

Abstract
While chemoselectivities in Pd0‐catalyzed coupling reactions are frequently non‐intuitive and a result of a complex interplay of ligand/catalyst, substrate, and reaction conditions, we herein report a general method based on PdI that allows for an a priori predictable chemoselective C −C coupling at C−Br in preference to C−OTf and C−Cl bonds, regardless of the electronic or steric bias of the substrate. The C−C bond formations are extremely rapid (<5 min at RT) and are catalyzed by an air‐ and moisture‐stable PdI dimer under open‐flask conditions.
Keywords: chemoselectivity, density-functional calculations, heterocycles, palladium, reaction mechanisms
The biaryl motif is not only present in numerous drug molecules, but it is also a ubiquitous building block in materials science and complex natural products. Its substitution pattern ultimately determines the function as pharmaceutical, agrochemical, electronic device, secondary metabolite, or even privileged ligand.1 Consequently, the development of a synthetic repertoire to selectively access diversely functionalized arenes is of considerable interest. Owing to the relative mildness, palladium‐catalyzed chemoselective cross‐coupling strategies of poly(pseudo)halogenated arenes represent a strategy of considerable interest.2 In this context, the oxidative addition step is generally selectivity‐controlling. While the relative ease of oxidative addition is frequently referred to as C−I>C−OTf≈C−Br>C−Cl,2 these rough trends by no means allow an a priori prediction of favored coupling site. Instead, it is a result of subtle interplay between substrate (electronic/steric bias and bond strength), catalyst (ligand/ligation state), solvent, and additive effects (Figure 1).2, 3 For example, while [PdCl2{P(o‐tol)3}2]‐catalyzed Kumada coupling of A gave selective C−Br functionalization, the introduction of two methyl groups ortho to C−Br (B) diminished selectivities, thus resulting in mixtures under otherwise identical reaction conditions (Figure 1).4 A similar substrate dependence was observed for [Pd(PPh3)4]‐catalyzed Suzuki couplings: while C−Br was the favored coupling site for C, analogous reaction conditions resulted in preferential coupling at the C−OTf site for D.5 Although progress has been made in the development of predictive models6 and in the mechanistic understanding of the factors that dictate site selectivity,2, 3, 7 selective couplings frequently remain a result of rigorous screening activities. However, modern research programs not only seek for greater sustainability (i.e. less waste), but also frequently involve iterative, programmable synthetic approaches to increase chemical diversity.8 As such, a general and predictive coupling protocol would be highly desired, particularly if paired with operational simplicity.
Figure 1.
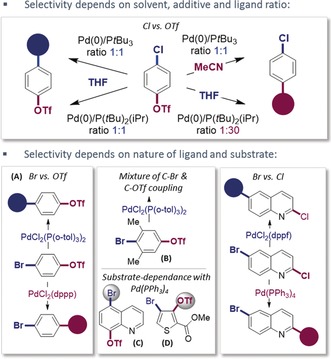
Examples of divergent selectivities.3, 4 Tf=trifluoromethanesulfonyl.
