

Abstract
Oligodendrocyte progenitor cells (OPCs) undergo marked morphological changes to become mature oligodendrocytes, but the metabolic resources for this process have not been fully elucidated. Although lactate, a metabolic derivative of glycogen, has been reported to be consumed in oligodendrocytes as a metabolite, and to ameliorate hypomyelination induced by low glucose conditions, it is not clear about the direct contribution of lactate to cell cycling and differentiation of OPCs, and the source of lactate for remyelination. Therefore, we evaluated the effect of 1,4‐dideoxy‐1,4‐imino‐d‐arabinitol (DAB), an inhibitor of the glycogen catabolic enzyme glycogen phosphorylase, in a mouse cuprizone model. Cuprizone induced demyelination in the corpus callosum and remyelination occurred after cuprizone treatment ceased. This remyelination was inhibited by the administration of DAB. To further examine whether lactate affects proliferation or differentiation of OPCs, we cultured mouse primary OPC‐rich cells and analyzed the effect of lactate. Lactate rescued the slowed cell cycling induced by 0.4 mM glucose, as assessed by the BrdU‐positive cell ratio. Lactate also promoted OPC differentiation detected by monitoring the mature oligodendrocyte marker myelin basic protein, in the presence of both 36.6 mM and 0.4 mM glucose. Furthermore, these lactate‐mediated effects were suppressed by the reported monocarboxylate transporter inhibitor, α‐cyano‐4‐hydroxy‐cinnamate. These results suggest that lactate directly promotes the cell cycling rate and differentiation of OPCs, and that glycogen, one of the sources of lactate, contributes to remyelination in vivo. J. Cell. Physiol. 232: 986–995, 2017. © 2016 The Authors. Journal of Cellular Physiology Published by Wiley Periodicals, Inc.
Abbreviations
- 4‐CIN
α‐cyano‐4‐hydroxy‐cinnamate
- BrdU
bromodeoxyuridine
- CNS
central nervous systems
- CNTF
ciliary neurotrophic factor
- DAB
1,4‐dideoxy‐1,4‐imino‐d‐arabinitol
- FGF
fibroblast growth factor
- GFAP
glial fibrillary acidic protein
- GPR81
G‐protein‐coupled receptor 81
- GSTπ
glutathione‐S‐transferase π
- Iba1
ionized calcium‐binding adaptor molecule 1
- LFB
luxol fast blue
- LIF
leukemia inhibitory factor
- MAG
myelin‐associated glycoprotein
- MBP
myelin basic protein
- MCT
monocarboxylate transporter
- NG2
neural/glial antigen 2
- OLIG2
oligodendrocyte lineage transcription factor 2
- OPC
oligodendrocyte progenitor cell
- PDGF
platelet‐derived growth factor
- PDGFRα
platelet‐derived growth factor receptor α
- SOX10
SRY (sex determining region Y)‐box 10
- TUJ1
neuron‐specific class III β‐tubulin
The myelin sheath is an axon‐surrounding component that allows saltatory conduction and preserves axonal integrity (Nave and Trapp, 2008; Bruce et al., 2010; Lee et al., 2012; Nave and Werner, 2014). In the central nervous systems (CNS), developmental myelination as well as remyelination after pathological conditions requires the proliferation of oligodendrocyte progenitor cells (OPCs), which eventually differentiate into mature oligodendrocytes to form the myelin structure. These processes include marked morphological changes in the membrane area to provide myelin segmentation (Baron and Hoekstra, 2010; Chong et al., 2012) and expend a vast amount of metabolic energy (Chrast et al., 2011; Harris and Attwell, 2012; Nave and Werner, 2014). Glucose, one of the major energy substrates in the brain, has been reported to play crucial roles in myelination in cerebellar slice cultures (Rinholm et al., 2011) and in myelin gene expression in primary OPC cultures (Yan and Rivkees, 2006). Moreover, neurologically impaired children suffering from neonatal hypoglycemia exhibit abnormal or delayed myelination (Murakami et al., 1999). Although metabolic conditions may also be important in remyelination after CNS diseases, little is known about the contribution of nutrient substances and source during remyelination.
Remyelination by oligodendrocytes is regulated by both intrinsic mechanisms and extrinsic factors from cells surrounding oligodendrocytes (Miron et al., 2011; Boulanger and Messier, 2014; El Waly et al., 2014; Tanaka and Yoshida, 2014), in the same manner as myelination by Schwann cells (Yamauchi et al., 2012; Miyamoto et al., 2015). Astrocytes function as cellular mediators of myelination and remyelination of oligodendrocytes by releasing various factors (PDGF, FGF2, CNTF, LIF, extracellular matrix‐related molecules, etc.) that modulate OPC proliferation, cell cycling, and differentiation (Jiang et al., 2001; Moore et al., 2011; Boulanger and Messier, 2014; Tanaka and Yoshida, 2014). Furthermore, astrocytes control energy conditions in the CNS by transferring energy substrates from circulating blood and stored glycogen, which is an energy pool for neural cells (Belanger et al., 2011; Dinuzzo et al., 2012; Evans et al., 2013). In astrocytes, glycogen is catabolized to lactate, which is released via monocarboxylate transporters (MCTs) and used by neurons as metabolic substrates (Belanger et al., 2011; Suzuki et al., 2011; Evans et al., 2013). Although the lactate produced from glycogen in astrocytes contributes to neural function, such as long‐term memory, by upregulation of mRNA expression in neuronal cells (Suzuki et al., 2011), the contribution of glycogen and lactate to remyelination of oligodendrocytes has not been examined.
Recently, lactate has been reported to act as a mediator in energy transfer between cells. Mature oligodendrocytes transport lactate to axons and preserve axonal integrity (Funfschilling et al., 2012; Lee et al., 2012). On the other hand, it has been shown that oligodendrocytes themselves utilize lactate as a metabolite in vitro (Sanchez‐Abarca et al., 2001). Furthermore, it has been demonstrated that lactate contributes to myelination in cerebellar slice cultures grown under low glucose conditions (Rinholm et al., 2011), suggesting the importance of lactate in the process of myelination. However, whether OPCs utilize lactate directly for their proliferation and differentiation remains to be elucidated.
Therefore, we here assessed the impact of an inhibitor of glycogen phosphorylase, which is a glycogen catalyzing enzyme, in a mouse remyelination model, as well as the direct effect of lactate on the proliferation and differentiation of mouse primary OPCs‐rich cells in culture.
Materials and Methods
Mice
Male C57BL/6J mice and pregnant ICR mice were obtained from Clea Japan (Tokyo, Japan). All experiments involving animals were approved by the Institutional Animal Care and Use Committee of National Rehabilitation Center for Persons with Disabilities. All mice were maintained on a 12‐h light/dark cycle at 22 ± 2°C, with constant humidity (55 ± 10%), and had free access to water and standard rodent chow.
