

Abstract
Myocardial fibrosis refers to a variety of quantitative and qualitative changes in the interstitial myocardial collagen network that occur in response to cardiac ischaemic insults, systemic diseases, drugs, or any other harmful stimulus affecting the circulatory system or the heart itself. Myocardial fibrosis alters the architecture of the myocardium, facilitating the development of cardiac dysfunction, also inducing arrhythmias, influencing the clinical course and outcome of heart failure patients. Focusing on myocardial fibrosis may potentially improve patient care through the targeted diagnosis and treatment of emerging fibrotic pathways. The European Commission funded the FIBROTARGETS consortium as a multinational academic and industrial consortium with the primary aim of performing a systematic and collaborative search of targets of myocardial fibrosis, and then translating these mechanisms into individualized diagnostic tools and specific therapeutic pharmacological options for heart failure. This review focuses on those methodological and technological aspects considered and developed by the consortium to facilitate the transfer of the new mechanistic knowledge on myocardial fibrosis into potential biomedical applications.
Keywords: Myocardial fibrosis, Animal models, Biomarkers, Cardiac imaging
Introduction
Cardiomyocytes, fibroblasts, and vascular cells in the heart are connected by a complex matrix principally composed of fibrillar collagen, which is instrumental in preserving structural integrity and plasticity. In the diseased heart, the matrix undergoes structural and subcellular changes that progressively influence heart function.1 Beyond the cardiomyocyte‐centric view of heart injury, it is now accepted that alterations of the cardiac extracellular matrix (ECM) and cardiac remodelling play a major role in the development and evolution of cardiac diseases leading to heart failure (HF).1 These ECM alterations result in cardiac fibrosis. At the site of myocardial infarction (MI), acute focal fibrotic scarring provides myocardial healing and prevents rupture.2 In contrast, chronic diffuse or focal reactive myocardial fibrosis is a consequence of either pressure or volume overload due to persisting hypertension, metabolic disorders, valvular heart diseases, ischaemic injury (in areas remote from the infarction), or diffuse myocardial diseases, such as cardiomyopathies. Myocardial fibrosis is characterized by dysregulated collagen turnover (increased synthesis predominates over unchanged or decreased degradation)3, 4 and excessive diffuse collagen accumulation in the interstitial and perivascular spaces.5 The dysregulation of distinct pro‐ and antifibrotic factors, including cytokines and chemokines, growth factors, proteases, hormones, and reactive oxygen species, is responsible for the alteration of the collagen matrix6 (Figure 1). This dysregulation of collagen turnover takes place mainly in phenotypically transformed fibroblasts, termed myofibroblasts.1, 7 The phenoconversion of fibroblasts into myofibroblasts involves the expression of alpha‐smooth muscle actin, a characteristic of smooth muscle cells, as well as the appearance of an extensive, synthetically active endoplasmic reticulum, and is stimulated by a number of bioactive effectors.1, 7, 8, 9, 10, 11, 12 Fibroblasts and particularly myofibroblasts secrete extracellular procollagen chains that assemble into collagen type I and III fibrils and become cross‐linked to form the final fibres.3 Collagen cross‐linking is an important post‐translational modification because it increases myocardial tensile strength and the resistance of collagen fibres to degradation by matrix metalloproteinases.13, 14
Figure 1.
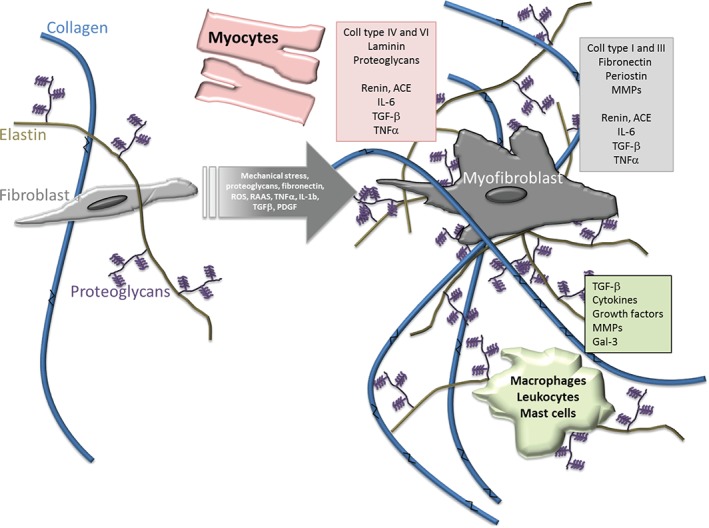
Schematic representation of biochemical and cellular mechanisms of cardiac fibrosis. Under physiological conditions (left), fibroblasts secrete extracellular procollagen chains into the interstitium that assemble into fibrils and are cross‐linked by lysyl oxidase. Several cell types are implicated in fibrotic remodelling of the heart either directly by producing matrix proteins (fibroblasts), or indirectly by secreting fibrogenic mediators (macrophages, mast cells, lymphocytes, cardiomyocytes, and vascular cells). Under pathological conditions (right), alterations in the matrix environment, induction and release of growth factors and cytokines, and increase of mechanical stress dynamically modulate fibroblast transdifferentiation into myofibroblasts. Higher collagen cross‐linking results in increased myocardial tensile strength. Resistance to degradation by matrix metalloproteinases (MMPs) increases cross‐linked collagen, which favours matrisome expansion. Pink, grey, and green boxes list part of the secretome of mycocytes, myofibroblasts, and macrophages/leucocytes/mast cells, respectively, that trigger and maintain fibrosis. Gal‐3, galectin‐3; IL, interleukin; PDGF, platelet‐derived growth factor; RAAS, renin–angiotensin–aldosterone system; ROS, reactive oxygen species; TGF, transforming growth factor; TNF, tumour necrosis factor.
Myocardial fibrosis disrupts the myocardial architecture, contributes to myocardial disarray, and determines mechanical,15, 16 electrical,17, 18, 19 and vasomotor20 dysfunction, thus promoting the progression of cardiac diseases to HF.21 Of note, fibrosis persists in the myocardium of HF patients under the current treatment regimen recommended by the official guidelines;22 thus, the treatment of HF patients improves clinical symptoms, but does not reverse fibrosis. In aortic stenosis patients, aortic valve replacements result in regression of LV hypertrophy (LVH), indicating that hypertrophy and fibrosis are reversible. Furthermore, the severity of histologically proven myocardial fibrosis has been reported to be associated with higher long‐term mortality in patients with cardiac diseases, particularly those with HF.23, 24 In this regard, the detection, prevention, and regression of myocardial fibrosis have emerged as important targets for improving HF therapy.25, 26
In order to achieve significant diagnostic and therapeutic advances, it appears to be critical to identify new profibrotic mechanisms not yet targeted by currently available therapies, and to translate these mechanisms into individualized diagnostic tools and specific therapeutic targets. Besides meaningful functional and therapeutic outcomes, valid molecular targets must not induce serious adverse effects, such as influencing inflammation processes or wound healing. Therefore, there is a need for a systematic collaboration between clinical investigators and basic scientists, together with industry, to allow integration of data from computer biomedical (in silico) and basic (in vitro) studies with pre‐clinical (in vivo) research findings to be distilled into clinically actionable information for the fight against myocardial fibrosis. The FIBROTARGETS consortium is a multinational consortium with industrial and academic partners, funded by the European Commission and primarily aimed at characterizing novel emerging mechanisms of myocardial fibrosis.6 Targets and biomarkers under investigation include proteins, proteoglycans, and microRNAs (miRNAs), and have been reviewed previously.6 This review highlights the methodological issues considered by the consortium to transfer these mechanisms into diagnostic biomarkers and therapeutic agents amenable to improve patient care.
Translational animal models of myocardial fibrosis
Sophisticated in vitro models (‘organ‐on‐a‐chip’) for cardiac tissue based on induced pluripotent stem cells have recently been developed and allow, for example, cardiotoxicity screening.27 Human cells adequately represent human biology, and thus will probably improve pre‐clinical drug screening. However, these cell culture models can currently not sufficiently mirror complex physiological and pathological processes and the interplay of distinct cell types in the heart. Thus, animal models are essential in reliably assessing pathological features and for evaluation of potential new drugs. As the development of myocardial fibrosis is characterized by a complex dysregulation of a number of different factors including inflammatory chemokines, angiotensin II, and endothelin signalling, there is a need for accurate and adequate animal models, which ideally closely reflect the pathological mechanisms found in humans. These animal models are instrumental in investigating molecular mechanisms of myocardial fibrosis, for identification and validation of disease targets, and for efficient pre‐clinical drug development and testing. Fibrosis is induced by various genetic dispositions, pressure or volume stress, heart injuries, and diseases. There is evidence that depending on the particular trigger, distinct molecular pathways have varying importance for the individual types of fibrosis. Consequently, the type of fibrosis induction in animal models is important for their translational value for distinct diseases. Several aspects of HF and myocardial fibrosis are not fully reproducible in animal models: often, a cluster of risk factors, such as metabolic syndrome, is essential in the development of HF. CAD is characterized by gradual narrowing of arteries due to atherosclerosis, but infarction in animal models is triggered by sudden artery occlusion.