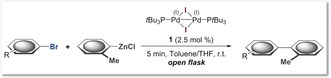
Clearly, the ultimate site selectivity is strongly affected by subtle changes in substrate, ligand, or reaction conditions in the context of palladium(0) catalysis. This selectivity is likely due to modifications of the electron‐richness of the active palladium(0) catalyst. Depending on the nature of the ligand and reaction conditions, multiple potentially reactive species may coexist, ranging from PdLn to anionic [PdLnX]− (n=1 or 2)3, 7, 9 and could in turn trigger divergent selectivities.3d We therefore envisioned that utilizing a different oxidation state (I), may be advantageous and potentially allow more pronounced chemoselectivities. We recently showed that the iodine‐bridged palladium(I) dimer 1 (shown in Table 1) is an air‐ and moisture‐stable, as well as thermally stable species which functions as and efficient catalyst for C−SCF3, C−SeCF3, and C−Br bond formations with aryl iodides and bromides.10, 11 Our mechanistic data indicated that the catalysis proceeded by dinuclear palladium(I) cycles. In this context, the cross‐coupling partner was employed as a nucleophile, which was incorporated as bridging unit in the PdI–PdI framework and subsequently exchanged with an aryl halide to give the functionalized arene. Whether carbon‐based nucleophiles, such as aryl Grignard or organozinc species, could also function as bridges in dinuclear palladium(I) catalysis, was unclear. Our initial studies on Kumada couplings suggested that high reactvities could be achieved with 1 however.12 Efficient Kumada and Negishi couplings of aryl (pseudo)halides have otherwise been developed for single coupling possibilities, using air‐sensitive palladium(0) [or nickel(0)] catalysis, with reaction times of several hours.13 Notably, Knochel and co‐workers observed more rapid Negishi and Kumada cross‐coupling reactions under modified palladium(0) catalysis conditions, which was ascribed to the potential involvement of radicals.14 A general method for the realization of chemoselective couplings has not been reported.15 To test whether selective cross‐couplings would be possible, we subjected phenyl Grignard along with 2.5 mol % of the 1 in toluene to the densely functionalized substrate 2, which has competitive possibilities for C−Br, C−OTf, and C−Cl couplings (Scheme 1 a). All reagents were handled in air and the reaction was performed in an open flask. Pleasingly, we saw exclusive coupling at C−Br. The reaction was remarkably rapid and completed in less than 5 minutes at room temperature. In stark contrast, when we subjected other air‐stable complexes based on palladium(II), we observed sluggish reactivity after 5–24 hours, thus obtaining low conversion and no pronounced selectivity under open‐flask or inert reaction conditions (Scheme 1 a).
Table 1.
C−Br selective Kumada cross‐couplings.[a]
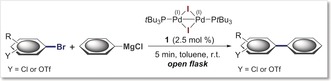
|
[a] Reaction conditions: 1 (4.4 mg, 0.005 mmol), ArBr (0.2 mmol), PhMgCl (2 m in THF, 150 μL, 0.3 mmol), toluene (3.0 mL). Yield is that of isolated product. [b] 5 mol % of 1 was used. [c] Used 1.8 equiv of PhMgCl. [d] Used 2 equiv of PhMgCl. [e] Used 2.5 equiv of PhMgCl.
Scheme 1.
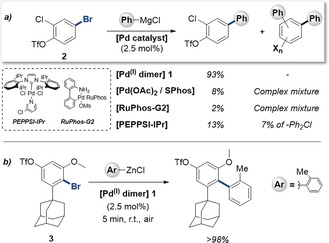
Stern tests: Palladium(I) dimer performance versus alternative air‐stable palladium complexes in either Kumada or Negishi couplings of challenging substrates.
We next focused on the sterically demanding aryl bromo triflate 3 (Scheme 1 b). This substrate is a stern test as it features an electronically deactivated (by ortho methoxy) and sterically shielded (by ortho adamantyl) C−Br site in competition with an accessible C−OTf site. Despite these challenges, upon using an organozinc as the coupling partner we observed strikingly rapid and selective C−Br coupling. As such, the bromo selectivity appears general for palladium(I) catalysis, and is independent of both the steric and electronic bias of the substrate, as well as the cross‐coupling partner.
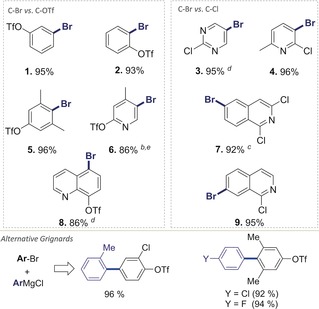
We subsequently investigated a wider range of substrates for the preferred coupling selectivities, including pharmaceutically relevant heterocycles. Table 1 summarizes the open‐flask Kumada cross‐coupling reactions performed with phenyl magnesium chloride at room temperature to test for C−Br versus C−OTf and C−Br versus C−Cl selectivities. In all cases, the coupling was completely selective for C−Br, thus allowing isolation of the coupling products in good to excellent yields. This selectivity was once again found to be independent of the respective relative positioning of the potential leaving groups (ortho, meta, para to each other) or the presence of additional (de)activating substituents or heterocycles (3, 4, 6–9) (see Table 1). Notably, the pharmaceutically relevant quinoline 9 was previously reported to give C−Cl coupling under Pd0/Xantphos catalysis.16 In our case, orthogonal and exclusive C−Br functionalization took place. The coupling was also efficient with alternative aryl Grignards, thus allowing the installation of additional functionality, such as either C−Cl or C−F using a Grignard (Table 1, bottom).