Materials
All chemicals and compounds, including 1, 4‐dideoxy‐1, 4‐imino‐d‐arabinitol (DAB), Bis(cyclohexanone)oxaldihydrazone (cuprizone), and sodium L‐lactate, were purchased from Sigma (St Louis, MO) or Wako (Osaka, Japan). Monoclonal anti‐glutathione‐S‐transferase π (GSTπ; 1:500) and monoclonal anti‐platelet‐derived growth factor receptor α (PDGFRα; 1:500) were purchased from BD Transduction Laboratories (Lexington, KY). Monoclonal or polyclonal anti‐glial fibrillary acidic protein (GFAP; 1:500), polyclonal anti‐neural/glial antigen 2 (NG2; 1:500), polyclonal anti‐oligodendrocyte lineage transcription factor 2 (OLIG2; 1:1,000), monoclonal anti‐myelin‐associated glycoprotein (MAG; 1:500), and monoclonal anti‐O1 (1:500) were obtained from Millipore (Billerica, MA). Polyclonal anti‐SRY (sex‐determining region Y)‐box 10 (SOX10; 1:500) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal anti‐ionized calcium‐binding adaptor molecule 1 (Iba1; 1:500) was obtained from Wako. Monoclonal anti‐neuron‐specific class III β‐tubulin (TUJ1; 1:500) was purchased from Covance (Berkeley, CA). Monoclonal anti‐myelin basic protein (MBP; 1:200) and monoclonal anti‐bromodeoxyuridine (BrdU; 1:500) were obtained from AbD Serotec (Raleigh, NC). Polyclonal anti‐Ki67 (1:500) was purchased from Abcam (Cambridge, MA). Polyclonal anti‐cleaved caspase 3 and monoclonal anti‐β‐actin were obtained from Cell Signaling Technology (Danvers, MA).
Cuprizone model
To assess the extent of remyelination, a cuprizone model was used (Lindner et al., 2008; Gudi et al., 2014). A diet containing 0.2% (w/w) cuprizone mixed into milled chow pellets was given to 8‐week‐old male C57BL/6J mice for 6 weeks to induce demyelination in the corpus callosum (week 6). To induce remyelination, the mice were then fed a normal diet for additional 2 weeks. To determine the effect of DAB on remyelination, chronic intracerebroventricular administration of saline or DAB (10 μg/kg/day) was started at the point of change‐over to the normal diet (week 8 saline or week 8 DAB). Chronic DAB injection for 2 weeks into the right lateral ventricle (0.9 mm lateral, 0.3 mm posterior, and 2.5 mm below the skull) was performed by means of an osmotic infusion pump (Alzet Brain Infusion Kit 3, Alzet Osmotic pump model 1002; DURECT, Cupertino, CA).
Immunohistochemistry
Mice were anesthetized with pentobarbital and perfused transcardially with buffered 4% paraformaldehyde (PFA). Brains were removed, postfixed in 4% PFA at 4°C overnight, cryoprotected in buffered 20% sucrose at 4°C for 24 h and 30% sucrose for another 24 h, and then embedded in optimal cutting temperature compound (Sakura Finetek, Torrance, CA). Frozen tissues were cut into 16‐μm coronal sections from bregma +0.8 mm to +1.8 mm, using a cryostat, and mounted onto glass slides. Sections were then blocked with 2% donkey serum in Tris‐buffered saline with 0.1% Tween 20 (TBS‐T) for 2 h at room temperature and incubated overnight at room temperature with a blocking solution containing the diluted primary antibodies. Subsequently, the sections were incubated with the Alexa Fluor 488 or 568 conjugated secondary antibodies (Invitrogen, Carlsbad, CA) diluted in blocking solution for 2 h at room temperature. Slides were then mounted with ProLong Gold (Invitrogen), and images were captured with a BZ‐9000 (Keyence, Osaka, Japan).
Histological analysis
To determine the extent of demyelination and remyelination in the corpus callosum, brain sections were stained with Luxol fast blue (LFB) and scored in a blinded manner on a scale from 0 (complete demyelination) to 3 (normal myelin) (Skripuletz et al., 2013). Positive cell counts of GSTπ, as a marker of mature oligodendrocytes, and Sox10 expressed intranuclearly, as an oligodendrocyte linage marker in the corpus callosum, determined immunohistochemically, were compared in each section. To observe gliosis, we stained for GFAP, an astrocyte marker, and Iba1, a microglia marker, and measured the GFAP‐ or Iba1‐positive area in the corpus callosum by ImageJ (NIH).
Primary OPC‐rich culture
Mouse primary OPC‐rich cultures were established by a modification of the method previously reported (Seiwa et al., 2004; Ogata et al., 2011). Briefly, the forebrains from E16.5 ICR mice were dissociated and mixed cells were cultured in Poly‐d‐Lysine (PDL)‐coated T‐75 flasks (Corning, NY, NY) in MEM‐E medium supplemented with 10% fetal bovine serum (FBS) and 0.5% glucose. After 5 days, the flasks were shaken at 450 rpm for 5 min and the supernatants were replaced with fresh medium. After a 2‐h incubation at 37°C, the flasks were shaken overnight at 200 rpm to dislodge less adherent cells (OPC‐rich cells) from the mixed‐cell cultures. In order to further remove astrocytes from the OPC‐rich cells, the harvested cells were plated onto Petri dishes and incubated for 30 min. Non‐adherent OPCs were collected and plated onto PDL‐coated culture dishes (Corning). The collected OPCs were further incubated in modified BS medium (DMEM containing 36.6 mM glucose, 1 mM sodium pyruvate, 0.01% BSA, 5 µg/ml insulin, 5 µg/ml transferrin, 0.6 ng/ml progesterone, 16 µg/ml putrescine, 40 ng/ml sodium selenate [Sigma], 0.4% ASF104 [Ajinomoto, Tokyo, Japan], and antibiotics), supplemented with 10 ng/ml PDGF‐AA (PDGF), 10 ng/ml FGF2, and 5% Nerve‐Cell Culture Medium (serum‐free conditioned medium from rat astrocyte confluent cultures grown in DMEM/F‐12 supplemented with N2; Sumitomo Bakelite, Akita, Japan). After 4 days, OPCs were re‐seeded and used for experiments.
In order to maintain their undifferentiated condition, the medium was switched to growth medium (DMEM supplemented with 0.01% BSA, N2 supplement, B27 supplement, 10 ng/ml PDGF‐AA, 10 ng/ml FGF2). To promote differentiation of OPCs, the medium was switched to differentiation medium (DMEM supplemented with 0.01% BSA, N2 supplement, B27 supplement, 40 ng/ml triiodothyronine, and 400 ng/ml thyroxine). DMEM containing 36.6 mM glucose was prepared by addition of glucose to low‐glucose DMEM (5.5 mM, Life Technologies, Carlsbad, CA). Culture media containing 5.4 mM and 0.4 mM glucose were prepared by mixing of 36.6 mM glucose DMEM and 0 mM glucose DMEM with 18.3 mM NaCl to compensate for the osmolality change. For culturing primary astrocyte‐rich cells, adherent cells were trypsinized after shaking the mixed culture from E16.5 ICR mice at day 5. The collected cells were cultured in MEM‐E containing 10% FBS.
Assessment of proliferation, cell death, and differentiation of OPCs
To assess OPC proliferation, OPC‐rich cultures were incubated with growth medium containing a defined concentration of glucose and lactate, and analyses were performed at day 0 (before medium change), 1 day and 3 days after. To evaluate the living cells at each time point, cells were stained with Calcein AM (Trevigen, Gaithersburg, MD) and propidium iodide (PI) and quantified. For BrdU (S‐phase marker) and Ki67 (pan‐cell cycle marker) analyses, cells were incubated in each condition for 16 h and were treated with 10 μM BrdU for 1 h before fixation. After fixation with 4% PFA for 30 min, BrdU, Ki67, and DAPI staining was performed and the positive cell rate then counted. Ki67‐positive cells were considered as the sum of both strongly and weakly positive cells were regarded as positive, since Ki67 is a marker for all stages of the cell cycle (G1/S/G2/M), but its expression level differs in each stage (Scholzen and Gerdes, 2000). To detect cell death, LDH activity was determined in growth medium after a 1‐day or 3‐day incubation using the LDH Cytotoxicity Detection Kit (Takara Bio, Shiga, Japan), according to the manufacturer's instructions. To evaluate OPC differentiation, Mbp mRNA expression and the MBP‐positive cell rate in OPC‐rich culture were measured after incubation for 3 days in differentiation medium. To determine the effect of the MCT inhibitor, α‐cyano‐4‐hydroxy‐cinnamate (4‐CIN; final concentration 1 mM; Sigma) or vehicle (0.1% DMSO) was added to medium at the time of medium change. After changing to each medium containing a defined concentration of glucose and lactate, the medium was renewed once daily.
