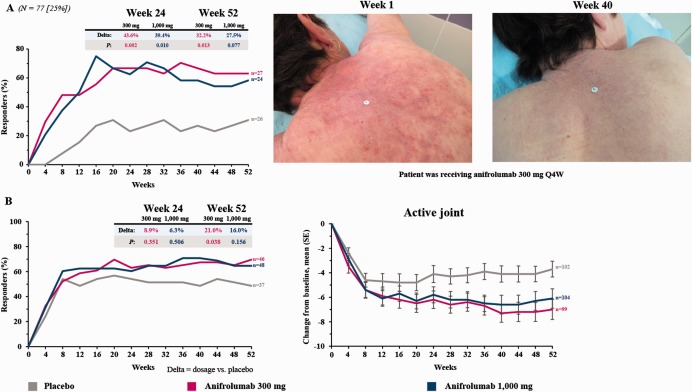
Organ‐specific efficacy results over time in patients with systemic lupus erythematosus (SLE) receiving anifrolumab 300 mg, anifrolumab 1,000 mg, or placebo. Treatment was given every 4 weeks from week 1 to week 48. A, Left, Proportion of patients with a Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI) activity score of ≥10 at baseline (n = 77) who had ≥50% improvement in the CLASI score. Right, A representative example of skin response following anifrolumab treatment. B, Left, Proportion of patients with ≥8 swollen and ≥8 tender joints at baseline (n = 37 in the placebo group, n = 46 in the anifrolumab 300‐mg group, and n = 48 in the anifrolumab 1,000‐mg group) who had ≥50% improvement in the swollen and tender joint count. Right, Mean ± SEM change from baseline in the active joint count in the modified intent‐to‐treat population. Joint counts are based on the assessment of 28 joints.
Assessments of biologic parameters were undertaken to investigate response following administration of anifrolumab. In patients with anti‐dsDNA antibodies detectable by the AtheNA Multi‐Lyte ANA‐II Plus assay at baseline, a numerically greater decrease in anti‐dsDNA antibodies was observed with anifrolumab than with placebo (mean ± SD −24.4 ± 177.6 for placebo, −70.3 ± 166.9 for anifrolumab 300 mg [P = 0.067], and −43.8 ± 86.3 for anifrolumab 1,000 mg [P = 0.144]). Similarly, in patients with low complement C3 concentrations at baseline, a nonsignificant numerically greater increase in complement C3 concentrations was observed at week 52 in patients treated with anifrolumab compared with those treated with placebo (mean ± SD 7.7 ± 18.9 for placebo, 12.9 ± 19.1 for anifrolumab 300 mg [P = 0.277], and 12.0 ± 16.5 for anifrolumab 1,000 mg [P = 0.242]) (see Supplementary Figure 6, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39962/abstract). No effect on complement C4 was observed. The median neutralization ratios of a 21‐gene type I IFN signature in the IFN‐high subpopulation at week 24 were 89.7 and 91.7 for the anifrolumab 300‐mg and anifrolumab 1,000‐mg groups, respectively. This degree of suppression was maintained until week 52. No neutralization in gene expression was observed with placebo. The rates of antidrug antibodies at any time postbaseline were low, with no difference between treatment groups (3% for placebo, 5.1% for anifrolumab 300 mg, and 2.0% for anifrolumab 1,000 mg).
Safety
The safety population consisted of the 305 patients who received at least 1 dose of study drug. One patient randomized to the placebo group mistakenly received a single dose of anifrolumab (1,000 mg). This patient was included in the 1,000‐mg anifrolumab group for the safety analyses. The percentages of patients with at least 1 adverse event (77.2% in the placebo group, 84.8% in the anifrolumab 300‐mg group, and 85.7% in the anifrolumab 1,000‐mg group) and serious adverse events (18.8% in the placebo group, 16.2% in the anifrolumab 300‐mg group, and 17.1% in the anifrolumab 1,000‐mg group) were similar across all treatment groups. Adverse events led to treatment discontinuation in 7.9% of the patients receiving placebo and 3.0% and 9.5% of the patients receiving anifrolumab 300 mg and anifrolumab 1,000 mg, respectively. The most frequently reported adverse events were headache, upper respiratory tract infection, nasopharyngitis, and urinary tract infection (Table 3). There was 1 death (due to sepsis syndrome) prior to randomization, and 1 death during the study in a patient who received 1 dose of anifrolumab 1,000 mg. The patient had acute colitis with a rapidly deteriorating clinical course and had features of macrophage activation syndrome prior to death. No infectious agent was identified. Autopsy confirmed the cause of death to be acute colitis and revealed portal vein thrombosis and increased numbers of phagocytic macrophages in the bone marrow, consistent with macrophage activation syndrome. This was not considered related to the study drug by the investigator.