To further test this methodology, we also undertook a coupling reaction between PhMgCl and 2‐bromophenyl triflate (see Table 1) on a 1 gram scale (3.28 mmol) using 1 mol % 1 under otherwise identical open‐flask reaction conditions. The coupling reaction was found to be equally rapid and completely selective for C−Br. The corresponding coupling product was isolated in 95 % yield after column chromatography, thus showcasing that these coupling reactions can also be performed under lower catalyst loadings or significantly larger scale.17
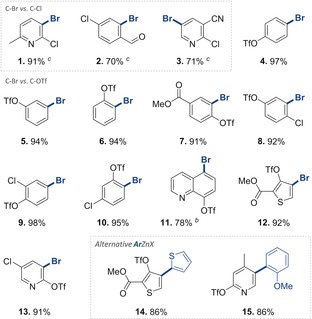
We subsequently explored the generality for site‐selective Negishi cross‐coupling and subjected a range of polysubstituted arenes and heterocycles to the catalyst 1 under open‐flask coupling conditions with ortho‐tolyl zinc chloride in toluene/THF. Table 2 summarizes the results. Once again, we observed that the reactions were completed within 5 minutes at room temperature. In analogy to the results in Table 1, also for Negishi cross‐coupling reactions, exclusive functionalizations of the C−Br bonds were seen in competition with C−Cl and/or C−OTf sites, regardless of the steric accessibility or electronic bias imposed by the substrate. Several pharmaceutically and agrochemically relevant pyridine, thiophene, and quinolone heterocycles were also studied, and the analogous exclusive C−Br selectivity was seen in all cases.18 Alternative arenes (14 and 15) could also be introduced in an equally efficient manner. As such, this catalytic system provides consistent and predictable C−Br selectivity, thus offering orthogonal and programmable synthetic approaches of high convenience (air‐stable catalyst, room temperature, rapid reaction speed, open flask).
Table 2.
C−Br selective Negishi cross‐couplings.[a]
|
[a] 1 (8.7 mg, 0.01 mmol), ArBr (0.4 mmol), o‐tolyl‐MgCl (1 m in THF, 600 μL, 0.6 mmol), ZnCl2 (1 m in THF, 640 μL, 0.64 mmol), toluene (1.5 mL). Yield is that of isolated product. [b] Used 1.1 equiv of ArMgCl and 1.2 equiv of ZnCl2. [c] Used 1.3 equiv of ArMgCl and 1.4 equiv of ZnCl2.
Intrigued by the observed reactivities, we subsequently performed additional studies to shed light on the origins of the selectivity and reactivity. There is the possibility that the coupling proceeds by direct reactivity of the PdI dimer10 or, alternatively, 1 functions as a precatalyst to a highly reactive monophosphine palladium(0) complex (Pd0PtBu3) upon activation by the nucleophilic organometallic coupling partner.11, 12 While palladium(0)‐based catalysis would generally be incompatible with open‐flask reaction conditions because of oxidation of the catalyst/ligand, the impressive speed of the present coupling reactions may simply exceed the rates of the competing oxidation of any potentially released palladium(0) species. Our in situ ReactIR monitoring showed that the palladium(I)‐dimer‐catalyzed cross‐coupling reaction of 4‐bromoaryl triflate with o‐tolyl‐MgCl reaches completion in just over a minute (Figure 2), displaying a non‐exponential pseudo‐first‐order growth.19 In contrast, when we subjected [Pd0(PtBu3)2] to air for 30 minutes, we saw a significant amount of [Pd0(PtBu3)2] remaining (as judged by 31P NMR data; see the Supporting Information). Interestingly, upon subsequent addition of (nBu4N)I along with PhMgCl (2 equiv relative to Pd) to the latter mixture, we observed the formation of 1 along with biphenyl.20 To the best of our knowledge, this is the first observation of an aerobically induced oxidation of a palladium(0) complex to a palladium(I) dimer.
Figure 2.

Top: ReactIR study of the Kumada coupling of 4‐bromoaryl triflate (Ar=o‐tol) catalyzed by 1. Bottom: aerobic oxidation of Pd0 to PdI–PdI.