Small animals
Several rodent models are available that reproduce some of the main causes of chronic HF such as hypertension, diabetes, metabolic syndrome, or a combination of several of these factors (Table 1). The various animal models differ not only in their availability and ease of use, but also importantly in the mechanism, time course, and severity of cardiac fibrosis. Transgenic and knockout mouse models are used with the aim to model genetic phenotypes and predispositions to cardiac hypertrophy and HF.28 Mutations or depletion of type I collagen,29 and α‐ or β‐cardiac myosin heavy chain30, 31 have been introduced in mice to model fibrosis or hypertrophic cardiomyopathy, respectively. The respective animals are used to simulate congenital cardiac hypertrophy, and they develop severe cardiac hypertrophy with substantial development of interstitial fibrosis and collagen deposition.32 While a more gradual development of pressure overload can be considered to be more clinically relevant, maintenance over a relative long period of time is necessary for development of severe HF.
Table 1.
Animal models for studying myocardial fibrosis
| Model | Species | Fibrosis generation | Degree of fibrosis | Mechanism | Advantages | Limitations | Fibrosis‐affected organs | Examples | Relevance for human therapy | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Volume and/or pressure overload‐induced fibrosis | ||||||||||
| Genetic | M, R | Spontaneous mutation | Varying degree of fibrosis and collagen accumulation, with/without cardiac hypertrophy | Altered signalling pathways, according to gene defect | Commercially available, reproducible, long‐term progressive interstitial fibrosis | Expensive, long‐term treatment needed to expect prevention/decrease of MIF (at least 3 months) | Not restricted to myocardium | SHR, Dahl salt‐sensitive rat | Life‐long follow‐up for treatment effect possible, symptomatic HF treatment | (28, 77) |
| M | Transgenesis, homologous recombination, inducible null | Varying degree of fibrosis and collagen accumulation, with/without cardiac hypertrophy | Altered signalling pathways, according to gene defect | Programmed cardiac hypertrophy with accompanying fibrosis | Difficult to obtain; extensive gene manipulation | Mainly myocardium | Muscle lim protein KO; mutation or depletion of type I collagen or alpha‐ or beta‐cardiac myosin heavy chain | Life‐long follow‐up for treatment effect possible, symptomatic HF treatment | (30, 31) | |
| R | Transgenesis | Diffuse or focal, indirectly associated with cardiac fibrosis | Altered signalling pathways, according to actual gene manipulation | Mostly commercially available, reproducible | Non‐specificity and non‐pathological levels of expression, indirectly associated with fibrosis generation | Not restricted to myocardium | Ren‐2 gene, and TGR(mREN2)27, ACE2, and several others | Life‐long follow‐up for treatment effect possible, symptomatic HF treatment | (78) | |
| Pharmacological | M, R, GP, D, P, S | NOS inhibitors; activation of RAAS; isoproterenol infusion | Mild, eventually focal fibrosis | Depending on the pharmacological agent, proliferation of non‐myocyte cells | Good reproducibility, non‐invasive | Questioned applicability | Not restricted to myocardium | Infusion of angiotensin II | Highly relevant for therapeutic target search | (79) |
| Surgical | M, R | Ascending aortic constriction | Mild, eventually focal fibrosis of the left ventricle | Activated renin–angiotensin system | Resembling human disease, quick onset of hypertension and related fibrosis | High mortality, technically challenging | Left ventricular myocardium | Severe aortic stenosis and cardiac hypertrophy induced cardiac fibrosis | Primarily preventive antihypertensive treatment | (54) |
| GP, D, P, S | Descending aortic constriction | Mild, eventually focal fibrosis of the left ventricle | Activated renin–angiotensin system | Reproducible, leads to diffuse severe cardiac hypertrophy | Surgical procedure, development of fibrosis from hypertrophy requires longer time | Left ventricular myocardium | Severe aortic stenosis and cardiac hypertrophy‐induced cardiac fibrosis | Primarily preventive antihypertensive treatment | (80) | |
| M, R, | Pulmonary artery constriction | Right ventricular hypertrophy and dilation, mild, eventually focal fibrosis of the right ventricle | Sarcoplasmic reticulum Ca‐ATP‐ase and phospholamban down‐regulation | Reproducible, leads to diffuse severe hypertrophy of the right ventricle | Surgical procedure, development of fibrosis from hypertrophy requires longer time | Right ventricular myocardium | Severe pulmonary stenosis and right heart insufficiency | Primarily preventive antihypertensive treatment | (81) | |
| D | Arteriovenous shunt; disruption of mitral cord; gradual constriction of the renal artery | Mild, eventually focal fibrosis of the left ventricle | Eccentric or concentric cardiac hypertrophy | Diffuse mild to moderate cardiac hypertrophy | Surgical procedure, development of fibrosis from hypertrophy requires longer time | Not restricted to myocardium | Cardiac hypertrophy | Causative treatment, difficult to treat with medicines | (40) | |
| P | Percutaneous artificial aortic isthmus stenosis | Severe and diffuse | Activated renin–angiotensin system | Very similar cardiac anatomy compared with human | Surgical procedure, development of fibrosis from hypertrophy requires longer time | Left and right ventricular myocardium | Severe aortic stenosis and cardiac hypertrophy induced cardiac fibrosis | Primarily preventive antihypertensive treatment | (56) | |
| Metabolic disease‐related cardiac fibrosis | ||||||||||
| Genetic | M, R | Spontaneous mutation (colonies) | Varying degree of fibrosis and collagen accumulation, with/without cardiac hypertrophy | Altered signalling pathways, according to gene defect | Commercially available, reproducible, long‐term progressive interstitial fibrosis | Expensive, dramatic phenotypes | Not restricted to myocardium | SHHF, Zucker rat, ZSF1‐LeprfaLeprcp/Crl, ZDF‐Leprfa/crl rats, Lep°b and Leprdb mice | Life‐long follow‐up for treatment effect possible, symptomatic HF treatment | (82, 83) |
| M, R | Transgenesis, homologous recombination, inducible null | Varying degree of fibrosis and collagen accumulation, with/without cardiac hypertrophy | Altered signalling pathways, according to gene defect | Reproduces gene expression alteration seen in disease | Developmental effect of mutant allele | Not restricted to myocardium | Conditional gene targeting in mice | Life‐long follow‐up for treatment effect possible, symptomatic HF treatment | (90) | |
| Pharmacological | M, R, P | Streptozotocin‐induced diabetes | Mild, or no | Non‐specific metabolic syndrome | Relative reproducibility, non‐invasive | High mortality, dramatic phenotypes; difficult method for large animals | Not restricted to myocardium | Similar to human metabolic syndrome | Treatment of metabolic syndrome | (84) |
| Dietary‐induced | M, R, P | High fat diet, western diet | Mild, or no | Non‐specific metabolic syndrome | Resembling human disease | Long and uneven results requiring large number of animals | Not restricted to myocardium | Similar to human metabolic syndrome | Treatment of metabolic syndrome | (85) |
| Myocardial ischaemia‐associated myocardial fibrosis | ||||||||||
| Surgical | M, R, D, and S | Coronary ligation (ischaemia w/o reperfusion): AMI‐related ventricular remodelling | Localized to the ischaemia site | Ischaemic necrosis and reparative fibrous scar formation, loss of myocytes in the ischaemia‐affected region, compensatory hypertrophy in the contralateral areas, later adverse remodelling | Commercially available, reproducible, mid‐term progressive interstitial fibrosis distant from the infarct zone (posteroinferior wall) | Moderately expensive; technically challenging; high mortality; mid‐term treatment needed to expect prevention/decrease of MIF | Part of the left ventricular myocardium | Pre‐clinical chronic coronary artery occlusion model | Anti‐ischaemic medication, primary and secondary prevention | (33, 34) |
| D | Coronary artery microembolization | Localized to the ischaemia site | Ischaemic necrosis and reparative fibrous scar formation, loss of myocytes in the ischaemia‐affected region, compensatory hypertrophy in the contralateral areas, later adverse remodelling | Mimics diffuse small‐vessel disease of the heart | Focal myocardial ischaemia | Part of the left ventricular myocardium | Pre‐clinical chronic coronary artery occlusion model | Anti‐ischaemic medication, primary and secondary prevention | (39) | |
| P | Surgical placement of ameroid constrictor for coronary ligation; closed chest ischaemia/reperfusion model | Localized to the ischaemia site | Ischaemic necrosis and reparative fibrous scar formation, loss of myocytes in the ischaemia‐affected region, compensatory hypertrophy in the contralateral areas, adverse remodelling | Very similar cardiac anatomy compared with human | Focal myocardial ischaemia | Part of the left ventricular myocardium | Pre‐clinical chronic coronary artery occlusion, or reperfused infarction model | Anti‐ischaemic medication, primary and secondary prevention | (49, 50) | |
| Dilated cardiomyopathy‐related cardiac fibrosis | ||||||||||
| Genetic | M | KO of certain genes inducing dilated cardiomyopathy | Mild to severe | Altered signalling pathways, according to knockout gene | Commercially available, reproducible, long‐term progressive interstitial fibrosis | Difficult to obtain; extensive gene manipulation | Mainly myocardium | MybpC | Life‐long follow‐up for treatment effect possible, symptomatic HF treatment | (86) |
| Pharmacological | R, Rab | Systemic cytotoxic therapy | Mild to severe, induced by myocyte apoptosis | Mitochondrial, endoplasmic/sarcoplasmic reticulum pathways | Reproducible, leads to dilated CMP with diffuse fibrosis | Highly relevant for humans, through anticancer therapy | Diffuse, biventricular fibrosis with normal heart size | Cardiotoxicity after cytotoxic treatment | Currently palliative therapy, standard HF therapy | (87) |
| Immunological | R | Viral or autoimmune myocarditis | Severe, based on acute severe interstitial inflammation | Diffuse inflammation, myocyte loss, and replacement with reactive fibrosis | Reproducible, leads to dilated CMP with diffuse fibrosis | Acute inflammation‐induced persistent myocardial fibrosis | Myocardial, biventricular enlargement of the heart, thin wall, hypertrophic or atrophic fibres, infiltrating mononuclear cells | Persistent viral infection, immunization with cardiac myosin fraction | Primarily antiinflammatory agents as therapy, chronic phase is similar to human dilated CMP | (88) |
| Surgical | D, P, Rab, S | Tachycardia pacing | Mild, diffuse, extracellular matrix remodelling | Myocardial energy depletion, abnormal Ca‐channel activity and excitation‐contraction coupling | Reproduces congestive HF by low output | Non‐specific for structural fibrosis | Left and right ventricular myocardium with enlargement of the heart | Tachycardia pacing | Primarily preventive antitachycardia therapy | (89) |
AMI, acute myocardial infarction; CMP, cardiomyopathy; D, dog; GP, guinea pig; HF, heart failure; KO, knockout; Lep, leptin gene; Lepr, leptin receptor gene; M, mouse; MIF, myocardial interstitial fibrosis; MybpC, myosin‐binding protein C; NOS, nitric oxide synthase; P, pig; R, rat; RAAS, renin–angiotensin–aldosterone system; Rab, rabbit; Ren, renin; S, swan; SHHF, spontaneously hypertensive heart failure rat; SHR, spontaneously hypertensive rat.