Immunocytochemistry
Cultured cells were washed with phosphate‐buffered saline (PBS) and fixed with 4% PFA for 30 min. Cells were then blocked with 10% FBS and 0.1% Triton‐X in PBS for 2 h at room temperature and then incubated for a further 2 h at room temperature with a blocking solution containing the diluted primary antibodies. Subsequently, the cells were incubated with the Alexa Fluor 488 or 568 conjugated secondary antibodies diluted in blocking solution for 1 h at room temperature. In order to stain BrdU, the cells were treated in 2 M HCl at room temperature for 15 min prior to the blocking process. Images were captured using a BZ‐9000 (Keyence, Osaka, Japan) and the positive cells were counted. More than three images per condition were counted and the average values were used to calculate the positive cell ratio in the experiment. We show the means of at least three independent experiments in graphs, unless otherwise indicated.
Quantitative real‐time reverse transcriptase‐polymerase chain reaction (RT‐PCR)
Total RNA from OPC‐ or astrocyte‐rich culture cells was isolated using an RNeasy mini kit (Qiagen, CA) and reverse transcribed using a PrimeScript RT Reagent Kit (Takara Bio, Shiga, Japan) according to the manufacturer's instructions. This cDNA was used as a template in real‐time PCR assays employing the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). The primer pairs used were as follows: Mbp, forward primer 5′‐ACTCACACACGAGAACTACCCA‐3′ and reverse primer 5′‐TGGTGTTCGAGGTGTCACAA‐3′; Gfap, forward primer 5′‐AGAAAACCGCATCACCATTC‐3′ and reverse primer 5′‐TCCTTAATGACCTCGCCATC‐3′; Mct1, forward primer 5′‐TTGGACCCCAGAGGTTCTCC‐3′ and reverse primer 5′‐AGGCGGCCTAAAAGTGGTG‐3′; Mct2, forward primer 5′‐CAGCAACAGCGTGATAGAGCT‐3′; and reverse primer 5′‐TGGTTGCAGGTTGAATGCTAA‐3′; Mct4, forward primer 5′‐CAGCTTTGCCATGTTCTTCA‐3′ and reverse primer 5′‐AGCCATGAGCACCTCAAACT‐3′; Gapdh forward primer 5′‐TGCACCACCAACTGCTTAGC‐3′ and reverse primer 5′‐GGATGCAGGGATGATGTTCT‐3′; β‐actin forward primer 5′‐ GGCTGTATTCCCCTCCATCG‐3′ and reverse primer 5′‐CCAGTTGGTAACAATGCCATGT‐3′. The expression levels of each amplicon were normalized against β‐actin expression, and were expressed as relative values compared with control samples in each experiment. All reactions were performed in duplicate.
Western blot
Whole cell lysates from OPCs were prepared and subjected to Western blot analysis. Briefly, cells were lysed in RIPA buffer containing 50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 0.1% SDS, 1% NP‐40, 0.5% sodium deoxycholate, 1 mM β glycerophosphate, 10 mM sodium pyrophosphate, 2 mM Na3VO4, and Halt protease inhibitor cocktail (Thermo Fisher Scientific, Inc., Waltham, MA), incubated on ice for 15 min, and centrifuged at 14,000 rpm for 15 min. Equal amounts of protein lysate were separated by SDS–PAGE, transferred onto membranes, and immunoblotted, as previously described (Ichihara et al., 2013). The images of the band corresponding to the cleaved caspase 3 or β‐actin were obtained by using the Odyssey imaging system (LI‐COR Biosciences).
Statistical analysis
All values are presented as the mean ± SEM. The significance of differences between two groups was determined by Student's t‐test, and among more than three groups by one‐way ANOVA with a post‐hoc test (Bonferroni's correction). To calculate the significance of the test for the myelin score, we used the Mann–Whitney U test. P < 0.05 was considered significant.
Results
DAB suppresses remyelination after cuprizone‐induced demyelination in the corpus callosum
To investigate the contribution of glycogen to remyelination, we administered an inhibitor of glycogen phosphorylase, which is a glycogen catabolic enzyme, to the cuprizone remyelination model mice (Lindner et al., 2008; Gudi et al., 2014). We used DAB, which is known to inhibit glycogen phosphorylase and lactate release in the brain (Suzuki et al., 2011). LFB‐staining showed reduced myelin staining after 6 weeks of cuprizone treatment (week 6) and recovery of myelin after an additional 2 weeks of feeding normal chow in the control group (week 8 saline; Fig. 1A and B). This recovery of LFB‐stained myelin after 2 weeks on a normal diet was inhibited by chronic DAB administration (week 8 DAB; Fig. 1A and B). In line with this, the count of GSTπ‐positive mature oligodendrocytes decreased after feeding a cuprizone diet and was ameliorated after 2 weeks of normal chow with saline injection; this recovery was inhibited in DAB‐administered mice compared to saline‐injected mice (Fig. 1C and D). Although nuclear SOX10‐positive cells were reduced after cuprizone treatment, as previously reported (Ye et al., 2013), DAB did not affect the nuclear SOX10‐positive cell count (Fig. 1E and F).
Figure 1.
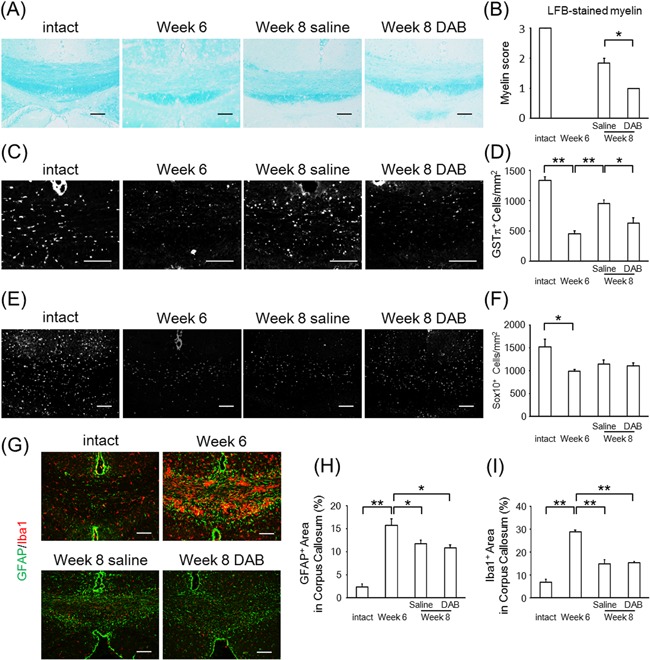
DAB suppresses remyelination in the cuprizone model. (A) Representative images of the corpus callosum stained by Luxol fast blue (LFB). Groups of mice were treated with no cuprizone (intact), cuprizone for 6 weeks (week 6), cuprizone for 6 weeks followed by 2 weeks of normal chow with injected saline (week 8 saline), or cuprizone for 6 weeks followed by 2 weeks of normal chow with injected DAB (week 8 DAB). (B) The myelin score of LFB‐stained corpus callosum of intact, week 6, week 8 saline, and week 8 DAB mice. The values represent the mean ± SEM (n = 3–7; *P < 0.05). (C and D) Representative image of GSTπ stain (C) and the count of positive cells in the corpus callosum (mean ± SEM, n = 3–6; *P < 0.05, **P < 0.01) (D). (E and F) Representative image of SOX10 immunohistochemistry (E) and the count of nuclear SOX10‐positive cells in the corpus callosum (mean ± SEM, n = 3–7; *P < 0.05) (F). (G–I) Representative image of GFAP (green) and Iba1 (red) immunohistochemistry (G) and the percentage of GFAP‐positive (H) and Iba1‐positive (I) area in corpus callosum (mean ± SEM, n = 3–6; *P < 0.05, **P < 0.01). Scale bars: (A, C, E, G) 100 μm.