These data indicate that any palladium(0) released in the reaction mixture could ultimately be reversibly transformed back into the palladium(I) dimer. On the basis of these data, neither palladium(0)‐ nor palladium(I)‐based catalysis can be excluded. Thus, we subsequently set out to computationally study the chemoselectivities for oxidative additions at Ph−Br versus Ph−Cl versus Ph−OTf for both catalysis regimes using the method CPCM (toluene) M06/def2‐TZVP//ωB97XD/6‐31G(d)/SDD.21 The computational data suggest a clear preference for oxidative addition at C−Br for both, a monoligated [Pd0PtBu3] and a PdI–PdI derived species (by ΔΔG ≠≥6.8 kcal mol−1 for [Pd0PtBu3] and ΔΔG ≠≥3.4 kcal mol−1 for PdI‐PdI relative to C−Cl). C−OTf addition is predicted to be even less favored (see the Supporting Information). In contrast, an anionic palladium(0) species [Pd0(PtBu3)X−] with X=halogen or aryl, which may form under conditions with nucleophilic additives,2a, 3a,3f, 9 particularly Grignard reagents,22 is predicted to add preferentially to C−OTf (by ΔΔG ≠≥3.5 kcal mol−1 for X=Ph and 2.8 kcal mol−1 for X=Cl). With monoligated [Pd0PtBu3] and PdI–PdI being formally electron‐deficient species (as opposed to PdLX−), selection occurs for the coupling site with lowest distortion energies.7b Key to overall high chemoselectivities therefore appears to be the avoidance of co‐existing reactive species, which are more likely encountered under prolonged reaction times.
In summary, an a priori predictable chemoselective C −C bond formation of poly(pseudo)halogenated arenes has been developed. The coupling reactions are triggered by the bench‐stable dinuclear palladium(I) complex [{(PtBu3)PdI}2}, and are completed in less than 5 minutes at room temperature in air.17 Exclusive bromo selectivity was observed in the presence of C−OTf and C−Cl sites, that is independent of any electronic or steric bias imposed by the substrate. The method proved to be compatible with heterocycles and functional groups, thus tolerating C−Cl, C−OTf, C−F, C−CN, aldehydes, esters, and sterically demanding groups (ortho‐adamantyl) as well as both, aryl zinc, and Grignard coupling partners. The larger scale applicability of the present method was also showcased (on 1 g scale with 1 mol % catalyst loading).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the RWTH Aachen, the MIWF NRW and the European Research Council (ERC‐637993) for funding. Calculations were performed with computing resources granted by JARA‐HPC from RWTH Aachen University under project “jara0091”.
I. Kalvet, G. Magnin, F. Schoenebeck, Angew. Chem. Int. Ed. 2017, 56, 1581.
Contributor Information
Indrek Kalvet, http://www.schoenebeck.oc.rwth‐aachen.de/.
Prof. Dr. Franziska Schoenebeck, Email: franziska.schoenebeck@rwth-aachen.de
References
- 1.For reviews, see:
- 1a. Horton D. A., Bourne G. T., Smythe M. L., Chem. Rev. 2003, 103, 893; [DOI] [PubMed] [Google Scholar]
- 1b. Hassan J., Sévignon M., Gozzi C., Schulz E., Lemaire M., Chem. Rev. 2002, 102, 1359; [DOI] [PubMed] [Google Scholar]
- 1c. Transition Metal-Catalyzed Couplings in Process Chemistry: Case Studies From the Pharmaceutical Industry (Eds.: J. Magano, J. R. Dunetz), Wiley, Hoboken, 2013; [Google Scholar]
- 1d. New Trends in Cross-Coupling: Theory and Applications (Ed.: T. Colacot), RSC Catalysis Series, Cambridge, UK, 2015. [Google Scholar]
- 2.For reviews, see:
- 2a. Almond-Thynne J., Blakemore D. C., Pryde D. C., Spivey A. C., Chem. Sci. 2017, 8, 40–62; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Rossi R., Bellina F., Lessi M., Adv. Synth. Catal. 2012, 354, 1181; [Google Scholar]
- 2c. Fairlamb I., Chem. Soc. Rev. 2007, 36, 1036; [DOI] [PubMed] [Google Scholar]
- 2d. Schröter S., Stock C., Bach T., Tetrahedron 2005, 61, 2245. [Google Scholar]
- 3.