Table 3.
Adverse events (safety population)a
| Placebo (n = 101) | Anifrolumab 300 mg (n = 99) | Anifrolumab 1,000 mg (n = 105) | Both anifrolumab doses (n = 204) | |
|---|---|---|---|---|
| Any adverse event | 78 (77.2) | 84 (84.8) | 90 (85.7) | 174 (85.3) |
| Serious adverse events | 19 (18.8) | 16 (16.2) | 18 (17.1) | 34 (16.7) |
| Death | 0 (0.0) | 0 (0.0) | 1 (1.0) | 1 (0.5) |
| Adverse events leading to discontinuation | 8 (7.9) | 3 (3.0) | 10 (9.5) | 13 (6.4) |
| Treatment‐related adverse events leading to discontinuation | 2 (2.0) | 1 (1.0) | 4 (3.8) | 5 (2.5) |
| Adverse events of special interestb | 12 (11.9) | 10 (10.1) | 15 (14.3) | 25 (12.3) |
| Most common adverse eventsc | ||||
| Headache | 13 (12.9) | 12 (12.1) | 12 (11.4) | 24 (11.8) |
| Upper respiratory tract infection | 10 (9.9) | 13 (13.1) | 11 (10.5) | 24 (11.8) |
| Nasopharyngitis | 4 (4.0) | 12 (12.1) | 12 (11.4) | 24 (11.8) |
| Urinary tract infection | 11 (10.9) | 15 (15.2) | 7 (6.7) | 22 (10.8) |
| Bronchitis | 4 (4.0) | 7 (7.1) | 9 (8.6) | 16 (7.8) |
| Herpes zoster | 2 (2.0) | 5 (5.1)d | 10 (9.5) | 15 (7.4) |
| Influenza | 2 (2.0) | 6 (6.1) | 8 (7.6) | 14 (6.9) |
| Diarrhea | 4 (4.0) | 4 (4.0) | 8 (7.6) | 12 (5.9) |
| Sinusitis | 3 (3.0) | 6 (6.1) | 6 (5.7) | 12 (5.9) |
| Cough | 2 (2.0) | 3 (3.0) | 8 (7.6) | 11 (5.4) |
Values are the number (%). Treatment was given every 4 weeks from week 1 to week 48.
New or reactivated tuberculosis infection, herpes zoster infection, malignancy, or reactions associated with infusion, hypersensitivity, or anaphylaxis.
Adverse events (preferred term) reported by >5% of patients in the total (both doses) anifrolumab group.
One patient also had transverse myelitis with a quantitatively positive test result for varicella‐zoster virus in the cerebrospinal fluid.
Herpes zoster infections were reported in 5.1% and 9.5% of the patients in the anifrolumab 300‐mg and 1,000‐mg groups, respectively, compared with 2.0% in the placebo group. All patients responded promptly to antiviral treatment. One patient in the 300‐mg group discontinued treatment due to transverse myelitis with a positive PCR test for herpes zoster in the cerebrospinal fluid; the patient responded to antiviral and high‐dose corticosteroid treatment. The percentages of patients who were reported as developing influenza were greater in the anifrolumab 300‐mg group (6.1%) and anifrolumab 1,000‐mg group (7.6%) than in the placebo group (2.0%). Infusion‐related reactions were reported in 6 (5.9%), 2 (2.0%), and 4 (3.8%) of the patients in the placebo, anifrolumab 300‐mg, and anifrolumab 1,000‐mg groups, respectively. Upper respiratory infections (reported under various terms) occurred more frequently in patients treated with anifrolumab (36.4% in the anifrolumab 300‐mg group and 41.9% in the anifrolumab 1,000‐mg group) compared with placebo (28.7%). No clinically important worsening was observed in hematology or chemistry panels, urinalysis, vital signs, lipid parameters, or electrocardiograms in any of the treatment groups.