The immunoreactivity for GFAP and Iba1 increased after cuprizone treatment and partially recovered after feeding mice a normal diet, in both the saline‐ and DAB‐injected mice (Fig. 1G–I). These data demonstrated that DAB inhibited remyelination after cuprizone‐induced demyelination without affecting activation of astrocytes and microglia or the cell number of oligodendrocyte‐linage cells.
Characterizations of OPCs‐rich culture
Our data suggested that glycogen plays a role in remyelination. Although glycogen in the brain has been reportedly catabolized to lactate, it was unclear whether lactate directly contributes to OPC proliferation, cell cycling, and differentiation, which is required for remyelination. Therefore, we prepared primary OPC‐rich cultures and tested the effect of lactate on proliferation, cell cycling, and differentiation in such an OPC‐rich population. Primary OPC‐rich cultures consisted of 88.9% OLIG2‐positive, 90.1% PDGFRα‐positive, and 85.5% NG2‐positive oligodendrocyte progenitors, and 2.2% TUJ1‐positive neurons (Fig. 2A and B). We could not find apparent GFAP‐positive astrocytes and Iba1‐positive microglia in this culture (Fig. 2B, and data not shown).
Figure 2.
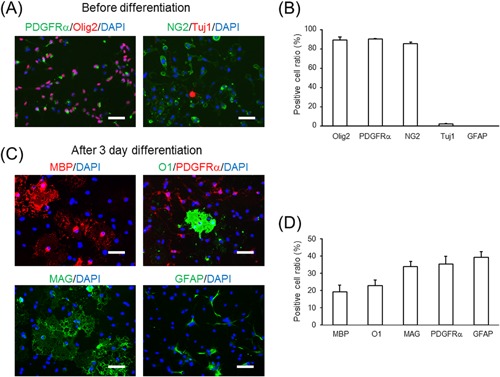
Characterization of mouse primary oligodendrocyte progenitor cell (OPC)‐rich cultures. (A) Representative images of PDGFRα (left, green), OLIG2 (left, red), NG2 (right, green), TUJ1 (right, red), and DAPI (blue) stains of OPC‐rich cultures in growth medium. (B) Positive cell ratio for each of the above marker proteins of OPC‐rich cultures in growth medium. The values represent the mean ± SEM (n = 3–4). (C) Representative images of MBP (upper left, red), O1 (upper right, green), PDGFRα (upper right, red), MAG (lower left, green), GFAP (lower right, green), and DAPI (blue) stains of OPC‐rich cultures in differentiation medium. (D) Positive cell ratio for each marker protein in OPC‐rich cultures in differentiation medium. The values represent mean ± SEM (n = 3–5). Scale bars: (A and C) 50 μm.
Three days after incubation in differentiation medium, the Mbp mRNA expression was 260‐fold increased compared to the values at day 0 (data not shown), which indicates differentiation of the cells into oligodendrocytes. Similarly, 19.4% MBP‐positive, 23.0% O1‐positive, and 34.0% MAG‐positive differentiated oligodendrocytes appeared after 3 days of culture in differentiation medium (Fig. 2C and D), while 35.4% of cells remained PDGFRα‐positive OPCs (Fig. 2C and D). Furthermore, 39.4% cells were GFAP‐positive after culturing in differentiation conditions (Fig. 2C and D). Consistent with this, Gfap mRNA expression was elevated after 3 days of differentiation, but this expression level was markedly lower compared to that in primary astrocyte cultures (primary OPCs in differentiation medium:primary astrocytes = 1:66.7). We used this OPC‐rich culture to evaluate the effect of glucose and lactate concentration on OPC proliferation, cell cycling, and differentiation.
Effects of lactate on 0.4 mM glucose‐induced slowed cell cycling and increased cell death in OPC‐rich cultures
To assess the effect of lactate on OPC proliferation and cell death, we first counted the Calcein AM‐positive live cells after 1 day or 3 days of incubation under several experimental conditions (growth medium containing defined glucose and lactate concentrations). Since it has been reported that the glucose concentration in the brain is 0.4–2.5 mM (Levin et al., 2011), we tested the effect of 0.4 mM glucose, the lowest physiological glucose concentration in the brain, on cell proliferation, cell cycling, and cell death. The lactate concentration in the brain has been reported to be 1–3 mM (Levin et al., 2011) and to increase to 10 mM during hypoxia and stroke in the brain (Schurr et al., 1997a,1997b; Chen et al., 2000). The number of Calcein AM‐positive cells at day 1 was increased with a similar amount in the 36.6, 5.4, and 0.4 mM glucose condition compared to that at day 0. However, at day 3, it was reduced in the 0.4 mM glucose condition as compared to that in the 36.6 mM and 5.4 mM glucose conditions. Lactate increased the Calcein AM‐positive cell number in the 0.4 mM glucose condition, but surprisingly, lactate did not affect this cell number in the 0 mM glucose condition (Fig. 3A). This indicates OPCs could not survive in a no glucose condition even in the presence of lactate.
Figure 3.
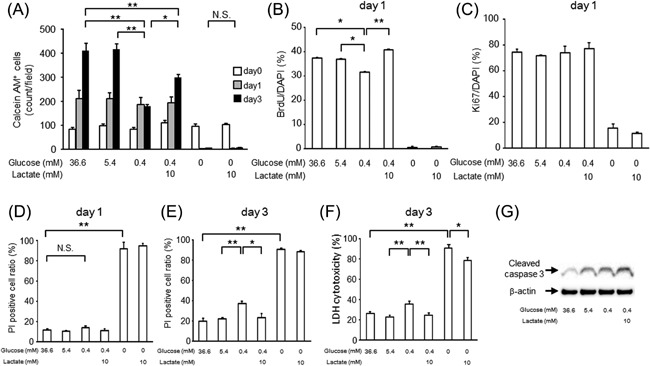
The effects of lactate on cell cycling and survival in OPC‐rich cultures in growth medium containing 0.4 mM glucose. (A) Calcein AM‐positive cell counts in growth medium containing 36.6 mM glucose, 5.4 mM glucose, 0.4 mM glucose, 0.4 mM glucose with 10 mM lactate, 0 mM glucose, or 0 mM glucose with 10 mM lactate at day 0 (white bar), day 1 (gray bar), and day 3 (black bar). Data are representative of three independent experiments and values are expressed as mean ± SEM. (n = 3–4; **P < 0.01). (B and C) BrdU‐positive (B) and Ki67‐positive (C) cell ratio in each type of growth medium at day 1 (mean ± SEM, n = 4; *P < 0.05, **P < 0.01). (D and E) Dead cell ratio measured based on PI (dead)‐positive and Calcein AM (live)‐positive cell counts in each type of growth medium at day 1 (D) and day 3 (E) (mean ± SEM, n = 4 [D] and n = 3 [E]; *P < 0.05, **P < 0.01). (F) Cell damage measured by LDH assay in each type of growth medium at day 3 (mean ± SEM, n = 9; *P < 0.05, **P < 0.01). (G) Representative images of cleaved caspase 3 and β‐actin detected by Western blot in each type of growth medium at day 3.