- 3a. Proutiere F., Lyngvi E., Aufiero M., Sanhueza I. A., Schoenebeck F., Organometallics 2014, 33, 6879; [Google Scholar]
- 3b. Tran T. D., et al., ChemMedChem 2014, 9, 1378; [DOI] [PubMed] [Google Scholar]
- 3c. Hassan Z., Hussain M., Villinger A., Langer P., Tetrahedron 2012, 68, 6305; [Google Scholar]
- 3d. Proutiere F., Schoenebeck F., Angew. Chem. Int. Ed. 2011, 50, 8192; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8342; [Google Scholar]
- 3e. Milbank J. B. J., Pryde D. C., Tran T. D., WO2011004276, 2011;
- 3f. Espino G., Kurbangalieva A., Brown J. M., Chem. Commun. 2007, 1742; [DOI] [PubMed] [Google Scholar]
- 3g. Kamikawa T., Hayashi T., Tetrahedron Lett. 1997, 38, 7087; [Google Scholar]
- 3h. Littke A. F., Dai C. Y., Fu G. C., J. Am. Chem. Soc. 2000, 122, 4020. [Google Scholar]
- 4.See the Supporting Information.
- 5.
- 5a. Khaddour Z., Akrawi O. A., Hamdy A. M., Suleiman A., Jamous K., Villinger A., Langer P., Tetrahedron Lett. 2015, 56, 554; [Google Scholar]
- 5b. Wang J., Seefeld M. A., Luengo J., Tetrahedron Lett. 2011, 52, 6346. [Google Scholar]
- 6.A multiparameter approach:
- 6a. Niemeyer Z. L., Milo A., Hickey D. P., Sigman M. S., Nat. Chem. 2016, 8, 610. Computations: [DOI] [PubMed] [Google Scholar]
- 6b. Garcia Y., Schoenebeck F., Legault C. Y., Merlic C. A., Houk K. N., J. Am. Chem. Soc. 2009, 131, 6632 1H-NMR: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Handy S. T., Zhang Y., Chem. Commun. 2006, 299. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Lyngvi E., Sanhueza I. A., Schoenebeck F., Organometallics 2015, 34, 805; [Google Scholar]
- 7b. Schoenebeck F., Houk K. N., J. Am. Chem. Soc. 2010, 132, 2496; [DOI] [PubMed] [Google Scholar]
- 7c. Legault C. Y., Garcia Y., Merlic C. A., Houk K. N., J. Am. Chem. Soc. 2007, 129, 12664. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Li J., Ballmer S. G., Gillis E. P., Fujii S., Schmidt M. J., Palazzolo A. M. E., Lehmann J. W., Morehouse G. F., Burke M. D., Science 2015, 347, 1221; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Crudden C. M., Ziebenhaus C., Rygus J. P. G., Ghozati K., Unsworth P. J., Nambo M., Voth S., Hutchinson M., Laberge V. S., Maekawa Y., Imao D., Nat. Commun. 2016, 7, 11065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Jutand A., Mosleh A., Organometallics 1995, 14, 1810; [Google Scholar]
- 9b. Roy A. H., Hartwig J. F., Organometallics 2004, 23, 194. [Google Scholar]
- 10.