DISCUSSION
The primary and secondary end points of this study were met. Furthermore, compared with placebo, anifrolumab treatment resulted in significantly greater rates of improvement across a broad range of composite and organ‐specific disease activity measures as well as in the achievement and maintenance of low disease activity, corticosteroid tapering (in the 300‐mg group), and a trend toward flare rate reduction. These results provide compelling evidence that blocking IFNAR is a promising strategy in the treatment of SLE.
The analysis of the primary end point in 2 populations (the modified ITT population and a modified ITT subpopulation of patients with a high IFN gene signature at screening) was based on the mechanism of action of anifrolumab and the assumption that demonstrating statistically significant benefit in the primary end point at a 2‐sided alpha level of 0.1 in either of the populations would provide sufficient preliminary evidence of efficacy to continue phase III studies in the appropriate population. A 2‐sided alpha level of 0.1 represents a 5% chance of declaring a positive study result when there is no treatment effect (risk of proceeding with an ineffective drug). The present study had a power of 88%, representing a 12% chance of declaring a negative study result when there was a positive treatment effect (risk of discontinuing development of a potentially efficacious drug). This combination of statistical risks was chosen to balance the continuation and discontinuation risks while maintaining a feasible phase IIb study. Further, to keep the sample size within reasonable limits, no adjustment was made for the multiplicity of testing the primary end point in the 2 populations. Therefore, the results of this study are clinically relevant, but are not definitive until prospectively replicated in larger studies with a more stringent alpha level and a more robust control for multiplicity.
The anifrolumab dosages were selected based on pharmacokinetic and pharmacodynamic modeling and simulation from a previous study 18, 19. Both the 300‐mg and 1,000‐mg dosages were more efficacious than placebo, with the lower dosage showing greater numerical response rates at week 52 for many end points (Table 2). The fact that the degrees of IFN gene signature inhibition were similar with the low and high anifrolumab dosages supports a plateau effect rather than an inverse dosage response. However, other mechanisms, such as an increased response in inducible immunologic feedback leading to a lower rate of response with the greater anifrolumab dosage, cannot be excluded. Biomarker studies to address this question are currently underway.
The fact that greater efficacy was seen in all end points in patients with high baseline IFN gene signatures compared with those with low baseline IFN gene signatures supports the pathobiology of this treatment strategy. The lack of treatment effect in the IFN‐low subgroup was due to a greater response to placebo and standard therapy and not to a lower response to anifrolumab, suggesting that the IFN‐high subgroup represents a subpopulation more likely to benefit from the addition of anifrolumab to standard therapy. The interpretation of the results in the IFN‐low subgroup is limited by its small size, and thus larger studies are needed to fully evaluate the effect of anifrolumab in this population.
Anifrolumab treatment was well tolerated, and adverse events reported were similar across the 3 treatment groups. Consistent with the mechanism of action of anifrolumab, a dosage‐related increase was observed in the occurrence of upper respiratory infections and reactivation of herpes zoster. Importantly, responses to antiviral treatment were prompt in the herpes zoster cases. In addition, there were more cases reported as influenza in patients treated with anifrolumab; however, most of these cases were not confirmed by laboratory testing or the application of strict clinical criteria. To accurately assess the true risk of influenza, future studies should require laboratory confirmation of influenza infections. Ensuring that patients’ immunizations are up to date prior to administration of anifrolumab may decrease the risk of influenza and other infections. The dosage‐related increase in these infections, together with the fact that increasing the dosage from 300 mg to 1,000 mg did not lead to an increase in efficacy, indicates a more favorable risk–benefit profile for the 300‐mg dosage.
This is the most successful phase II study in SLE to date. The benefits achieved across multiple global and organ‐specific disease activity measures compare favorably to those observed in other recent studies of potential lupus therapies 15, 16, 29, 31. The phase IIb study evaluating sifalimumab in a similar cohort of patients met its primary and some of its secondary end points, albeit with smaller treatment effects 16. The greater efficacy and broader impact demonstrated by anifrolumab are likely the result of achieving greater suppression of the type I IFN pathway. The positive results of the present study suggest that targeting IFNAR is a promising and novel therapeutic approach for patients with SLE whose disease does not respond to currently available therapies.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Furie had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Furie, Drappa, Wang, Yoo.