To examine the cause of the increase in live cells by lactate, we counted the BrdU‐positive cells in each type of glucose and lactate concentration condition after 1 day, to evaluate the effect of lactate on cell cycling. The BrdU‐positive cell ratio was less in the 0.4 mM glucose condition compared to that in 36.6 mM or 5.5 mM glucose condition, and lactate increased this cell ratio (Fig. 3B). The Ki67‐positive cell ratio was comparable in 36.6 mM, 5.4 mM, and 0.4 mM glucose conditions, regardless of the lactate concentration, whereas such cells were rare in the no‐glucose condition (Fig. 3C). This suggests that the 0.4 mM glucose condition slows cell cycling without promoting cell cycle exit, and that lactate restores cell cycle progression.
Cell survival, tested by PI staining or LDH release into the culture medium, did not differ among the 0.4 mM, 5.4 mM, and 36.6 mM glucose conditions at 1 day (Fig. 3D). At 3 days, the PI‐positive cell ratio or LDH release was increased in the 0.4 mM glucose condition as compared to the 5.4 mM glucose condition, and lactate prevented this increase (Fig. 3E and F). As for the apoptosis pathways in this experimental condition, the cleaved caspase‐3 level at day 3 increased in the 0.4 mM glucose condition as compared to that in the 36.6 mM and 5.4 mM glucose conditions. We found that lactate did not affect cleaved caspase‐3 expression in the 0.4 mM glucose condition (Fig. 3G).
Overall, these data demonstrated that the 0.4 mM glucose condition acutely (within 1 day) induced slower cell cycling of cells and after longer culturing (3 days), this condition caused cell damage. In this condition, we found that the addition of lactate exerted effects on cell cycling without an effect on apoptosis.
Lactate upregulates OPC differentiation in both 0.4 mM and 36.6 mM glucose conditions in vitro
To evaluate the effect of lactate on OPC differentiation, we induced differentiation of OPC‐rich cultures in differentiation medium containing glucose and lactate at defined concentrations for 3 days. Mbp mRNA expression was decreased in 0.4 mM glucose compared to 36.6 mM glucose conditions, and this was rescued by lactate dose‐dependently (Fig. 4A). The MBP‐positive cell ratio was also significantly decreased in 5.4 mM and 0.4 mM glucose medium, as compared to 36.6 mM glucose medium, and this was rescued by lactate (Fig. 4B and C). Lactate increased Mbp mRNA expression and the MBP‐positive cell rate even in the 36.6 mM glucose condition (Fig. 4D–F). On the other hand, the cleaved caspase‐3 level was increased even in 36.6 mM glucose conditions compared to that in growth medium, and the trend was not changed by lower glucose or lactate addition (Fig. 4G). These data suggest that lactate upregulated OPC differentiation in both low and high glucose conditions without affecting apoptosis in vitro.
Figure 4.
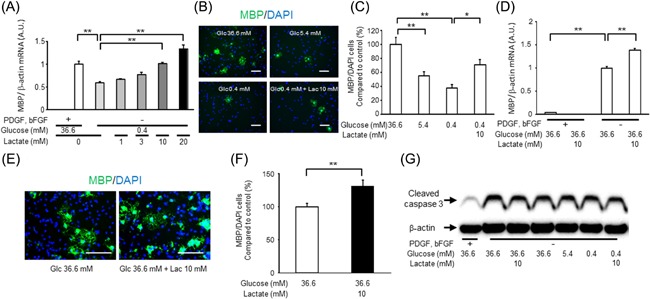
The effect of lactate on the differentiation of OPC‐rich cultures. (A) Mbp mRNA expression in each type of medium after 3 days. Data are representative of three independent experiments and values are expressed as mean ± SEM (n = 3; **P < 0.01). (B) Representative images of MBP (green) and DAPI (blue) in each type of differentiation medium at 3 days. (C) MBP‐positive cell ratio in each type of differentiation medium at 3 days (mean ± SEM, n = 5; *P < 0.05, **P < 0.01). (D) Mbp mRNA expression in each type of medium after 3 days (mean ± SEM, n = 6; **P < 0.01). (E) Representative images of MBP (green) and DAPI (blue) in each type of differentiation medium at 3 days. (F) MBP‐positive cell ratio in each type of differentiation medium at 3 days (mean ± SEM, n = 3; **P < 0.01). (G) Representative images of cleaved caspase 3 and β‐actin detected by Western blot in each type of medium at day 3. Scale bars: (B and E) 100 μm.
Suppressive effects of an MCT inhibitor on OPC‐rich cultures in the presence of lactate
To determine the direct uptake of lactate into OPCs, we assessed the effect of an inhibitor of MCTs, which is known as a lactate transporter (Halestrap and Meredith, 2004). A previous report showed that MCT1, MCT2, and MCT4 expressed in the brain cells (Bergersen, 2007), and we detected mRNA expression of MCTs in our OPC‐rich culture (Fig. 5A).
Figure 5.
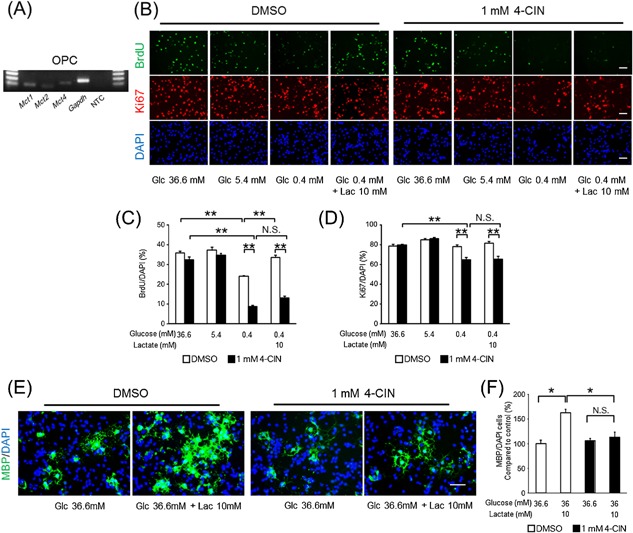
The effects of a monocarboxylate transporter (MCT) inhibitor, 4‐CIN, on OPC‐rich cells in the presence of lactate. (A) Gel imaging of PCR products of Mct1, Mct2, Mct4, Gapdh, and a non‐template control (NTC) of OPC‐rich cultures in growth medium (OPC, 30 cycles). (B) Representative images of BrdU (green), Ki67 (red), and DAPI (blue) staining in each type of growth medium at 1 day. (C) BrdU‐positive and (D) Ki67‐positive cell ratio in each type of growth medium supplemented with 0.1% DMSO (white) or 1 mM 4‐CIN (black) at 1 day (mean ± SEM, n = 3; **P < 0.01). (E) Representative images of MBP (green) and DAPI (blue) staining in each type of differentiation medium at 3 days. (F) MBP‐positive cell ratio in each type of differentiation medium supplemented with 0.1% DMSO (white) or 1 mM 4‐CIN (black) at 3 days (mean ± SEM, n = 4; *P < 0.05). Scale bars: (B and E) 50 μm.