Infusion of angiotensin II induces severe cardiac interstitial fibrosis in mice. More frequently, surgical interventions in rodents33 are used as models of fibrosis. Artificial MI by ligation of the coronary artery in mice eventually followed by reperfusion results in scarring, cardiac remodelling, and fibrosis.34 Repeated brief ischaemia and reperfusion resulted in chemokine induction, inflammation, cardiac dysfunction, and fibrosis in the absence of MI.35 A partial occlusion of the ascending or descending aorta by a ligature or clip (aortic banding) which is followed by an abrupt increase in pre‐occlusion pressure is commonly used to model LVH in rodents.28, 36 Cardiomyocyte hypertrophy and extensive diffuse fibrotic remodelling occur after several days. However, surgical partial occlusion of the aorta requires an open‐chest procedure, and causes an immediate compromise of the circulation and a sudden increase in pressure stress, in contrast to the more gradual development of myocardial hypertrophy and fibrosis within pathological settings. Furthermore, the sudden pressure increase can cause myocardial injury. A gradual increase of overload is difficult to achieve in rodents. In addition, there are important deviations between rodent and human hearts on the macroscopic (e.g. size, beating frequency) and molecular levels (e.g. relative predominant expression of major histocompatibility complex isoforms and the importance of distinct signalling pathways).
Large animals
Direct translation of murine or rat models to the clinic is problematic, and large animal models are essential for successful translational aspects. In general, large animals used for translational research share a higher extent of genetic homology with humans as compared with rodents. A number of physiological and pharmacological parameters in large animal models are closer to humans, and they have a longer life span, which facilitates longitudinal studies. Dog, pig, and sheep models of HF and fibrosis have been developed, and these species resemble the human pathophysiology more closely.37 Dogs have long been studied in cardiology as a model for MI, and ischaemia and reperfusion results in cardiac remodelling and fibrosis. In this dog MI/reperfusion model, the ARB valsartan resulted in decreased infarct size, increased EF, and improved diastolic function.38 Ischaemic cardiomyopathies can be simulated in canines through coronary microembolization,39 resulting in reduction of LVEF to <35%. Over the course of a few months, progressive LV dysfunction with neurohumoral activation occurred. For simulating the volume overload HF phenotype, mitral chordae have been disrupted with arterially placed grasping forceps in a closed‐chest procedure in dogs.40 Using this model allowed the discovery that beta‐adrenergic receptor blockage attenuates sympathomimetic stimulation by the renin–angiotensin system.41 Gradual constriction of a renal artery in dogs induced LVH42 as a model for chronic pressure overload. A similar model used banding of the ascending aorta of dogs with gradual increase of aortic constriction at 2‐week intervals.43, 44
Because several confounding factors such as coronary collateral circulation complicate the translation of results gathered in canine models, pigs and sheep have recently been increasingly used as animals of translational models.37
In sheep, procedures for causing MI have been elaborated. Selective coronary ligation triggers infarction, followed by progressive cardiac remodelling.45, 46 A couple of issues with sheep, such as zoonotic diseases and anatomical characteristics that complicate detailed imaging, limit the use and translational value of sheep models in cardiology.37
Pigs have a very similar cardiac anatomy, circulation physiology, and distribution of blood supply to humans,47 and are a well‐suited species for translational cardiology.48 In porcine models, cardiac remodelling and HF are most commonly triggered by MI through occlusion of coronary arteries placing ameroid constrictors during open heart surgery.49, 50 For several years, the closed‐chest reperfused MI model has been used, by percutaneous occlusion of either the left anterior or left circumflex coronary arteries, followed by balloon deflation resulting in reperfusion; the sudden reopening of the coronary artery closely resembles primary PCI in patients with acute MI.51 Besides testing of drugs to improve outcome of MI, the swine model is increasingly used for evaluating cardiac regenerative therapies in general, including gene‐ and cell‐based therapies.52, 53 LVH caused by surgical aortic banding is another possibility to provoke cardiac remodelling and HF.54 Besides the close anatomical resemblance to humans, the adaptation of modern multimodal imaging has enabled detailed evaluation of progressing HF in pig models, and thus this species has emerged as having the best translational value for developing novel treatments for HF and fibrosis.
Few large animal models of pressure overload have been described. An ideal model is based on a gradual increase of LV aortic pressure by a slowly evolving gradient, characterized by an initially preserved EF and cardiomyocyte hypertrophy, and progressive development of fibrosis, diastolic dysfunction, and eventual systolic dysfunction.55, 56 Such models are instrumental in effective and rapid translation from basic research to therapy.
Connecting pre‐clinical research with patient care
The field of myocardial fibrosis is continually evolving with regard to the ongoing acquisition of new knowledge on mechanisms and pathways linked to its development. How these novel targets can be utilized as diagnostic tools, for disease monitoring, or for therapeutic targeting is challenging because of two major issues: the cardiac specificity of the target and the complexity and high cost of the process of drug discovery, respectively. Several developments, however, may help overcome these two challenges for the translation of discovery to routine clinical practice. First, the application of cardiac imaging, and the adoption of a circulating biomarker approach for developing myocardial fibrosis‐specific diagnostic assays, and secondly, the use of screening methodologies for early reduction of the number of potential candidates to be further explored as targets with therapeutic potential.
Detection of myocardial fibrosis: imaging and biomarkers
Imaging of myocardial fibrosis
Several single or multimodal imaging technologies have been used to assess the extent and type of myocardial fibrosis (Table 2). The distinct imaging modalities reflect specific changes on the molecular, cellular, or functional level, and appropriate selection should be based on the degree and mechanisms of fibrosis. Besides the direct morphological display of the fibrotic tissue, indirect cardiac functional imaging may evidence fibrosis associated with loss of systolic function integrity and increased myocardial stiffness with diastolic dysfunction.2 Non‐invasive morphological multimodal imaging has the advantage that it can be carried out serially, enabling visualization of the turnover of the fibrotic tissue during the pathological processes. The assessment of ECM volume and molecular targeting of essential mechanisms involved in collagen deposition and degradation may eventually also find a role in development of personalized patient treatment.57
Table 2.