Acquisition of data
Furie, Khamashta, Merrill, Werth, Kalunian, Brohawn, Wang, Yoo.
Analysis and interpretation of data
Furie, Khamashta, Merrill, Werth, Kalunian, Brohawn, Illei, Drappa, Wang, Yoo.
ROLE OF THE STUDY SPONSOR
The study was funded by MedImmune. All authors interpreted the data, critically reviewed the manuscript for important intellectual content, approved the final draft, and agreed to its submission. Publication of the article was contingent upon approval by MedImmune. Writing assistance was provided by Katie Alexander, PhD (QXV Communications, an Ashfield business, part of UDG Healthcare, Macclesfield, UK) and fully funded by MedImmune.
ADDITIONAL DISCLOSURES
Author Yoo is an employee of Regenxbio.
Supporting information
Supplementary Figure S1. Patient disposition.
Supplementary Figure S2. SRI(4) including OCS taper responses at Week 24 and Week 52 in the mITT population and in the IFN‐low and IFN‐high subpopulations.
IFN = interferon; mITT = modified intention‐to‐treat; OCS = oral corticosteroid; Q4W = every 4 weeks; SRI(4) = Systemic Lupus Erythematosus Responder Index.
Supplementary Figure S3. Percentage of patients achieving a modified SRI (mSRI) response (requiring SLEDAI–2K reductions of 5–8 points) over time.
Q4W = every 4 weeks; SRI = Systemic Lupus Erythematosus Responder Index.
Supplementary Figure S4. Panel A shows joint count over time. ≥20% improvement in the swollen and tender joint count in patients with ≥8 swollen and ≥8 tender joints at baseline (placebo: n=37; anifrolumab 300 mg: n=46; anifrolumab 1,000 mg: n=48). Panel B shows mean change (SE) from baseline in swollen joint count (middle) and tender joint count (right) in the mITT population. The joint counts are based on the assessment of 28 joints.
Q4W = every 4 weeks; SE = standard error; mITT = modified intention‐to‐treat.
Supplementary Figure S5. Achieving low disease activity at Week 52. Patients achieving a SLEDAI‐2K score of ≤2 (left). Major clinical response, defined as BILAG 2004 “C” or better in all organ domains at Week 24 with maintenance of this response through Week 52 (right).
BILAG = British Isles Lupus Assessment Group; mITT = modified intention‐to‐treat; Q4W = every 4 weeks; SLEDAI–2K = Systemic Lupus Erythematosus Disease Activity Index 2000.
Supplementary Figure S6. Change in serologies. Mean (SE) decrease in anti‐dsDNA antibodies in patients with detectable anti‐dsDNA antibodies by the AtheNA Multi‐Lyte® ANA‐II Plus assay at baseline (left). Mean (SE) increase in complement C3 and C4 concentrations in patients with low complement C3 and C4 concentrations, respectively at baseline.dsDNA = double‐stranded DNA; mITT = modified intention‐to‐treat; Q4W = every 4 weeks; SE = standard error.
ACKNOWLEDGMENTS
The authors would like to thank the patients and investigators and their site personnel who contributed to the study.
ClinicalTrials.gov identifier: NCT01438489.
Supported by MedImmune.
Dr. Furie has received consulting fees (more than $10,000) and research grants from MedImmune. Professor Khamashta has received consulting fees from MedImmune, GlaxoSmithKline, and UCB and speaking fees from Inova Diagnostics (less than $10,000 each) and research grants from Bayer. Dr. Merrill has received consulting fees from MedImmune and Genentech/Roche (less than $10,000 each) and research grants from Genentech. Dr. Werth has received consulting fees from MedImmune (less than $10,000) and holds a patent for the Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI; owned by the University of Pennsylvania) used in this study. Dr. Kalunian has received consulting fees from MedImmune and AstraZeneca (less than $10,000 each) and research grants from MedImmune. Mr. Brohawn and Drs. Illei, Wang, and Yoo own stock or stock options in AstraZeneca. Dr. Drappa owns stock options in AstraZeneca and holds a patent relating to type I IFN in systemic lupus erythematosus.