Given the expression of MCTs in our cultures, we tested the effect of an MCT inhibitor on OPC cell cycling and differentiation. The MCT inhibitor, 1 mM 4‐CIN, blocked the effect of lactate on BrdU uptake in the 0.4 mM glucose condition (Fig. 5B and C). Although the Ki67‐positive cell ratio remained unchanged by 4‐CIN in 36.6 mM and 5.4 mM glucose conditions, 4‐CIN reduced the Ki67‐positive cell ratio in the 0.4 mM‐glucose condition (Fig. 5B and D). Lactate did not affect the Ki67‐positive cell ratio, which was reduced in the 0.4 mM glucose condition (Fig. 5B and D). Furthermore, 4‐CIN also inhibited the increase of the MBP‐positive cell ratio by lactate in differentiation medium (Fig. 5E and F). Overall, the MCT inhibitor blocked the effect of lactate on cell cycling and differentiation.
Discussion
In the present study, lactate directly induced faster cell cycling in 0.4 mM glucose medium, and increased differentiation in both the 36.6 mM and 0.4 mM glucose conditions in OPC‐rich cultures. This effect of lactate was blocked by an MCT inhibitor. Moreover, a glycogen phosphorylase inhibitor blocked remyelination in the corpus callosum after cuprizone‐induced demyelination. These data demonstrated the impact of lactate on remyelination via cell cycling and differentiation of OPCs (Fig. 6).
Figure 6.
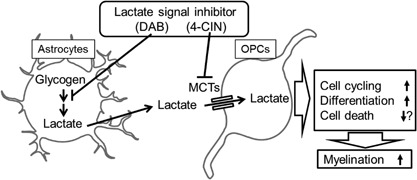
Illustrating the current working hypothesis regarding the mechanism of glycogen and lactate to contribute to myelination.
We showed that intracerebroventricular injection of DAB inhibited remyelination in the cuprizone‐induced demyelination model (Fig. 1A–D). DAB has been reported to suppress glycogenolysis in the brain as well as lactate release (Andersen et al., 1999; Gibbs et al., 2006; Suzuki et al., 2011). In addition to this, the glycogen content is more highly enriched in the corpus callosum than in the other brain region (Kong et al., 2002), and the corpus callosum has a high myelin content. These reports imply that glycogen functions as a glucose‐derived energy‐complementing substrate for myelination and remyelination. In the present study, DAB may affect the differentiation, but not the proliferation, of OPCs in the corpus callosum, since DAB decreased the GSTπ positive cell ratio and myelin score without changing SOX10‐positive cell counts (Fig. 1). Recently, not only astrocytes, but also neurons, are reported to contain glycogen, contributing to tolerance to hypoxia (Saez et al., 2014). As we could not measure the glycogen content and lactate concentration in the corpus callosum, our data do not reveal the cell source for glycogen. Moreover, we could not show whether a glycogen shunt does occur in our model (Walls et al., 2008). These are the limitations of this study. However the suppression of remyelination by a glycogen phosphorylase inhibitor might indicate that a vast amount of energy substrates, glucose and related substrates, need to be utilized for remyelination.
We used a primary OPC‐rich culture to test the effect of lactate on cell cycling and differentiation. In this condition, the possibility of indirect effects of lactate via astrocytes is not completely excluded, especially in differentiation tests. Although we used about 90% purified OPCs, which is detected by the OPC markers NG2 and PDGFRα, NG2 cells (marker of OPCs) also differentiate to astrocytes in vitro as reported (Honsa et al., 2016). Therefore, this is the limitation of our culture experiments.
As for the effect of lactate on proliferation, lactate rescued the decrease in the BrdU‐positive cell ratio (an indicator for S‐phase progression) in the 0.4 mM glucose condition, although the Ki67‐positive cell rate (an indicator for G1/S/G2/M phase) was comparable among the various glucose and lactate concentration conditions (Fig. 3B and C). This suggests that lactate restored cell cycle progression in the low glucose condition. On the other hand, lactate ameliorated the decreased differentiation in the 0.4 mM glucose condition (Fig. 4A–C). Moreover, lactate promoted differentiation even in the 36.6 mM glucose condition (Fig. 4D–F). It has been reported that decreased intracellular ATP (energy) levels due to mitochondrial respiration defects caused by mitochondrial DNA with a pathogenic 4696‐bp deletion impairs the differentiation of B lymphoid cells, but not the proliferation of hematopoietic stem and progenitor cells (Inoue et al., 2010). These findings and our results suggested that energy demand may be higher during differentiation than during proliferation in some types of progenitor cells, and the demand during differentiation might not be sufficient even in high glucose condition in OPCs. Our results suggest that lactate may be an additional energy source for OPCs when available.
Although our result indicates lactate increased differentiation even in high glucose conditions in OPC‐rich culture, previous report using cerebral slice culture showed lactate did not promote further myelination in high glucose condition (Rinholm et al., 2011). We assume that, in OPC‐rich condition, oligodendrocyte differentiation could not be supported by surrounding cells such as neurons and astrocytes via neuronal activity, growth factor, and cholesterol (Gibson et al., 2014; Kiray et al., 2016). The lack of those supports may allow lactate to promote differentiation. To determine this, we need further investigation in the future.
The effects of lactate we showed may occur physiologically as well as pathologically in the CNS, since the concentration we used as a low glucose condition (0.4 mM) and the concentration of the added lactate (10 mM) was physiologically relevant (Schurr et al., 1997a,1997b; Chen et al., 2000; Levin et al., 2011). Radiolabeled lactate in carbon skeletons is metabolized and transformed into carbon dioxide (CO2) or lipids during oligodendrocyte culture (Sanchez‐Abarca et al., 2001). Aside from being a metabolite, it has been reported recently that lactate acts as a ligand of G‐protein‐coupled receptor 81 (GPR81) and signal transduction via this molecule also mediates the effects of lactate (Ahmed et al., 2009). In the CNS, GPR81 is expressed in neurons, astrocytes, and capillaries, but the expression is very low in oligodendrocytes (Zhang et al., 2014). Moreover, we did not detect Gpr81 mRNA in our OPC‐rich cultures (data not shown). These reports are consistent with our results, which showed the effects of lactate as a metabolite, acting via a transporter in OPCs, and not as a ligand of GPR81.
As candidates for the transporters, we detected Mct1, Mct2, and Mct4 mRNA expression by real‐time RT‐PCR in OPC‐rich cultures (Fig. 5A). MCT1 expression has been detected in myelin at higher levels than in the axons (Lee et al., 2012). MCT2 has been reported to be expressed at lower levels in myelin than in the axons and postsynaptic density (Rinholm et al., 2011). Although myelinating Schwann cells in the peripheral nervous system express MCT4 (Domenech‐Estevez et al., 2015), MCT4 is not co‐expressed with MBP in oligodendrocytes in the CNS (Rinholm et al., 2011). Astrocytes express MCT4, and lactate transported via MCT4 from astrocytes contributes to neuronal function (Suzuki et al., 2011; Gao et al., 2015). The effects of lactate on cell cycling and differentiation of OPCs were blocked by an MCT inhibitor, 4‐CIN (Fig. 5). The IC50 of 4‐CIN is 425 µM for MCT1, 24 µM for MCT2, and 900 µM for MCT4 (Broer et al., 1999; Erlichman et al., 2008). We used a concentration of 1 mM of 4‐CIN, which is considered to inhibit all isoforms of MCTs in oligodendrocytes. Therefore, our results suggested that OPCs take up lactate via MCTs, at least in part. Further experiments are needed to show which isoform is crucial for this uptake of lactate into OPCs.