Non‐invasive techniques for assessment of myocardial fibrosis (adapted from Jellis et al. 2)
| Technique for fibrosis detection | Specificity | Fibrosis characterization | Fibrotic disease diagnosis | Localization of fibrosis | Description | Availability | Technical challenge |
|---|---|---|---|---|---|---|---|
| Echocardiography | |||||||
| backscatter | Low | Increased acoustic brightness, backscatter techniques | Hypertrophy, muscular dystrophy, systemic sclerosis | Transmural trend of fibrosis, diffuse | Quantitative assessment | Good | Easy |
| Tissue Doppler imaging | Low | Impairment of longitudinal function of the left ventricle, strain and strain rate | Non‐ischaemic or ischaemic heart disease | Diffuse | Functional assessment | Good | Easy |
| Nuclear imaging | |||||||
| SPECT myocardial perfusion scintigraphy | Low | Indirect, perfusion defect reflects myocardial scar | Myocardial infarction | Segmental | Indirect proof of collagenous scar | Good | Easy |
| SPECT myofibroblast labelling | High | Targeted myofibroblast receptor labelling | Myocardial scar | Segmental | Only experimental | Specialized institution | Complicated |
| SPECT collagelin labelling | High | Localization of collagen‐producing myofibroblasts | Left ventricular remodelling and prediction of heart failure | Infarct area, peri‐infarct zone and remote areas | Only experimental | Specialized institution | Complicated |
| PET perfusable tissue index | High | Calculated indirect marker, correlates with reduced circumferential shortening in MRI tissue tagging | Ischaemic and non‐ischaemic cardiomyopathy | Segmental or diffuse | Quantitative assessment | Moderate | Easy |
| PET 15O‐labelled water | High | Calculation of perfusable tissue index | Ischaemic and non‐ischaemic cardiomyopathy | Segmental or diffuse | Only experimental | Specialized institution | Complicated |
| Cardiac magnetic resonance | |||||||
| Delayed enhancement with T1 imaging | High | High intensity signal in late enhancement image using inversion recovery gradient‐echo sequences, shortening of the inversion time (T1) | Myocardial infarction | Ischaemic area: subendocardial or transmural localization; non‐ischaemic fibrosis is rather irregular and intramural, often subepicardial | Quantitative assessment | Good | Easy |
| T1 mapping | High | Contrast‐enhanced T1 mapping, use of modified Look‐Locker inversion–recovery prototype sequence | Non‐ischaemic cardiomyopathy | Diffuse | Quantitative assessment | Good | Easy |
| T2 mapping | High | T2‐weighted sequences | Isolated LV non‐compaction | Localized collagen fraction | To be validated | Moderate | Complicated |
| Tissue tagging | Low | Tagging of myocardial tissue with a matrix of radiofrequency saturation | Abnormal cardiac torsion and motion | Diffuse | Functional assessment | Moderate | Easy |
| Fused PET–MRI | |||||||
| Delayed enhancement + [18F]FDG PET | High | Combined PET and MRI techniques and data | Myocardial infarction, correlation between viablility/non‐viability and segmental wall motion and infarct scar location and size | Segmental and diffuse fibrosis | Quantitative assessment | Moderate | Easy to moderate |
[18F]FDG, 2‐[18F]fluoro‐2‐deoxy‐d‐glucose; MRI, magnetic resonance imaging; PET, positron emission tomography; SPECT, single photon emission computed tomography.
Cardiac magnetic resonance imaging (MRI) provides detailed tissue characterization, identifying focal myocardial fibrotic scars with late gadolinium enhancement (ventricular LGE) and an estimation of diffuse myocardial fibrosis with post‐contrast enhanced T1 and T2 mapping (Figure 2).58 For molecular imaging of fibrosis, a collagen‐targeted MRI contrast agent (EP‐3533, a cyclic peptide with specific binding to type I collagen) was successfully used to characterize myocardial fibrosis and collagen in a murine model of MI.59
Figure 2.
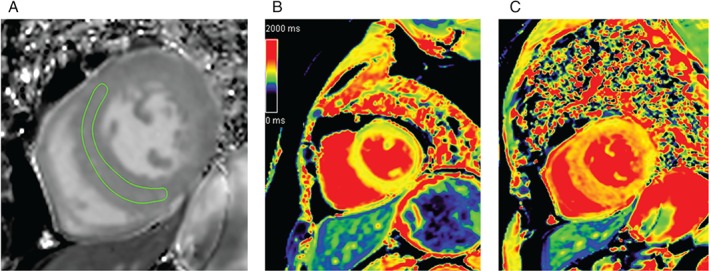
Representative native and T1 cardiac magnetic resonance imaging (cMRI) of diffuse myocardial fibrosis. (A) Diffuse myocardial fibrosis on the short‐axis view of the cMRI image, with the circumference of the anteroseptal myocardial area (region of interest). (B) cMRI T1 map of a patient with moderate aortic stenosis and moderate diffuse myocardial fibrosis. (C) cMRI T1 map of another patient with severe aortic stenosis and severe diffuse fibrosis of the left ventricle. Reproduced with permission from the Radiological Society of North America from Lee et al. 76
Myocardial perfusion scintigraphy is the preferred method to diagnose viable and non‐viable cardiac tissue with reduced coronary flow. Extensive scarring identified on perfusion scan is associated with increased mortality.60 Radiolabelling of myofibroblasts with technetium‐99 m‐labelled Cy5.5‐RGD imaging peptide (CRIP) and positron emission tomography (PET) imaging enabled a non‐invasive indirect assessment of collagen deposition in mice with infarct‐based cardiac remodelling.61 Non‐invasive molecular imaging of fibrosis was demonstrated using collagelin, a peptidomimetic of the platelet collagen receptor glycoprotein VI.62 The suitability of collagelin as an in vivo probe was tested in a rat model of healed MIs. Injecting Tc‐99 m‐labelled collagelin, scintigraphy imaging showed that uptake of the probe occurred in the cardiac area of rats with infarction, but not in controls.62
Positron emission tomography imaging performed by using 15O‐labelled water (H2 15O) and carbon monoxide (C15O) allowed the non‐invasive quantification of both myocardial perfusion and fibrosis.63 Myocardial fibrosis can be indirectly assessed through calculation of the perfusable tissue index (PTI), separating perfusable and non‐perfusable tissues. A reduction in PTI serves as an estimate of fibrosis in a chronic MI model and in human dilated cardiomyopathy.64
Combining PET and MRI has the potential for sensitive and quantitative imaging of cardiovascular anatomy and function with detection of molecular events at the same time.65, 66 A fused PET–MRI (Biograph mMRI, Siemens AG) image allows the simultaneous detection of myocardial global and regional function, extracellular volume, and tissue perfusion and metabolism.67
Circulating biomarkers of myocardial fibrosis
Histopathological analysis of endomyocardial biopsy specimens is the current gold standard for diagnosis and assessment of cardiac fibrosis. A number of circulating biomarkers, including (pro‐)collagen cleavage products, processing enzymes, but also miRNAs (Table 3), have been proposed and analysed. The most consistent results have been found for the C‐terminal propeptide of procollagen type I (PICP) and the N‐terminal propeptide of procollagen type III (PIIINP). In many other cases, however, direct correlation to the extent of cardiac fibrosis is lacking or inconclusive.68 Recent work suggests that urinary peptidomics could provide a further promising alternative to circulating biomarkers of fibrosis.69, 70
Table 3.
Potential circulating biomarkers for assessment of cardiac fibrosis
| Biomarker candidates | Role and correlation to fibrosis | Evidence of association with myocardial fibrosis |
|---|---|---|
| ECM formation | ||
| Procollagen type I C‐terminal propeptide (PICP) | Cleaved enzymatically from procollagen I (collagen biosynthesis) | Yes |
| Procollagen type I N‐terminal propeptide (PINP) | Unknown | |
| Procollagen type III N‐terminal propeptide (PIIINP) | Cleaved enzymatically from procollagen III (collagen biosynthesis) | Yes |
| Collagen type I C‐terminal telopeptide (CITP) | Cleaved by MMP‐1 (collagen I degradation), PICP:CITP ratio corresponds to collagen turnover | Inconclusive |
| Fibrolytic enzymes | ||
| MMP‐1 and other MMPs | Degrades collagens I, II, and III | Unknown |
| TIMP‐1 and other TIMPs | Inhibits MMPs | No (TIMP‐1), unknown (others) |
| miRNAs | ||
| miR‐21 | Correlation with fibrosis in aortic stenosis | Inconclusive |
| miR‐29a | Correlation of plasma levels with hypertrophy and fibrosis in HCM, reduced cardiac expression | Unknown |
| miRNA panels | Concomitant quantification of several miRNAs increases the diagnostic and prognostic value | Unknown |
| Others | ||
| TGF‐β1 | Promotes myofibroblast transactivation and ECM synthesis, deactivates macrophages | Inconclusive |
| Osteopontin | Matricellular protein involved in macrophage regulation | No association |
| Galectin‐3 | Galactosamine binding protein associated with collagen deposition of fibroblasts | Inconclusive |
| Cardiotrophin‐1 | Cytokine associated with cardiac fibrosis | No association |
| Natriuretic peptides | Triggered by myocardial stretch, correlate with HF | Unknown |
ECM, extracellular matrix; HF, heart failure; HCM, hypertrophic cardiomyopathy; miRNA, microRNA; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinases; TGF, transforming growth factor.
Biomarkers of mechanistic pathways involved in myocardial fibrosis
Beyond detecting ongoing myocardial fibrosis and monitoring overall ECM turnover, one aim of the FIBROTARGETS programne is the identification and clinical validation of circulating biomarkers that inform about the alteration and relative importance of the distinct mechanistic pathways involved in myocardial interstitial fibrosis. Details of these biomarkers and potential targets have been described previously.6 These include proteins (cardiotrophin‐1, galectin‐3, NAD phosphate oxidases, neutrophil gelatinase‐associated lipocalin, osteonectin, and lysyl oxidase) and proteoglycans (osteoglycin) that impact fibrosis, and miRNAs that act as upstream regulators or downstream effectors of the fibrotic process.6 For use as biomarkers, it seems reasonable that a combination of several of these increases the predictive and mechanistic power, particularly in the case of miRNAs.71, 72 Stratifying patients according to their myocardial fibrosis bioprofile is an attractive approach for identifying patients with specifically altered expression levels of distinct targets that can be mitigated with respective therapeutic agents.73 Laying the ground for an antifibrotic precision medicine strategy is the ultimate aim of FIBROTARGETS, all the way from validation of biotargets to identifying mechanistically designed antifibrotic therapeutic agents to biomarker profiling for identification of patients most likely to respond to these agents.
Combination of imaging and circulating biomarkers
A multibiomarker‐based strategy should allow for the maximization of the performance of diagnostic tests, and its application at the earliest detectable stage within the disease spectrum is an ultimate goal to support timely interventions and enhance HF prevention.74 Recently, a combination of some specific circulating and imaging biomarkers of myocardial fibrosis was proposed as a useful tool to assess this lesion non‐invasively in HF patients.68
Developments in drug screening
According to Pharmaceutical Research and Manufacturers of America, developing a single new drug takes 10–15 years, thousands of researchers, and costs approximately US$1 billion, with a success rate of only ∼20% (Figures 3 and 4). The failure factor is mainly caused by poor in vivo efficacy and serious adverse events. Improvement in pre‐clinical research strategies with careful selection of drug candidates for clinical evaluation would increase success rates and lower the financial burden. Therefore, it is important to rationalize drug discovery by using meaningful in vitro models to discard irrelevant molecules in terms of efficacy, and pharmacokinetic and toxicological profiles at an early stage. Drug screening technologies are widely used for identifying new potential drug candidates. They comprise protein binding assays and sophisticated cell models in which disease‐relevant biomarkers are measured.75 These technologies termed high throughput screening (HTS) are now miniaturized to allow automatized testing of several thousand compounds per day and measurements of multiple biological parameters simultaneously (high content screening; HCS). With the increasing calculation power of computers, cheminformatics is gaining importance. It is possible to predict biological activities, ADME (absorption, distribution, metabolism, and excretion), and toxicological profiles of molecules based on their chemical structure. For example, this allows the estimation of the affinity of a molecule for a target protein, reducing experimental evaluation to only compounds predicted as most promising.
Figure 3.
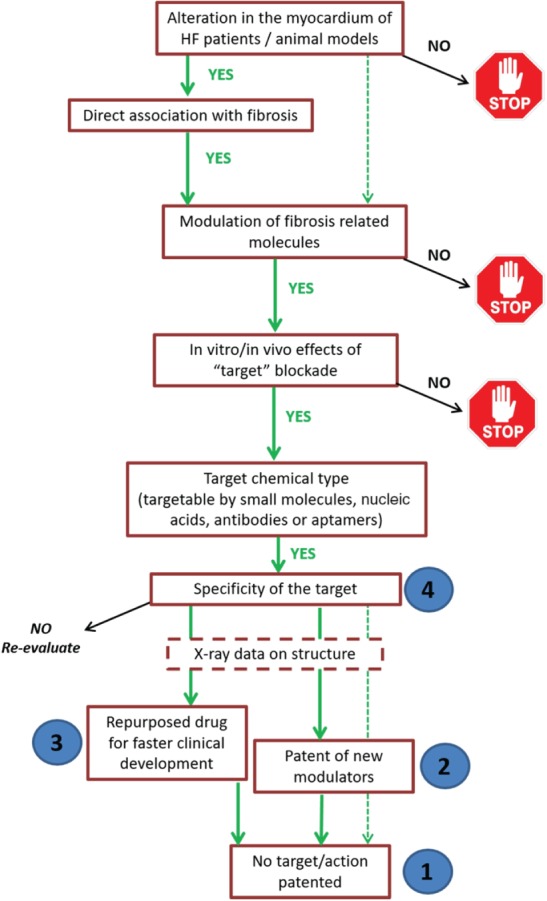
Algorithm for selection of new antifibrotic factors to be further tested as potential therapeutic targets. In order to prioritize the potential antifibrotic targets currently under study in the FIBROTARGETS consortium, and select those to be evaluated in depth from a therapeutic point of view, a number of aspects will be considered in a step‐by‐step process. Targets need to fulfil the stated criteria, otherwise they will be discarded (stop signs). Numbers in blue circles indicate the prioritization of potential therapeutic agents according to their properties. HF, heart failure.
Figure 4.
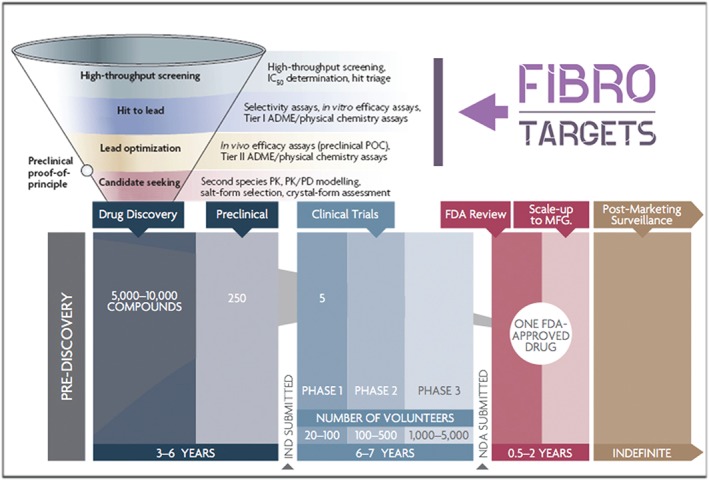
Drug development pipeline highlighting the phases developed by the FIBROTARGETS consortium (modified from Phrma.com). The activities developed by the consortium cover the first steps of the drug discovery strategy; high throughput screening (HTS), hit to lead phase, and lead optimization. By the end of the project, we aim to have identified a set of promising candidates for further evaluation. ADME, absorption, distribution, metabolism, and excretion; FDA, Food and Drug Administration; IC50, half‐maximal inhibitory concentration; HCS, high content screening; IND, investigational new drug; MFG, manufacturing; NDA, new drug application; PD, pharmacodynamics; PK, pharmacokinetics; POC, proof of concept.
FIBROTARGETS aims to find promising hits for further development into drugs targeting cardiac fibrosis. The starting points are several potential targets for two major pathways and biological entities involved in myocardial interstitial fibrosis: the mineralocorticoid and transforming growth factor‐β (TGF‐β) pathways, and non‐structural matrix proteins and miRNAs.6 One target of each group is selected and validated according to the criteria illustrated in Figure 3. Screening of commercially available (drug) compound libraries supplemented by in silico modelling will provide lead structures that are consequently further screened with high content methodologies in relevant cardiac in vitro assays. Toxicity, ADME, and the mechanisms of the molecules in the fibroblast physiology are determined in order to ascertain the therapeutic potential in myocardium interstitial fibrosis treatment. For facilitating further pre‐clinical and clinical drug development, preference will be given to novel molecular targets and/or drug repurposing, i.e. the evaluation of therapeutics that have already been tested and approved for other indications (Figure 3).
Conclusions and perspectives
The FIBROTARGETS consortium has identified novel factors potentially involved in the effector mechanisms of diffuse myocardial interstitial fibrosis. An excess or deficiency of these individual molecules is hypothesized to contribute significantly to fibrillary collagen turnover. The FIBROTARGETS consortium is now validating and qualifying these factors as imaging and/or circulating biomarkers of myocardial fibrosis in HF, as well as developing effective and safe antifibrotic therapies for HF prevention or treatment of HF patients with the aim of fibrosis regression (Figure 5). Target selection and prioritization is based on pathophysiological properties, but also takes drug development aspects into account, including the accessibility for chemical compounds (drugability) and intellectual property opportunities (Table 4).
Figure 5.
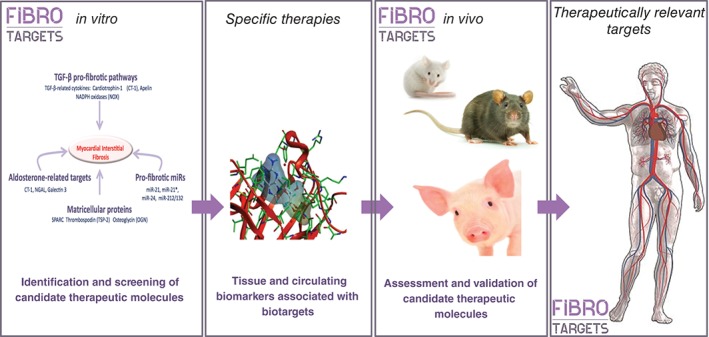
Schematic workflow and aims of FIBROTARGETS.
Table 4.