REFERENCES
- 1. Mathian A, Hie M, Cohen‐Aubart F, Amoura Z. Targeting interferons in systemic lupus erythematosus: current and future prospects. Drugs 2015;75:835–46. [DOI] [PubMed] [Google Scholar]
- 2. Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum 2006;54:2550–7. [DOI] [PubMed] [Google Scholar]
- 3. Panopalis P, Clarke AE, Yelin E. The economic burden of systemic lupus erythematosus. Best Pract Res Clin Rheumatol 2012;26:695–704. [DOI] [PubMed] [Google Scholar]
- 4. Yurkovich M, Vostretsova K, Chen W, Aviña‐Zubieta JA. Overall and cause‐specific mortality in patients with systemic lupus erythematosus: a meta‐analysis of observational studies. Arthritis Care Res (Hoboken) 2014;66:608–16. [DOI] [PubMed] [Google Scholar]
- 5. Petri M. Long‐term outcomes in lupus. Am J Manag Care 2001;7 Suppl:S480–5. [PubMed] [Google Scholar]
- 6. Doria A, Briani C. Lupus: improving long‐term prognosis. Lupus 2008;17:166–70. [DOI] [PubMed] [Google Scholar]
- 7. Ruiz‐Irastorza G, Danza A, Khamashta M. Glucocorticoid use and abuse in SLE. Rheumatology (Oxford) 2012;51:1145–53. [DOI] [PubMed] [Google Scholar]
- 8. Lauwerys BR, Ducreux J, Houssiau FA. Type I interferon blockade in systemic lupus erythematosus: where do we stand? Rheumatology (Oxford) 2014;53:1369–76. [DOI] [PubMed] [Google Scholar]
- 9. Crow MK, Olferiev M, Kirou KA. Targeting of type I interferon in systemic autoimmune diseases. Transl Res 2015;165:296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol 2014;192:5459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rönnblom L, Alm GV, Eloranta ML. The type I interferon system in the development of lupus. Semin Immunol 2011;23:113–21. [DOI] [PubMed] [Google Scholar]
- 12. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014;14:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lichtman EI, Helfgott SM, Kriegel MA. Emerging therapies for systemic lupus erythematosus: focus on targeting interferon‐α. Clin Immunol 2012;143:210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crow MK. Advances in understanding the role of type I interferons in systemic lupus erythematosus. Curr Opin Rheumatol 2014;26:467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, et al. A phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon‐α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis 2016;75:196–202. [DOI] [PubMed] [Google Scholar]
- 16. Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti‐interferon‐α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double‐blind, placebo‐controlled study. Ann Rheum Dis 2016;75:1909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peng L, Oganesyan V, Wu H, Dall'Acqua WF, Damschroder MM. Molecular basis for antagonistic activity of anifrolumab, an anti‐interferon‐α receptor 1 antibody. MAbs 2015;7:428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goldberg A, Geppert T, Schiopu E, Frech T, Hsu V, Simms RW, et al. Dose‐escalation of human anti‐interferon‐α receptor monoclonal antibody MEDI‐546 in subjects with systemic sclerosis: a phase 1, multicenter, open label study. Arthritis Res Ther 2014;16:R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang B, Higgs BW, Chang L, Vainshtein I, Liu Z, Streicher K, et al. Pharmacogenomics and translational simulations to bridge indications for an anti‐interferon‐α receptor antibody. Clin Pharmacol Ther 2013;93:483–92. [DOI] [PubMed] [Google Scholar]
- 20. Hochberg MC, for the Diagnostic and Therapeutic Criteria Committee of the American College of Rheumatology . Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- 21. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982;25:1271–7. [DOI] [PubMed] [Google Scholar]
- 22. Gladman DD, Ibañez D, Urowitz MB. Systemic Lupus Erythematosus Disease Activity Index 2000. J Rheumatol 2002;29:288–91. [PubMed] [Google Scholar]
- 23. Isenberg DA, Rahman A, Allen E, Farewell V, Akil M, Bruce IN, et al. BILAG 2004: development and initial validation of an updated version of the British Isles Lupus Assessment Group's disease activity index for patients with systemic lupus erythematosus. Rheumatology (Oxford) 2005;44:902–6. [DOI] [PubMed] [Google Scholar]