It should be noted that 4‐CIN unexpectedly reduced the BrdU‐ and Ki67‐positive cell rates only in the 0.4 mM glucose condition but not in 36.6 mM condition (Fig. 5). The possible mechanisms may be (i) 4‐CIN inhibits the function of MCTs expressed in mitochondrial membrane as described previously (Halestrap, 1975; Halestrap and Denton, 1975); or (ii) 4‐CIN inhibits pyruvate transport through MCTs, since pyruvate is also known as a substrate of MCTs (Nakamichi et al., 2005). The first possibility would be ruled out by the fact that 4‐CIN did not affect proliferation at 36.6 mM glucose condition in which intracellular substrates for mitochondrial MCTs are abundant. The second possibility is also not likely because we observed 4‐CIN decreased the BrdU‐ and Ki67‐positive ratio even in no‐pyruvate medium (data not shown). Although there remain other possibilities that 4‐CIN disturbed cell function in unidentified mechanisms at low glucose condition, the current results support that OPCs utilize lactate through MCTs.
In the present study, we showed that lactate directly promotes cell cycling and differentiation in OPC‐rich cultures and is taken up into such cells via MCTs, at least in part. The blockade of glycogen phosphorylase, which may inhibit lactate production from glycogen, suppressed remyelination. These data suggested that glycogen and its metabolite, lactate, contribute to remyelination by inducing cell cycling and differentiation of OPCs. In pathological conditions of the CNS, such as spinal cord injury, in which demyelination occurs, an increased concentration of lactate in the spinal cord has been reported (Anderson et al., 1976). Lactate or glycogen in the CNS in pathological conditions may contribute to promoting recovery, for instance by remyelination in demyelinated tissue.
Authors' Contributions
Y.I. performed the experiments and analyzed the data; Y.I. and T.O. interpreted the results of the experiments; Y.I. prepared the figures; Y.I., T.D., Y.R., M.N., Y.S., and T.O. contributed to the discussion; Y.I. and T.O. designed the research; Y.I. and T.O. wrote the manuscript.
Acknowledgments
We thank Dr. K. Hayakawa at Tokyo University, Japan, for excellent technical assistances and Ms. N. Kume in our lab for animal cares. MEXT‐supported Program for the Strategic Research Foundation at Private Universities, 2015–2019 from the MEXT (S1511017).
Disclosures: No conflicts of interest, financial or otherwise, have been declared by the authors.
Literature Cited
- Ahmed K, Tunaru S, Offermanns S. 2009. GPR109A, GPR109B and GPR81, a family of hydroxy‐carboxylic acid receptors. Trends Pharmacol Sci 30:557–562. [DOI] [PubMed] [Google Scholar]
- Andersen B, Rassov A, Westergaard N, Lundgren K. 1999. Inhibition of glycogenolysis in primary rat hepatocytes by 1, 4‐dideoxy‐1, 4‐imino‐D‐arabinitol. Biochem J 342:545–550. [PMC free article] [PubMed] [Google Scholar]
- Anderson DK, Prockop LD, Means ED, Hartley LE. 1976. Cerebrospinal fluid lactate and electrolyte levels following experimental spinal cord injury. J Neurosurg 44:715–722. [DOI] [PubMed] [Google Scholar]
- Baron W, Hoekstra D. 2010. On the biogenesis of myelin membranes: Sorting, trafficking and cell polarity. FEBS Lett 584:1760–1770. [DOI] [PubMed] [Google Scholar]
- Belanger M, Allaman I, Magistretti PJ. 2011. Brain energy metabolism: Focus on astrocyte‐neuron metabolic cooperation. Cell Metab 14:724–738. [DOI] [PubMed] [Google Scholar]
- Bergersen LH. 2007. Is lactate food for neurons? Comparison of monocarboxylate transporter subtypes in brain and muscle. Neuroscience 145:11–19. [DOI] [PubMed] [Google Scholar]
- Boulanger JJ, Messier C. 2014. From precursors to myelinating oligodendrocytes: Contribution of intrinsic and extrinsic factors to white matter plasticity in the adult brain. Neuroscience 269:343–366. [DOI] [PubMed] [Google Scholar]
- Broer S, Broer A, Schneider HP, Stegen C, Halestrap AP, Deitmer JW. 1999. Characterization of the high‐affinity monocarboxylate transporter MCT2 in Xenopus laevis oocytes. Biochem J 341:529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CC, Zhao C, Franklin RJ. 2010. Remyelination—An effective means of neuroprotection. Horm Behav 57:56–62. [DOI] [PubMed] [Google Scholar]
- Chen T, Qian YZ, Rice A, Zhu JP, Di X, Bullock R. 2000. Brain lactate uptake increases at the site of impact after traumatic brain injury. Brain Res 861:281–287. [DOI] [PubMed] [Google Scholar]
- Chong SY, Rosenberg SS, Fancy SP, Zhao C, Shen YA, Hahn AT, McGee AW, Xu X, Zheng B, Zhang LI, Rowitch DH, Franklin RJ, Lu QR, Chan JR. 2012. Neurite outgrowth inhibitor Nogo‐A establishes spatial segregation and extent of oligodendrocyte myelination. Proc Natl Acad Sci USA 109:1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrast R, Saher G, Nave KA, Verheijen MH. 2011. Lipid metabolism in myelinating glial cells: Lessons from human inherited disorders and mouse models. J Lipid Res 52:419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinuzzo M, Mangia S, Maraviglia B, Giove F. 2012. The role of astrocytic glycogen in supporting the energetics of neuronal activity. Neurochem Res 37:2432–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech‐Estevez E, Baloui H, Repond C, Rosafio K, Medard JJ, Tricaud N, Pellerin L, Chrast R. 2015. Distribution of monocarboxylate transporters in the peripheral nervous system suggests putative roles in lactate shuttling and myelination. J Neurosci 35:4151–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Waly B, Macchi M, Cayre M, Durbec P. 2014. Oligodendrogenesis in the normal and pathological central nervous system. Front Neurosci 8:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlichman JS, Hewitt A, Damon TL, Hart M, Kurascz J, Li A, Leiter JC. 2008. Inhibition of monocarboxylate transporter 2 in the retrotrapezoid nucleus in rats: A test of the astrocyte‐neuron lactate‐shuttle hypothesis. J Neurosci 28:4888–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RD, Brown AM, Ransom BR. 2013. Glycogen function in adult central and peripheral nerves. J Neurosci Res 91:1044–1049. [DOI] [PubMed] [Google Scholar]
- Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Mobius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, Nave KA. 2012. Glycolytic oligodendrocytes maintain myelin and long‐term axonal integrity. Nature 485:517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Zhou L, Zhu W, Wang H, Wang R, He Y, Li Z. 2015. Monocarboxylate transporter‐dependent mechanism confers resistance to oxygen‐ and glucose‐deprivation injury in astrocyte‐neuron co‐cultures. Neurosci Lett 594:99–104. [DOI] [PubMed] [Google Scholar]
- Gibbs ME, Anderson DG, Hertz L. 2006. Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia 54:214–222. [DOI] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, Barres BA, Woo PJ, Vogel H, Monje M. 2014. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344:1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudi V, Gingele S, Skripuletz T, Stangel M. 2014. Glial response during cuprizone‐induced de‐ and remyelination in the CNS: Lessons learned. Front Cell Neurosci 8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP. 1975. The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem J 148:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Denton RM. 1975. The specificity and metabolic implications of the inhibition of pyruvate transport in isolated mitochondria and intact tissue preparations by alpha‐cyano‐4‐hydroxycinnamate and related compounds. Biochem J 148:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Meredith D. 2004. The SLC16 gene family‐from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch 447:619–628. [DOI] [PubMed] [Google Scholar]