Score to rank the relevance and interest of new potential targets of myocardial fibrosis
| Aspect to evaluate for each candidate | Score |
|---|---|
| 1. Pathophysiological aspects: | |
| 1.1. Alteration in the myocardium | (max 5) |
| Heart failure patients | 2.5 |
| Animal models | 1.5 |
| In more than one model | 1 |
| 1.2. Myocardial expression/activation associated with end‐point fibrosis | (max.5) |
| Association with fibrosis | 3.5 |
| Pleiotropic effects on other cell types | 1.5 |
| 1.3. Direct modulation of fibrosis‐related molecules | 4 |
| 1.4. Effect of the blockade on myocardial fibrosis | (max. 8) |
| In vitro data | 2 |
| In vivo data | 3 |
| Effects on other features of cardiac remodelling | 1.5 |
| No detrimental side effects (other pathways, tumour growth, etc.) | 1.5 |
| 2. Availability of a non‐invasive circulating biomarker | 2 |
| 3. Chemical properties of the target | (max. 2) |
| Enzyme | 2 |
| Receptor | 1.5 |
| Transporter | 1 |
| Protein‐protein interaction surface | 0.5 |
| microRNAs | 2 |
| 4. Drugability | (max. 5) |
| High specificity of the target | 2.5 |
| Repurposing of drugs that enable a faster clinical development | 2 |
| X‐ray data on the target structure | 0.5 |
| 5. Intellectual property | (max. 4) |
| Non‐patented target/action | 2 |
| Non‐patented modulators | 2 |
The development of fibrosis biomarkers and antifibrotic therapies comprises major challenges, such as lack of organ specificity of biomarkers and the occurrence of related side effects. Is it possible to identify a multibiomarker panel that correlates with the extent of cardiac fibrosis, without being interfered with by fibrosis in other organs such as liver fibrosis, immune processes, or scarring? One of the major challenges in targeting myocardial fibrosis is to avoid side effects such as tendinitis, abnormal wound healing, or adverse inflammation. Hence, a lack of organ specificity might also be an advantage in view of HF being a systemic disease with co‐morbidities of different organs. For example, metabolic risk‐induced HF with preserved EF is accompanied by renal failure, liver fibrosis, and systemic inflammation in patients with diabetes and hypertension, and finding the key hub for fibrosis in all these organs may lead to promising novel therapies and biomarkers. In particular, miRNAs tend to modulate common pathways in different organs, and are often disease and/or organ specific. The multibiomarker approach represents a way to circumvent this problem: combining different biomarkers may help to increase the specificity and positive predictive value in detecting myocardial fibrosis in HF patients.
Finally, FIBROTARGETS closely works together with industry to develop these novel biomarkers and therapeutic tools to be tested and validated in different phases. This interchange provides a unique opportunity to gain access to dual knowledge and to develop these small molecules and miRNA‐based therapies that later on could be applied in humans. Still, the road to the development of individualized therapies or multibiomarkers for human application is a long, expensive and bumpy one. In depth in silico screening, a viable strategy for protein, ligand, or miRNAs targeting, and further toxicological and (pre‐)clinical testing in translational animal models are mandatory before letting ourselves even dream of human application.
Acknowledgements
The authors want to express their gratitude to all the partners of the FIBROTARGETS consortium: Faiez Zannad and Frédéric Jaisser, Institut National de la Santé et de la Recherche Médicale, France; Catherine Clusel and Marie‐Alix Fauvel, Institut National de la Santé et de la Recherche Médicale‐Transfert SA, France; Javier Díez and Arantxa González, Fundación para la Investigación Médica Aplicada, Spain; Kenneth Mac Donald, University College Dublin, National University of Ireland, Ireland; Stephane Heymans and Anna Papageorgiou, Universiteit Maastricht, The Netherlands; Thomas Thum, Medizinische Hochschule Hannover, Germany; Mariann Gyöngyösi and Johannes Winkler, Medical University of Vienna, Austria; Quoc‐Tuan Do, Greenpharma S.A.S., France; Hueseyin Firat and Kaidre Bendjama, Firalis S.A.S., France; Isbaal Ramos and Clarisa Salado, Innovative Technologies in Biological Systems, Spain; and Natalia López‐Andrés, Fundación Pública Miguel Servet, Spain.
Funding
This work was supported by the European Commission FP7 Programme [FIBROTARGETS project grant HEALTH‐2013‐6029047].
Conflict of interest: T.T. filed and partly licensed patents regarding the diagnostic and therapeutic use of non‐coding RNAs. F.Z. reports personal fees from Janssen, Bayer, Pfizer, Novartis, Boston Scientific, Resmed, Amgen, CVRx, Quantum Genomics, EliLilly, Takeda, and General Electric, all outside the submitted work. The other authors declare no conflict of interest.
References
- 1. Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast‐mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol 2013;10:15–26. [DOI] [PubMed] [Google Scholar]
- 2. Jellis C, Martin J, Narula J, Marwick TH. Assessment of nonischemic myocardial fibrosis. J Am Coll Cardiol 2010;56:89–97. [DOI] [PubMed] [Google Scholar]
- 3. Kong P, Christia P, Frangogiannis N. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 2014;71:549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li AH, Liu PP, Villarreal FJ, Garcia RA. Dynamic changes in myocardial matrix and relevance to disease: translational perspectives. Circ Res 2014;114:916–927. [DOI] [PubMed] [Google Scholar]
- 5. Weber KT, Pick R, Jalil JE, Janicki JS, Carroll EP. Patterns of myocardial fibrosis. J Mol Cell Cardiol 1989;21:121–131. [DOI] [PubMed] [Google Scholar]
- 6. Heymans S, González A, Pizard A, Papageorgiou AP, López‐Andrés N, Jaisser F, Thum T, Zannad F, Díez J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur J Heart Fail 2015;17:764–771. [DOI] [PubMed] [Google Scholar]
- 7. Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res 2005;65:40–51. [DOI] [PubMed] [Google Scholar]
- 8. Lajiness JD, Conway SJ. Origin, development, and differentiation of cardiac fibroblasts. J Mol Cell Cardiol 2014;70:2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Watson CJ, Phelan D, Collier P, Horgan S, Glezeva N, Cooke G, Xu M, Ledwidge M, McDonald K, Baugh JA. Extracellular matrix sub‐types and mechanical stretch impact human cardiac fibroblast responses to transforming growth factor beta. Connect Tissue Res 2014;55:248–256. [DOI] [PubMed] [Google Scholar]
- 10. Brilla CG, Maisch B, Zhou G, Weber KT. Hormonal regulation of cardiac fibroblast function. Eur Heart J 1995;16:45–50. [DOI] [PubMed] [Google Scholar]
- 11. Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res 2004;64:24–31. [DOI] [PubMed] [Google Scholar]
- 12. Kumarswamy R, Thum T. Non‐coding RNAs in cardiac remodeling and heart failure. Circ Res 2013;113:676–689. [DOI] [PubMed] [Google Scholar]
- 13. Robins SP. Biochemistry and functional significance of collagen cross‐linking. Biochem Soc Trans 2007;35:849–852. [DOI] [PubMed] [Google Scholar]
- 14. Shoulders MD, Raines RT. Collagen structure and stability. Annu Rev Biochem 2009;78:929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. López B, Querejeta R, González A, Larman M, Díez J. Collagen cross‐linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension 2012;60:677–683. [DOI] [PubMed] [Google Scholar]
- 16. Izawa H, Murohara T, Nagata K, Isobe S, Asano H, Amano T, Ichihara S, Kato T, Ohshima S, Murase Y, Iino S, Obata K, Noda A, Okumura K, Yokota M. Mineralocorticoid receptor antagonism ameliorates left ventricular diastolic dysfunction and myocardial fibrosis in mildly symptomatic patients with idiopathic dilated cardiomyopathy: a pilot study. Circulation 2005;112:2940–2945. [DOI] [PubMed] [Google Scholar]
- 17. McLenachan JM, Dargie HJ. Ventricular arrhythmias in hypertensive left ventricular hypertrophy: relationship to coronary artery disease, left ventricular dysfunction, and myocardial fibrosis. Am J Hypertens 1990;3:735–740. [DOI] [PubMed] [Google Scholar]
- 18. Kawara T, Derksen R, de Groot JR, Coronel R, Tasseron S, Linnenbank AC, Hauer RN, Kirkels H, Janse MJ, de Bakker JM. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation 2001;104:3069–3075. [DOI] [PubMed] [Google Scholar]
- 19. Anderson KP, Walker R, Urie P, Ershler PR, Lux RL, Karwandee SV. Myocardial electrical propagation in patients with idiopathic dilated cardiomyopathy. J Clin Invest 1993;92:122–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwartzkopff B, Brehm M, Mundhenke M, Strauer BE. Repair of coronary arterioles after treatment with perindopril in hypertensive heart disease. Hypertension 2000;36:220–225. [DOI] [PubMed] [Google Scholar]
- 21. Segura A, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Fail Rev 2014;19:173–185. [DOI] [PubMed] [Google Scholar]
- 22. Querejeta R, López B, González A, Sánchez E, Larman M, Martínez Ubago JL, Díez J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation 2004;110:1263–1268. [DOI] [PubMed] [Google Scholar]
- 23. Azevedo CF, Nigri M, Higuchi ML, Pomerantzeff PM, Spina GS, Sampaio RO, Tarasoutchi F, Grinberg M, Rochitte CE. Prognostic significance of myocardial fibrosis quantification by histopathology and magnetic resonance imaging in patients with severe aortic valve disease. J Am Coll Cardiol 2010;56:278–287. [DOI] [PubMed] [Google Scholar]
- 24. Aoki T, Fukumoto Y, Sugimura K, Oikawa M, Satoh K, Nakano M, Nakayama M, Shimokawa H. Prognostic impact of myocardial interstitial fibrosis in non‐ischemic heart failure—comparison between preserved and reduced ejection fraction heart failure. Circ J 2011;75:2605–2613. [DOI] [PubMed] [Google Scholar]
- 25. González A, Ravassa S, Beaumont J, López B, Díez J. New targets to treat the structural remodeling of the myocardium. J Am Coll Cardiol 2011;58:1833–1843. [DOI] [PubMed] [Google Scholar]
- 26. Schelbert EB, Fonarow GC, Bonow RO, Butler J, Gheorghiade M. Therapeutic targets in heart failure: refocusing on the myocardial interstitium. J Am Coll Cardiol 2014;63:2188–2198. [DOI] [PubMed] [Google Scholar]
- 27. Mathur A, Loskill P, Shao K, Huebsch N, Hong S, Marcus SG, Marks N, Mandegar M, Conklin BR, Lee LP, Healy KE. Human iPSC‐based cardiac microphysiological system for drug screening applications. Sci Rep 2015;5:8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Breckenridge R. Heart failure and mouse models. Dis Model Mech 2010;3:138–143. [DOI] [PubMed] [Google Scholar]
- 29. Miller AD, Tyagi SC. Mutation in collagen gene induces cardiomyopathy in transgenic mice. J Cell Biochem 2002;85:259–267. [DOI] [PubMed] [Google Scholar]
- 30. Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE, Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non‐myocyte proliferation and requires TGF‐β;. J Clin Invest 2010;120:3520–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geisterfer‐Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science 1996;272:731–734. [DOI] [PubMed] [Google Scholar]
- 32. Marsiglia JD, Pereira AC. Hypertrophic cardiomyopathy: how do mutations lead to disease? Arq Bras Cardiol 2014;102:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patten RD, Hall‐Porter MR. Small animal models of heart failure: development of novel therapies, past and present. Circ Heart Fail 2009;2:138–144. [DOI] [PubMed] [Google Scholar]
- 34. Chen W, Frangogiannis NG. Fibroblasts in post‐infarction inflammation and cardiac repair. Biochim Biophys Acta 2013;1833:945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dewald O, Frangogiannis NG, Zoerlein M, Duerr GD, Klemm C, Knuefermann P, Taffet G, Michael LH, Crapo JD, Welz A, Entman ML. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc Natl Acad Sci USA 2003;100:2700–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wong K, Boheler KR, Petrou M, Yacoub MH. Pharmacological modulation of pressure‐overload cardiac hypertrophy: changes in ventricular function, extracellular matrix, and gene expression. Circulation 1997;96:2239–2246. [DOI] [PubMed] [Google Scholar]
- 37. Dixon JA, Spinale FG. Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail 2009;2:262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jugdutt BI, Menon V. Valsartan‐induced cardioprotection involves angiotensin II type 2 receptor upregulation in dog and rat models of in vivo reperfused myocardial infarction. J Card Fail 2004;10:74–82. [DOI] [PubMed] [Google Scholar]
- 39. Sabbah HN, Imai M, Cowart D, Amato A, Carminati P, Gheorghiade M. Hemodynamic properties of a new‐generation positive luso‐inotropic agent for the acute treatment of advanced heart failure. Am J Cardiol 2007;99:41A–46A. [DOI] [PubMed] [Google Scholar]
- 40. Kleaveland JP, Kussmaul WG, Vinciguerra T, Diters R, Carabello BA. Volume overload hypertrophy in a closed‐chest model of mitral regurgitation. Am J Physiol Heart Circ Physiol 1988;254:H1034–H1041. [DOI] [PubMed] [Google Scholar]
- 41. Tallaj J, Wei CC, Hankes GH, Holland M, Rynders P, Dillon AR, Ardell JL, Armour JA, Lucchesi PA, Dell'Italia LJ. β1‐Adrenergic receptor blockade attenuates angiotensin II‐mediated catecholamine release into the cardiac interstitium in mitral regurgitation. Circulation 2003;108:225–230. [DOI] [PubMed] [Google Scholar]
- 42. Nguyen TN, Chagas AC, Glantz SA. Left ventricular adaptation to gradual renovascular hypertension in dogs. Am J Physiol Heart Circ Physiol 1993;265:H22–H38. [DOI] [PubMed] [Google Scholar]
- 43. Koide M, Nagatsu M, Zile MR, Hamawaki M, Swindle MM, Keech G, DeFreyte G, Tagawa H, Cooper G, Carabello BA. Premorbid determinants of left ventricular dysfunction in a novel model of gradually induced pressure overload in the adult canine. Circulation 1997;95:1601–1610. [DOI] [PubMed] [Google Scholar]
- 44. Tagawa H, Koide M, Sato H, Zile MR, Carabello BA, Cooper G. Cytoskeletal role in the transition from compensated to decompensated hypertrophy during adult canine left ventricular pressure overloading. Circ Res 1998;82:751–761. [DOI] [PubMed] [Google Scholar]
- 45. Jackson BM, Gorman JH, Moainie SL, Guy TS, Narula N, Narula J, St. John‐Sutton MG, Edmunds LH Jr, Gorman RC. Extension of borderzone myocardium in postinfarction dilated cardiomyopathy. J Am Coll Cardiol 2002;40:1160–1167. [DOI] [PubMed] [Google Scholar]
- 46. Markovitz LJ, Savage EB, Ratcliffe MB, Bavaria JE, Kreiner G, Iozzo RV, Hargrove WC, Bogen DK, Edmunds LH. Large animal model of left ventricular aneurysm. Ann Thorac Surg 1989;48:838–845. [DOI] [PubMed] [Google Scholar]
- 47. Swindle MM, Makin A, Herron AJ, Clubb FJ, Frazier KS. Swine as models in biomedical research and toxicology testing. Vet Pathol 2012;49:344–356. [DOI] [PubMed] [Google Scholar]
- 48. Weaver ME, Pantely GA, Bristow JD, Ladley HD. A quantitative study of the anatomy and distribution of coronary arteries in swine in comparison with other animals and man. Cardiovasc Res 1986;20:907–917. [DOI] [PubMed] [Google Scholar]
- 49. Teramoto N, Koshino K, Yokoyama I, Miyagawa S, Zeniya T, Hirano Y, Fukuda H, Enmi J, Sawa Y, Knuuti J, Iida H. Experimental pig model of old myocardial infarction with long survival leading to chronic left ventricular dysfunction and remodeling as evaluated by PET. J Nucl Med 2011;52:761–768. [DOI] [PubMed] [Google Scholar]
- 50. Tarkia M, Stark C, Haavisto M, Kentala R, Vähäsilta T, Savunen T, Strandberg M, Hynninen VV, Saunavaara V, Tolvanen T, Teräs M, Rokka J, Pietilä M, Saukko P, Roivainen A, Saraste A, Knuuti J. Cardiac remodeling in a new pig model of chronic heart failure: assessment of left ventricular functional, metabolic, and structural changes using PET, CT, and echocardiography. J Nucl Cardiol 2015;22:655–665. [DOI] [PubMed] [Google Scholar]
- 51. McCall FC, Telukuntla KS, Karantalis V, Suncion VY, Heldman AW, Mushtaq M, Williams AR, Hare JM. Myocardial infarction and intramyocardial injection models in swine. Nature Prot 2012;7:1479–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pleger ST, Brinks H, Ritterhoff J, Raake P, Koch WJ, Katus HA, Most P. Heart failure gene therapy: the path to clinical practice. Circ Res 2013;113:792–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanazawa H, Tseliou E, Malliaras K, Yee K, Dawkins JF, De Couto G, Smith RR, Kreke M, Seinfeld J, Middleton RC, Gallet R, Cheng K, Luthringer D, Valle I, Chowdhury S, Fukuda K, Makkar RR, Marbán L, Marbán E. Cellular postconditioning: allogeneic cardiosphere‐derived cells reduce infarct size and attenuate microvascular obstruction when administered after reperfusion in pigs with acute myocardial infarction. Circ Heart Fail 2015;8:322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lunde S, Smerup M, Hasenkam JM, Sloth E. A model for left ventricular hypertrophy enabling non‐invasive assessment of cardiac function. Scand Cardiovasc J 2009;43:267–272. [DOI] [PubMed] [Google Scholar]
- 55. Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, Koch WJ. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res 2012;111:131–150. [DOI] [PubMed] [Google Scholar]
- 56. Winkler J, Pavo N, Zlabinger K, Macejovska D, Bergler‐Klein J, Maurer G, Gyongyosi M. Experimental model of left ventricular hypertrophy, diastolic dysfunction and secondary pulmonary hypertension for translational research. Wien Klin Wochenschr 2015;127:S2‐S3. [Google Scholar]
- 57. de Haas HJ, Arbustini E, Fuster V, Kramer CM, Narula J. Molecular imaging of the cardiac extracellular matrix. Circ Res 2014;114:903–915. [DOI] [PubMed] [Google Scholar]
- 58. van Oorschot JW, Gho JM, van Hout GP, Froeling M, Jansen of Lorkeers SJ, Hoefer IE, Doevendans PA, Luijten PR, Chamuleau SA, Zwanenburg JJ. Endogenous contrast MRI of cardiac fibrosis: beyond late gadolinium enhancement. J Magn Reson Imaging 2015;41:1181–1189. [DOI] [PubMed] [Google Scholar]
- 59. Helm PA, Caravan P, French BA, Jacques V, Shen L, Xu Y, Beyers RJ, Roy RJ, Kramer CM, Epstein FH. Postinfarction myocardial scarring in mice: molecular MR imaging with use of a collagen‐targeting contrast agent. Radiology 2008;247:788–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hachamovitch R, Rozanski A, Shaw LJ, Stone GW, Thomson LEJ, Friedman JD, Hayes SW, Cohen I, Germano G, Berman DS. Impact of ischaemia and scar on the therapeutic benefit derived from myocardial revascularization vs. medical therapy among patients undergoing stress–rest myocardial perfusion scintigraphy. Eur Heart J 2011;32:1012–1024. [DOI] [PubMed] [Google Scholar]
- 61. van den Borne SW, Isobe S, Verjans JW, Petrov A, Lovhaug D, Li P, Zandbergen HR, Ni Y, Frederik P, Zhou J, Arbo B, Rogstad A, Cuthbertson A, Chettibi S, Reutelingsperger C, Blankesteijn WM, Smits JF, Daemen MJ, Zannad F, Vannan MA, Narula N, Pitt B, Hofstra L, Narula J. Molecular imaging of interstitial alterations in remodeling myocardium after myocardial infarction. J Am Coll Cardiol 2008;52:2017–2028. [DOI] [PubMed] [Google Scholar]
- 62. Muzard J, Sarda‐Mantel L, Loyau S, Meulemans A, Louedec L, Bantsimba‐Malanda C, Hervatin F, Marchal‐Somme J, Michel JB, Le Guludec D, Billiald P, Jandrot‐Perrus M. Non‐invasive molecular imaging of fibrosis using a collagen‐targeted peptidomimetic of the platelet collagen receptor glycoprotein VI. PLoS One 2009;4:e5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sado DM, Flett AS, Moon JC. Novel imaging techniques for diffuse myocardial fibrosis. Future Cardiol 2011;7:643–650. [DOI] [PubMed] [Google Scholar]
- 64. Knaapen P, Götte MJ, Paulus WJ, Zwanenburg JJ, Dijkmans PA, Boellaard R, Marcus JT, Twisk JW, Visser CA, van Rossum AC, Lammertsma AA, Visser FC. Does myocardial fibrosis hinder contractile function and perfusion in idiopathic dilated cardiomyopathy? PET and MR imaging study. Radiology 2006;240:380–388. [DOI] [PubMed] [Google Scholar]
- 65. Gyöngyösi M, Blanco J, Marian T, Trón L, Petneházy Ö, Petrasi Z, Hemetsberger R, Rodriguez J, Font G, Pavo IJ, Kertész I, Balkay L, Pavo N, Posa A, Emri M, Galuska L, Kraitchman DL, Wojta J, Huber K, Glogar D. Serial noninvasive in vivo positron emission tomographic tracking of percutaneously intramyocardially injected autologous porcine mesenchymal stem cells modified for transgene reporter gene expression. Circ Cardiovasc Imaging 2008;1:94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rischpler C, Nekolla SG, Dregely I, Schwaiger M. Hybrid PET/MR imaging of the heart: potential, initial experiences, and future prospects. J Nucl Med 2013;54:402–415. [DOI] [PubMed] [Google Scholar]
- 67. Li XG, Roivainen A, Bergman J, Heinonen A, Bengel F, Thum T, Knuuti J. Enabling [18 F]‐bicyclo[6.1.0]nonyne for oligonucleotide conjugation for positron emission tomography applications: [18 F]‐anti‐microRNA‐21 as an example. Chem Commun 2015;51:9821–9824. [DOI] [PubMed] [Google Scholar]
- 68. López B, González A, Ravassa S, Beaumont J, Moreno MU, San José G, Querejeta R, Díez J. Circulating biomarkers of myocardial fibrosis: the need for a reappraisal. J Am Coll Cardiol 2015;65:2449–2456. [DOI] [PubMed] [Google Scholar]
- 69. Kuznetsova T, Mischak H, Mullen W, Staessen JA. Urinary proteome analysis in hypertensive patients with left ventricular diastolic dysfunction. Eur Heart J 2012;33:2342–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rossing K, Bosselmann HS, Gustafsson F, Zhang ZY, Gu YM, Kuznetsova T, Nkuipou‐Kenfack E, Mischak H, Staessen JA, Koeck T, Schou M. Urinary proteomics pilot study for biomarker discovery and diagnosis in heart failure with reduced ejection fraction. PLoS One 2016;11:e0157167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Watson CJ, Gupta SK, O'Connell E, Thum S, Glezeva N, Fendrich J, Gallagher J, Ledwidge M, Grote‐Levi L, McDonald K, Thum T. MicroRNA signatures differentiate preserved from reduced ejection fraction heart failure. Eur J Heart Fail 2015;17:405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fang L, Ellims A, Moore XL, White D, Taylor A, Chin‐Dusting J, Dart A. Circulating microRNAs as biomarkers for diffuse myocardial fibrosis in patients with hypertrophic cardiomyopathy. J Transl Med. 2015;13:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand J‐L, Cohen‐Tervaert JW, Drexler H, Filippatos G, Felix SB, Gullestad L, Hilfiker‐Kleiner D, Janssens S, Latini R, Neubauer G, Paulus WJ, Pieske B, Ponikowski P, Schroen B, Schultheiss H‐P, Tschöpe C, Van Bilsen M, Zannad F, McMurray J, Shah AM. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2009;11:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ahmad T, Fiuzat M, Felker GM, O'Connor C. Novel biomarkers in chronic heart failure. Nat Rev Cardiol 2012;9:347–359. [DOI] [PubMed] [Google Scholar]
- 75. Singh S, Carpenter AE, Genovesio A. Increasing the content of high‐content screening: an overview. J Biomol Screen 2014;19:640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lee SP, Lee W, Lee JM, Park EA, Kim HK, Kim YJ, Sohn DW. Assessment of diffuse myocardial fibrosis by using MR imaging in asymptomatic patients with aortic stenosis. Radiology 2015;274:359–369. [DOI] [PubMed] [Google Scholar]
- 77. Leong XF, Ng CY, Jaarin K. Animal models in cardiovascular research: hypertension and atherosclerosis. BioMed Res Int 2015;2015:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Marttila M, Vuolteenaho O, Ganten D, Nakao K, Ruskoaho H. Synthesis and secretion of natriuretic peptides in the hypertensive TGR(mREN‐2)27 transgenic rat. Hypertension 1996;28:995–1004. [DOI] [PubMed] [Google Scholar]
- 79. Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 2006;103:17985–17990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Chien KR. Segregation of atrial‐specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci USA 1991;88:8277–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rockman HA, Ono S, Ross RS, Jones LR, Karimi M, Bhargava V, Ross J, Chien KR. Molecular and physiological alterations in murine ventricular dysfunction. Proc Natl Acad Sci USA 1994;91:2694–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fellmann L, Nascimento AR, Tibiriça E, Bousquet P. Murine models for pharmacological studies of the metabolic syndrome. Pharmacol Ther. 2013;137:331–340. [DOI] [PubMed] [Google Scholar]
- 83. Youcef G, Olivier A, L'Huillier CP, Labat C, Fay R, Tabcheh L, Toupance S, Rodriguez‐Guéant RM, Bergerot D, Jaisser F, Lacolley P, Zannad F, Laurent V, Pizard A. Simultaneous characterization of metabolic, cardiac, vascular and renal phenotypes of lean and obese SHHF rats. PLoS One 2014;9:e96452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. King AJ. The use of animal models in diabetes research. Br J Pharmacol 2012;166:877–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Winzell MS, Ahrén B. The high‐fat diet‐fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004;53:S215–S219. [DOI] [PubMed] [Google Scholar]
- 86. Jiang J, Burgon PG, Wakimoto H, Onoue K, Gorham JM, O'Meara CC, Fomovsky G, McConnell BK, Lee RT, Seidman JG, Seidman CE. Cardiac myosin binding protein C regulates postnatal myocyte cytokinesis. Proc Natl Acad Sci USA 2015;112:9046–9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jang YM, Kendaiah S, Drew B, Phillips T, Selman C, Julian D, Leeuwenburgh C. Doxorubicin treatment in vivo activates caspase‐12 mediated cardiac apoptosis in both male and female rats. FEBS Lett 2004;577:483–490. [DOI] [PubMed] [Google Scholar]
- 88. Kodama M, Hanawa H, Saeki M, Hosono H, Inomata T, Suzuki K, Shibata A. Rat dilated cardiomyopathy after autoimmune giant cell myocarditis. Circ Res 1994;75:278–284. [DOI] [PubMed] [Google Scholar]
- 89. Shinbane JS, Wood MA, Jensen DN, Ellenbogen KA, Fitzpatrick AP, Scheinman MM. Tachycardia‐induced cardiomyopathy: a review of animal models and clinical studies. J Am Coll Cardiol 1997;29:709–715. [DOI] [PubMed] [Google Scholar]
- 90. Doetschman T, Azhar M. Cardiac‐specific inducible and conditional gene targeting in mice. Circ Res 2012;110:1498–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]