- 24. Yee CS, Cresswell L, Farewell V, Rahman A, Teh LS, Griffiths B, et al. Numerical scoring for the BILAG‐2004 index. Rheumatology (Oxford) 2010;49:1665–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yao Y, Higgs BW, Richman L, White B, Jallal B. Use of type I interferon‐inducible mRNAs as pharmacodynamic markers and potential diagnostic markers in trials with sifalimumab, an anti‐IFNα antibody, in systemic lupus erythematosus. Arthritis Res Ther 2010;12 Suppl 1:S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Furie RA, Petri MA, Wallace DJ, Ginzler EM, Merrill JT, Stohl W, et al. Novel evidence‐based systemic lupus erythematosus responder index. Arthritis Rheum 2009;61:1143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Albrecht J, Taylor L, Berlin JA, Dulay S, Ang G, Fakharzadeh S, et al. The CLASI (Cutaneous Lupus Erythematosus Disease Area and Severity Index): an outcome instrument for cutaneous lupus erythematosus. J Invest Dermatol 2005;125:889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klein R, Moghadam‐Kia S, LoMonico J, Okawa J, Coley C, Taylor L, et al. Development of the CLASI as a tool to measure disease severity and responsiveness to therapy in cutaneous lupus erythematosus. Arch Dermatol 2011;147:203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wallace DJ, Kalunian K, Petri MA, Strand V, Houssiau FA, Pike M, et al. Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: results from EMBLEM, a phase IIb, randomised, double‐blind, placebo‐controlled, multicentre study. Ann Rheum Dis 2014;73:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith E, Lai JS, Cella D. Building a measure of fatigue: the functional assessment of Chronic Illness Therapy Fatigue Scale. PM R 2010;2:359–63. [DOI] [PubMed] [Google Scholar]
- 31. Furie RA, Leon G, Thomas M, Petri MA, Chu AD, Hislop C, et al. A phase 2, randomised, placebo‐controlled clinical trial of blisibimod, an inhibitor of B cell activating factor, in patients with moderate‐to‐severe systemic lupus erythematosus, the PEARL‐SC study. Ann Rheum Dis 2015;74:1667–75. [DOI] [PubMed] [Google Scholar]
- 32. Khamashta MA, Illei GG, Yoo S, Wang L, Greth W. Decreased sensitivity of a commercial anti‐dsDNA assay in patients with moderately to severely active systemic lupus erythematosus [abstract]. Arthritis Rheum 2013;65 Suppl 10:S669–70. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Patient disposition.
Supplementary Figure S2. SRI(4) including OCS taper responses at Week 24 and Week 52 in the mITT population and in the IFN‐low and IFN‐high subpopulations.
IFN = interferon; mITT = modified intention‐to‐treat; OCS = oral corticosteroid; Q4W = every 4 weeks; SRI(4) = Systemic Lupus Erythematosus Responder Index.
Supplementary Figure S3. Percentage of patients achieving a modified SRI (mSRI) response (requiring SLEDAI–2K reductions of 5–8 points) over time.
Q4W = every 4 weeks; SRI = Systemic Lupus Erythematosus Responder Index.
Supplementary Figure S4. Panel A shows joint count over time. ≥20% improvement in the swollen and tender joint count in patients with ≥8 swollen and ≥8 tender joints at baseline (placebo: n=37; anifrolumab 300 mg: n=46; anifrolumab 1,000 mg: n=48). Panel B shows mean change (SE) from baseline in swollen joint count (middle) and tender joint count (right) in the mITT population. The joint counts are based on the assessment of 28 joints.
Q4W = every 4 weeks; SE = standard error; mITT = modified intention‐to‐treat.
Supplementary Figure S5. Achieving low disease activity at Week 52. Patients achieving a SLEDAI‐2K score of ≤2 (left). Major clinical response, defined as BILAG 2004 “C” or better in all organ domains at Week 24 with maintenance of this response through Week 52 (right).
BILAG = British Isles Lupus Assessment Group; mITT = modified intention‐to‐treat; Q4W = every 4 weeks; SLEDAI–2K = Systemic Lupus Erythematosus Disease Activity Index 2000.
Supplementary Figure S6. Change in serologies. Mean (SE) decrease in anti‐dsDNA antibodies in patients with detectable anti‐dsDNA antibodies by the AtheNA Multi‐Lyte® ANA‐II Plus assay at baseline (left). Mean (SE) increase in complement C3 and C4 concentrations in patients with low complement C3 and C4 concentrations, respectively at baseline.dsDNA = double‐stranded DNA; mITT = modified intention‐to‐treat; Q4W = every 4 weeks; SE = standard error.