- Harris JJ, Attwell D. 2012. The energetics of CNS white matter. J Neurosci 32:356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honsa P, Valny M, Kriska J, Matuskova H, Harantova L, Kirdajova D, Valihrach L, Androvic P, Kubista M, Anderova M. 2016. Generation of reactive astrocytes from NG2 cells is regulated by sonic hedgehog. Glia 64:1518–1531. [DOI] [PubMed] [Google Scholar]
- Ichihara Y, Fujimura R, Tsuneki H, Wada T, Okamoto K, Gouda H, Hirono S, Sugimoto K, Matsuya Y, Sasaoka T, Toyooka N. 2013. Rational design and synthesis of 4‐substituted 2‐pyridin‐2‐ylamides with inhibitory effects on SH2 domain‐containing inositol 5′‐phosphatase 2 (SHIP2). Eur J Med Chem 62:649–660. [DOI] [PubMed] [Google Scholar]
- Inoue S, Noda S, Kashima K, Nakada K, Hayashi J, Miyoshi H. 2010. Mitochondrial respiration defects modulate differentiation but not proliferation of hematopoietic stem and progenitor cells. FEBS Lett 584:3402–3409. [DOI] [PubMed] [Google Scholar]
- Jiang F, Frederick TJ, Wood TL. 2001. IGF‐I synergizes with FGF‐2 to stimulate oligodendrocyte progenitor entry into the cell cycle. Dev Biol 232:414–423. [DOI] [PubMed] [Google Scholar]
- Kiray H, Lindsay SL, Hosseinzadeh S, Barnett SC. 2016. The multifaceted role of astrocytes in regulating myelination. Exp Neurol 283:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Shepel PN, Holden CP, Mackiewicz M, Pack AI, Geiger JD. 2002. Brain glycogen decreases with increased periods of wakefulness: Implications for homeostatic drive to sleep. J Neurosci 22:5581–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. 2012. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin BE, Magnan C, Dunn‐Meynell A, Le Foll C. 2011. Metabolic sensing and the brain: Who, what, where, and how? Endocrinology 152:2552–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner M, Heine S, Haastert K, Garde N, Fokuhl J, Linsmeier F, Grothe C, Baumgartner W, Stangel M. 2008. Sequential myelin protein expression during remyelination reveals fast and efficient repair after central nervous system demyelination. Neuropathol Appl Neurobiol 34:105–114. [DOI] [PubMed] [Google Scholar]
- Miron VE, Kuhlmann T, Antel JP. 2011. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim Biophys Acta 1812:184–193. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Torii T, Takada S, Ohno N, Saitoh Y, Nakamura K, Ito A, Ogata T, Terada N, Tanoue A, Yamauchi J. 2015. Involvement of the Tyro3 receptor and its intracellular partner Fyn signaling in Schwann cell myelination. Mol Biol Cell 26:3489–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Abdullah SL, Brown A, Arulpragasam A, Crocker SJ. 2011. How factors secreted from astrocytes impact myelin repair. J Neurosci Res 89:13–21. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Yamashita Y, Matsuishi T, Utsunomiya H, Okudera T, Hashimoto T. 1999. Cranial MRI of neurologically impaired children suffering from neonatal hypoglycaemia. Pediatr Radiol 29:23–27. [DOI] [PubMed] [Google Scholar]
- Nakamichi N, Kambe Y, Oikawa H, Ogura M, Takano K, Tamaki K, Inoue M, Hinoi E, Yoneda Y. 2005. Protection by exogenous pyruvate through a mechanism related to monocarboxylate transporters against cell death induced by hydrogen peroxide in cultured rat cortical neurons. J Neurochem 93:84–93. [DOI] [PubMed] [Google Scholar]
- Nave K‐A, Trapp BD. 2008. Axon‐glial signaling and the glial support of axon function. Ann Rev Neurosci 31:535–561. [DOI] [PubMed] [Google Scholar]
- Nave KA, Werner HB. 2014. Myelination of the nervous system: Mechanisms and functions. Annu Rev Cell Dev Biol 30:503–533. [DOI] [PubMed] [Google Scholar]
- Ogata T, Ueno T, Hoshikawa S, Ito J, Okazaki R, Hayakawa K, Morioka K, Yamamoto S, Nakamura K, Tanaka S, Akai M. 2011. Hes1 functions downstream of growth factors to maintain oligodendrocyte lineage cells in the early progenitor stage. Neuroscience 176:132–141. [DOI] [PubMed] [Google Scholar]
- Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D. 2011. Regulation of oligodendrocyte development and myelination by glucose and lactate. J Neurosci 31:538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF, Martinez‐Pons C, Milan M, Guinovart JJ. 2014. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab 34:945–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Abarca LI, Tabernero A, Medina JM. 2001. Oligodendrocytes use lactate as a source of energy and as a precursor of lipids. Glia 36:321–329. [DOI] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J. 2000. The Ki‐67 protein: From the known and the unknown. J Cell Physiol 182:311–322. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Miller JJ, Rigor BM. 1997a. Brain lactate is an obligatory aerobic energy substrate for functional recovery after hypoxia: Further in vitro validation. J Neurochem 69:423–426. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Miller JJ, Rigor BM. 1997b. Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: An in vitro study. Brain Res. 744:105–111. [DOI] [PubMed] [Google Scholar]
- Seiwa C, Nakahara J, Komiyama T, Katsu Y, Iguchi T, Asou H. 2004. Bisphenol A exerts thyroid‐hormone‐like effects on mouse oligodendrocyte precursor cells. Neuroendocrinology 80:21–30. [DOI] [PubMed] [Google Scholar]
- Skripuletz T, Hackstette D, Bauer K, Gudi V, Pul R, Voss E, Berger K, Kipp M, Baumgartner W, Stangel M. 2013. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone‐induced demyelination. Brain 136:147–167. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. 2011. Astrocyte‐neuron lactate transport is required for long‐term memory formation. Cell 144:810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Yoshida S. 2014. Mechanisms of remyelination: Recent insight from experimental models. Biomol Concepts 5:289–298. [DOI] [PubMed] [Google Scholar]
- Walls AB, Sickmann HM, Brown A, Bouman SD, Ransom B, Schousboe A, Waagepetersen HS. 2008. Characterization of 1,4‐dideoxy‐1,4‐imino‐d‐arabinitol (DAB) as an inhibitor of brain glycogen shunt activity. J Neurochem 105:1462–1470. [DOI] [PubMed] [Google Scholar]
- Yamauchi J, Miyamoto Y, Torii T, Takashima S, Kondo K, Kawahara K, Nemoto N, Chan JR, Tsujimoto G, Tanoue A. 2012. Phosphorylation of cytohesin‐1 by Fyn is required for initiation of myelination and the extent of myelination during development. Sci Signal 5:ra69. [DOI] [PubMed] [Google Scholar]
- Yan H, Rivkees SA. 2006. Hypoglycemia influences oligodendrocyte development and myelin formation. Neuroreport 17:55–59. [DOI] [PubMed] [Google Scholar]
- Ye JN, Chen XS, Su L, Liu YL, Cai QY, Zhan XL, Xu Y, Zhao SF, Yao ZX. 2013. Progesterone alleviates neural behavioral deficits and demyelination with reduced degeneration of oligodendroglial cells in cuprizone‐induced mice. PLoS ONE 8:e54590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. 2014. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]