- 10a. Yin G., Kalvet I., Schoenebeck F., Angew. Chem. Int. Ed. 2015, 54, 6809; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6913; [Google Scholar]
- 10b. Aufiero M., Sperger T., Tsang A. S.-K., Schoenebeck F., Angew. Chem. Int. Ed. 2015, 54, 10322; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10462; [Google Scholar]
- 10c. Kalvet I., Bonney K. J., Schoenebeck F., J. Org. Chem. 2014, 79, 12041; [DOI] [PubMed] [Google Scholar]
- 10d. Bonney K. J., Proutiere F., Schoenebeck F., Chem. Sci. 2013, 4, 4434. [Google Scholar]
- 11.Labile PdI dimers have otherwise found applications as precatalysts:
- 11a. Melvin P. R., Nova A., Balcells D., Dai W., Hazari N., Hruszkewycz D. P., Shah H. P., Tudge M. T., ACS Catal. 2015, 5, 3680; [Google Scholar]
- 11b. Colacot T. J., Platinum Met. Rev. 2009, 53, 183; [Google Scholar]
- 11c. Christmann U., Pantazis D. A., Benet-Buchholz J., McGrady J. E., Maseras F., Vilar R., J. Am. Chem. Soc. 2006, 128, 6376; [DOI] [PubMed] [Google Scholar]
- 11d. Weissman H., Shimon L. J. W., Milstein D., Organometallics 2004, 23, 3931; [Google Scholar]
- 11e. Prashad M., Mak X. Y., Liu Y., Repic O., J. Org. Chem. 2003, 68, 1163; [DOI] [PubMed] [Google Scholar]
- 11f. Stambuli J., Kuwano R., Hartwig J. F., Angew. Chem. Int. Ed. 2002, 41, 4746; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 4940. [Google Scholar]
- 12. Aufiero M., Scattolin T., Proutière F., Schoenebeck F., Organometallics 2015, 34, 5191. [Google Scholar]
- 13.Selected examples:
- 13a. Iglesias M. J., Prieto A., Nicasio M. C., Org. Lett. 2012, 14, 4318; [DOI] [PubMed] [Google Scholar]
- 13b. Martin R., Buchwald S. L., J. Am. Chem. Soc. 2007, 129, 3844; [DOI] [PubMed] [Google Scholar]
- 13c. Organ M. G., Abdel-Hadi M., Avola S., Hadei N., Nasielski J., O'Brien C. J., Valente C., Chem. Eur. J. 2007, 13, 150; [DOI] [PubMed] [Google Scholar]
- 13d. Ackermann L., Born R., Spatz J. H., Meyer P., Angew. Chem. Int. Ed. 2005, 44, 7216; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7382; [Google Scholar]
- 13e. Limmert M. E., Roy A. H., Hartwig J. F., J. Org. Chem. 2005, 70, 9364; [DOI] [PubMed] [Google Scholar]
- 13f. Yoshikai N., Mashima H., Nakamura E., J. Am. Chem. Soc. 2005, 127, 17978; [DOI] [PubMed] [Google Scholar]
- 13g. Bonnet V., Mongin F., Trecourt F., Queguiner G., Knochel P., Tetrahedron 2002, 58, 4429; [Google Scholar]
- 13h. Huang J., Nolan S. P., J. Am. Chem. Soc. 1999, 121, 9889. For reviews, see: [Google Scholar]
- 13i. Haas D., Hammann J. M., Greiner R., Knochel P., ACS Catal. 2016, 6, 1540. [Google Scholar]
- 14.
- 14a. Kienle M., Knochel P., Org. Lett. 2010, 12, 2702; [DOI] [PubMed] [Google Scholar]
- 14b. Manolikakes G., Knochel P., Angew. Chem. Int. Ed. 2009, 48, 205; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 211. [Google Scholar]
- 15.While a general protocol for chemoselective couplings of poly(pseudo)-halogenated arenes have not been accomplished, halogenated arenes could be introduced by the Grignard reagent (Ref. [13b]).
- 16. Weiss M., Dimauro E. F., Dineen T., Graceffa R., Guzman-Perez A., Huang H., Kreisman C., Marx I. E., Nguyen H. N., Peterson E., WO2014201173 A1, 2014.
- 17.Note of safety: organozinc and Grignard reagents are moisture sensitive and may react violently in air. In our tests with the substrates 2 and 3 from Scheme 1 and substrate 4 from Table 2, the couplings were equally efficient also under an argon atmosphere. The presence of O2 therefore does not appear critical.
- 18.Although the coupling was selective and complete, some loss of material in purifications was experienced.
- 19.This could be indicative of reductive elimination being rate determining, or a potentially autocatalytic scenario.
- 20.Subjecting [Pd0(PtBu3)2] to oxygen and (nBu4N)I did not yield PdI dimer 1 However, mixing PhMgCl, [Pd0(PtBu3)2], and(nBu4N)I first under Ar (no change), followed by exposure to air, gave PdI dimer 1.
- 21.Gaussian 09, Revision D.01, M. J. Frisch, et al. [see SI for full reference]. For appropriateness of the chosen computational method, see: Sperger T., Sanhueza I. A., Kalvet I., Schoenebeck F., Chem. Rev. 2015, 115, 9532. [DOI] [PubMed] [Google Scholar]
- 22. Kolter M., Koszinowski K., Chem. Eur. J. 2016, 22, 15744–15750. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary