

Abstract
Aims
Schizophrenia is associated with cardiovascular co‐morbidity and a reduced life‐expectancy of up to 20 years. Antipsychotics are dopamine D2 receptor antagonists and are the standard of medical care in schizophrenia, but the drugs are associated with severe metabolic side effects such as obesity and diabetes. Glucagon‐like peptide‐1 receptor agonists (GLP‐1RAs) are registered for treatment of both obesity and type 2 diabetes. We investigated metabolic effects of the GLP‐1RA, exenatide once‐weekly, in non‐diabetic, antipsychotic‐treated, obese patients with schizophrenia.
Material and methods
Antipsychotic‐treated, obese, non‐diabetic, schizophrenia spectrum patients were randomized to double‐blinded adjunctive treatment with once‐weekly subcutaneous exenatide (n = 23) or placebo (n = 22) injections for 3 months. The primary outcome was loss of body weight after treatment and repeated measures analysis of variance was used as statistical analysis.
Results
Between March 2013 and June 2015, 40 patients completed the trial. At baseline, mean body weight was 118.3 ± 16.0 kg in the exenatide group and 111.7 ± 18.0 kg in the placebo group, with no group differences ( P = .23). The exenatide and placebo groups experienced significant ( P = .004), however similar ( P = .98), weight losses of 2.24 ± 3.3 and 2.23 ± 4.4 kg, respectively, after 3 months of treatment.
Conclusions
Treatment with exenatide once‐weekly did not promote weight loss in obese, antipsychotic‐treated patients with schizophrenia compared to placebo. Our results could suggest that the body weight‐lowering effect of GLP‐1RAs involves dopaminergic signaling, but blockade of other receptor systems may also play a role. Nevertheless, anti‐obesity regimens effective in the general population may not be readily implemented in antipsychotic‐treated patients with schizophrenia.
Keywords: antidiabetic drug, exenatide, GLP‐1 analogue, randomized trial schizophrenia
1. INTRODUCTION
Schizophrenia is a severe mental illness affecting approximately .5% of the world's population.1 Patients with schizophrenia have a 2‐ to 3‐fold increased risk of premature death, mainly resulting from cardiovascular diseases2 and, consequently, a reduced life expectancy of up to 20 years compared to the background population.1, 3 With the increasing use of so‐called second generation antipsychotics, the mortality gap between patients with schizophrenia and the background population appears to be expanding.4
Obesity is considered the single most important risk factor for development of diabetes and cardiovascular diseases. The prevalence of obesity (body mass index (BMI) ≥ 30 kg/m2) in the USA has been estimated as 60% in patients with schizophrenia compared to approximately 30% in the background population.5, 6 The etiology of obesity is multifactorial and influenced by, eg, genetic predisposition, physical inactivity and unhealthy dietary habits.7 In patients with severe mental illness, treatment with antipsychotics is an additional contributor to obesity.2, 4
The mechanisms underlying the association between antipsychotics and obesity are not fully understood. Notably, all marketed antipsychotics share the mechanism of dopamine D2 receptor antagonism.8 Dopamine is the main modulator of the brain's reward system, and dopaminergic signaling is pivotal in the regulation of appetite and food intake,9, 10 and central insulin‐receptors may also modulate striatal dopamine release.11 Recently, we reported that attenuated brain reward activity in antipsychotic‐naïve patients with schizophrenia was associated with individual susceptibility to gain weight prior to antipsychotic exposure.12 Moreover, brain activity after dopamine D2 receptor antagonism was associated with weight gain after treatment.12 Antipsychotics are effective in reducing psychotic symptoms; hence, clinical and ethical imperatives to alleviate these symptoms render antipsychotics, despite their severe metabolic side effects, the standard of medical care in schizophrenia and other psychotic disorders.13
Current strategies to reduce antipsychotic‐associated weight gain and obesity include antipsychotic dose reduction, change of antipsychotic compound, behavioral therapy including diet and exercise interventions, psychotherapy and add‐on pharmacological therapy, eg metformin. These regimens may result in up to 4 kg‐reductions in body weight, but sustainability is questionable, and clear recommendations are not available.14, 15, 16, 17, 18
Glucagon‐like peptide‐1 receptor agonists (GLP‐1RAs) constitute an antidiabetic and antiobesity drug class that mimicks the effects of the endogenous gut hormone GLP‐1. GLP‐1 regulates blood glucose homeostasis and appetite through peripheral and central actions. In addition to the blood glucose‐ and body weight‐lowering effects of GLP‐1RAs, these drugs moderately reduce blood pressure and blood lipids.19 The side effects of the GLP‐1RAs are generally tolerable, mostly comprising temporary, mild‐to‐moderate gastrointestinal symptoms (nausea, vomiting and diarrhea).19
Based on these favourable features, we previously proposed GLP‐1RAs as a potential body weight‐lowering treatment for obese patients with schizophrenia.20, 21 Here we report results from the first randomized trial investigating the effect of a GLP‐1RA on body weight in patients with schizophrenia and antipsychotic‐associated obesity. As secondary outcomes, we report effects on blood pressure, pulse rate, body composition and biochemical parameters.
2. MATERIALS AND METHODS
2.1. Study setting and design
The study was an investigator‐initiated, randomized, placebo‐controlled, double‐blinded, parallel group, 3‐month intervention trial. The trial protocol has been published previously.22 In short, antipsychotic‐treated, clinically stable schizophrenia spectrum patients (ICD‐10 diagnoses F20.x and F25.x) between 18 and 65 years of age with obesity were recruited from psychiatric clinics in the capital region of Copenhagen, Denmark. Exclusion criteria included diabetes, current substance dependency and pregnancy. Baseline examinations (week 0) were followed by a weekly injection of trial medication (12‐16 injections per participant) and 3 scheduled visits: at 1 week (7 ± 2 days), at 4 weeks (4‐6 weeks) and at end‐of‐trial (12‐6 weeks). A CONSORT‐flowchart of the study is provided in Figure 1.23
Figure 1.
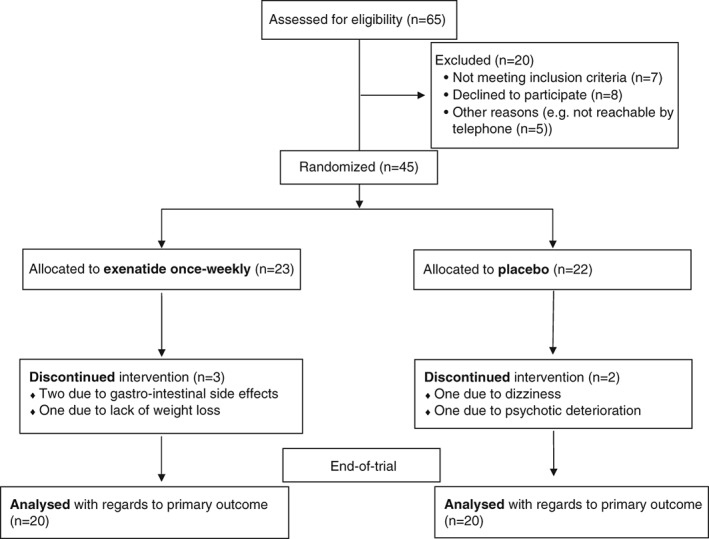
Study diagram of the patient flow according to CONSORT 2010 statement. Of the 65 patients originally referred to the study, 45 entered the subsequent 3‐month treatment period. Five patients withdrew from trial, resulting in a completion of n = 40 (n = 20 exenatide only weekly, n = 20 placebo).
2.2. Randomization and blinding
The randomization code was a computer‐generated randomization allocation sequence with a block size of initially 40 and subsequently 10 to reach the goal of 40 patients completing the trial (20 in each group). Allocation ratio was 1:1. Using the randomization code, 16 sealed allocation envelopes, one per week, were prepared and validated for each patient. For each injection, the staff responsible for administering medication collected the relevant envelope from a locked cabinet, not accessible to the principal investigator (PLI). To maintain blinding during administration of trial medication to patients, injection pens were prepared and covered with opaque labels in a separate room. Blinding was kept until analyses on the primary endpoint had been performed.24
2.3. Approvals and ethics
The study was approved by the National Committee on Health Research Ethics (project no.: 36378), the Danish Health and Medicines Authority (EudraCT no.: 2012‐005404‐17) and The Danish Data Protection Agency (project no.: RHP‐2012‐027) and was conducted according to the Declaration of Helsinki II (7th revision, 2013). The Good Clinical Practice (GCP) Unit at Copenhagen University Hospital monitored the trial according to ICH‐GCP guidelines. All referrals received an oral and written description of the trial and were screened by the principal investigator. All patients approved participation by written informed consent prior to enrolment.
2.4. Trial medication
Trial medication was the GLP‐1RA exenatide once‐weekly (Bydureon, AstraZeneca AB, Södertälje, Sweden). The once‐weekly administration profile of Bydureon was judged favourable for facilitating trial adherence in our psychiatric patient sample. A subcutaneous injection with either exenatide 2 mg (fixed dose) or placebo once‐weekly, was administered by unblinded trial personnel as described above. Placebo injections were solvent from the Bydureon kit (without exenatide). Tolerability was evaluated by giving the first two injections at the psychiatric research facility. Full medication compliance was ensured by giving all subsequent injections in the home of the patient. Adverse events were recorded at trial visits.
2.5. Primary outcome
The primary outcome was loss of body weight after 3 months of treatment with exenatide once‐weekly compared to placebo. Patients received no instructions on diet and exercise during the trial. Our power calculation was based on an expected difference in weight loss of 2.5 ± 2.5 kg in the exenatide group vs 0 ± 2.5 kg in the placebo group, and showed that, to reach a power of 0.8, a sample size of 16 patients in each group was needed.22 Patients were weighed at all 4 scheduled visits. Body weight measurement was obtained without shoes and coat, using an authorized and calibrated scale, Cardinal Detecto 750 (Webb City, Missouri).
2.6. Secondary outcomes
2.6.1. Waist and hip circumference measurements
At all visits, waist circumference was measured with the patient in a standing position and the measure tape positioned horizontally at the level of the iliac crest. To measure hip circumference, the measure tape was positioned horizontally at the widest point of the hip.
2.6.2. 24‐hour blood pressure measurement
At baseline and end‐of‐trial, we performed a 24‐hour (24 h) blood pressure recording with approximately 80 measurements, using the validated monitor, Mobil‐O‐Graph 24 hour PWA Monitor (I.E.M. GmbH, Stolberg, Germany).25 Here we report data on central (systolic and diastolic), peripheral (systolic and diastolic) blood pressures, pulse rate and aortic pulse wave velocity for arterial stiffness quantification.26 Quality and validity of the measurements were determined by the Mobil‐O‐Graph.
2.6.3. Dual‐energy X‐ray absorptiometry (DEXA)
To assess changes in body composition from baseline to end‐of‐trial, patients underwent a whole‐body DEXA scan in the fasting state using the Lunar Prodigy whole‐body scanner (General Electric Medical Systems, Madison, Wisconsin) in conjunction with the software enCORE version 14.1. Data on visceral adipose tissue, body fat percentage, muscle‐mass and android‐to‐gynoid fat mass ratio are reported.
2.6.4. Blood sampling
Blood sampling was performed in the fasting state at baseline, at 4 weeks and at end‐of‐trial visits and analysis was by the Department of Clinical Biochemistry, Rigshospitalet, University of Copenhagen (Glostrup, Denmark) using standard methods. We report on glycated hemoglobin A1c (HbA1c), an indicator of long‐term blood glucose levels (a value <42 mmol/mol indicates normoglycemia), fasting blood glucose and lipids (Table 3).
Table 3.
Biochemical fasting blood values
| Exenatide (n = 20) | Placebo (n = 20) | Time | Group | Time × Group | |||
|---|---|---|---|---|---|---|---|
| Baseline | End‐of‐trial | Baseline | End‐of‐trial | P value | P value | P value | |
| HbA1c (mmol/mol) | 33.3 ± 3.3 | 36.2 ± 3.4 | 34.7 ± 4.8 | 39.15 ± 13.4 | .002* | .33 | .53 |
| [28‐40] | [28‐42] | [25‐47] | [28‐93] | ||||
| Fasting plasma glucose (mmol/L) | 5.1 ± 0.3 | 5.8 ± 0.6 | 5.3 ± 0.5 | 6.1 ± 1.6 | .001* | .27 | .60 |
| [4.2‐5.9] | [5.0‐7.4] | [4.5‐6.4] | [4.9‐12.4) | ||||
| Plasma exenatide (pmol/L) | 3.4 ± 10.5 | 84.9 ± 29.6 | 0.0 ± 0 | 1.1 ± 3.2 | .002* | <.001* | .002* |
| [0‐40] | [65‐147] | [0‐0] | [0‐11] | ||||
| n = 17 | n = 7 | n = 18 | n = 20 | ||||
| Anti‐exenatide antibody binding (%) | 3.75 ± 13.1 | 17.8 ± 19.1 | 0.0 ± 0.0 | 0.0 ± 0.0 | .004* | <.001* | .004* |
| [0‐58] | [0‐50] | [0‐0] | [0‐0] | ||||
| Plasma glucagon (pmol/L) | 10.1 ± 4.8 | 10.5 ± 5.0 | 9.1 ± 3.8 | 9.1 ± 4.1 | .24 | .56 | .38 |
| [3‐21] | [1‐23] | [2‐14] | [2‐18] | ||||
| Triglyceride (mmol/L) | 1.5 ± 0.7 | 2.4 ± 1.0 | 1.4 ± 0.8 | 2.1 ± 1.1 | <.001* | .46 | .21 |
| [0.6‐3.2] | [0.9‐4.5] | [0.4‐3.1] | [0.8‐4.3] | ||||
| Total cholesterol, (mmol/L) | 4.5 ± 1.0 | 5.2 ± 1.1 | 4.2 ± 0.7 | 5.0 ± 0.9 | <.001* | .46 | .47 |
| [2.6‐7.0] | [3.1‐7.7] | [2.5‐5.1] | [3.2‐6.9] | ||||
| LDL cholesterol (mmol/L) | 2.6 ± 0.9 | 3.3 ± 0.9 | 2.4 ± 0.5 | 3.1 ± 0.7 | <.001* | .46 | .56 |
| [0.7‐4.4] | [1.1‐5.2] | [0.9‐3.0] | [1.2‐4.7] | ||||
| VLDL cholesterol (mmol/L) | 0.7 ± 0.3 | 1.1 ± 0.5 | 0.7 ± 0.4 | 1.0 ± 0.5 | <.001* | .48 | .17 |
| [0.3‐1.4] | [0.4‐2.0] | [0.2‐1.4] | [0.4‐1.9] | ||||
| HDL cholesterol (mmol/L) | 0.9 ± 0.2 | 1.1 ± 0.2 | 1.0 ± 0.3 | 1.1 ± 0.3 | <.001* | .47 | .46 |
| [0.6‐1.39] | [0.69‐1.6] | [0.6‐1.5] | [0.7‐1.8] | ||||
Abbreviations: HbA1c, hemoglobin A1c (<42 mmol/mol indicates normoglycemia; 42‐47 mmol/mol indicates prediabetes and >48 mmol/mol indicates diabetes); HDL, high density lipoprotein; LDL, low density lipoprotein; VLDL, very low density lipoprotein.
Mean, standard deviation (SD) and range (in square brackets) are presented as: mean ± SD (min‐max in the table. “n” is provided when the number of validated and/or obtained data points of a variable was <20. P values were analysed using repeated measures analysis of variance, and significant group differences are indicated by asterisks (*). Columns represent effects of Time, Group and Time × Group interaction (treatment).
2.6.5. Experimental procedures regarding exenatide, anti‐exenatide antibodies and glucagon
Plasma samples intended for analyses for exenatide, anti‐exenatide antibodies and glucagon were stored at −80°C as previously described.27, 28 The sensitivity of the exenatide assay is <1 pmol/L, but in order to avoid plasma interference, samples were diluted 10‐fold in assay buffer. For estimation of antibodies against exenatide, plasma samples were incubated with 125‐I‐labelled exendin 9‐39, which binds to the antiserum with same energy as full‐length exenatide, and tracer‐antibody complexes separated from the mixture using plasma‐coated charcoal as in the exenatide radioimmunoassay. Any increases in binding of the tracer, above that observed in plasma from subjects never exposed to exenatide, is indicative of the presence of antibodies and results are presented in percent binding of the tracer, a proxy of antibody titer. The glucagon assay employs a C‐terminally directed antiserum and therefore measures glucagon of mainly pancreatic origin. Sensitivity was <1 pmol/L.
2.7. Statistical methods
Data analyses on the primary outcome and most of the secondary outcomes were performed before un‐blinding and according to the per‐protocol principle. Analyses of adverse events were performed according to the intention‐to‐treat principle, including data from drop‐outs (n = 45) (Figure 1). Demographic variables and clinical characteristics are reported in frequency (percentage) for categorical data, and in mean values (with standard deviations and range) for normally distributed, continuous variables. Group comparisons for demographic data were performed using independent t‐tests for continuous variables and Chi‐square tests for nominal and ordinal variables. Primary and secondary outcomes were tested using rmANOVA. The between‐subject factor, ie exenatide vs placebo, was denoted “Group,” and the within‐subject factor between time points was denoted “Time.” A significant “Time × Group interaction” indicates a difference in response between the two treatment groups. Because of the exploratory nature of the analyses of secondary outcomes, we did not correct for multiple comparisons. All statistical tests were two‐tailed and .05 was set as the significance level throughout. IBM SPSS version 23 (IBM Corporation, Armonk, New York) was used for statistical analyses.
3. RESULTS
Between March 2013 and June 2015, a total of 65 subjects were referred to the trial. Among 65 patients, 57 were outpatients (87.7%). A total of 20 referrals met exclusion criteria (Figure 1). A total of 45 patients were enrolled in the trial and randomized to treatment with either exenatide or placebo and 5 patients dropped out, corresponding to an attrition rate of 11%. In the exenatide group, 3 patients dropped out, 2 because of intolerable gastrointestinal side effects and 1 because of dissatisfaction with the clinical effect, ie unchanged body weight after 9 weeks of treatment. In the placebo group, 1 patient dropped out because of severe nausea and dizziness and 1 because of worsening of psychotic symptoms. A total of 40 patients (20 in each group) completed the trial (Figure 1). Over the 3‐month intervention period the mean number of injections (± standard deviation [SD]) was 14.2 ± 1.0 in the exenatide group and 14.2 ± 1.2 in the placebo group (P = .89).
At baseline, we found no significant differences between groups concerning age, gender, ethnicity, socio‐economic status, lifetime drug dependency, diagnosis, weight, height, BMI or HbA1c (P values > .23) (Table 1). In the exenatide group, 7 patients were smokers compared to 1 patient in the placebo group (P = .02) (Table 1).
Table 1.
Demographics and baseline characteristics of patients
| Exenatide (n = 20) | Placebo (n = 20) | ||||||
|---|---|---|---|---|---|---|---|
| Mean | SD | Range | Mean | SD | Range | P value | |
| Age (years) | 37.4 | 10.7 | 19‐65 | 34.4 | 10.6 | 19‐56 | .37 |
| Gender (male/female) | 11/9 | 9/11 | .53 | ||||
| Ethnicity (%) | |||||||
| Caucasian | 47.5 | 45.0 | .55 | ||||
| Mongolian | 2.5 | 5.0 | |||||
| Education (years) | 12.2 | 2.6 | 12.2 | 2.7 | .93 | ||
| Smoking yes/no (%) | 7/13 (35) | 1/19 (5) | .02* | ||||
| Lifetime substance dependency yes/no (%) | 7/13 (35) | 4/16 (20) | .45 | ||||
| Diagnosis (%) | |||||||
| Schizophrenia, F20.x | 90 | 90 | N/A | ||||
| Schizoaffective, F25.x | 10 | 10 | N/A | ||||
| Body weight (kg) | 118.3 | 16.0 | 84.4‐150.7 | 111.7 | 18.0 | 88.2‐149.7 | .23 |
| Height (cm) | 172.8 | 8.4 | 162‐192 | 170.4 | 10.1 | 153‐190 | .42 |
| BMI (kg/m2) | 39.5 | 3.5 | 31.0‐48 | 38.6 | 6.3 | 30.1‐55.1 | .54 |
| HbA1c (mmol/mol) | 33.3 | 3.3 | 28‐40 | 34.7 | 4.8 | 25‐47 | .31 |
| Typical antipsychotic monotherapy (%) | 5 (n = 1) | 5 (n = 1) | 1 | ||||
| Atypical antipsychotic monotherapy (%) | 60 (n = 12) | 45 (n = 9) | .36 | ||||
| Antipsychotic polypharmacy (%) | 35 (n = 7) | 50 (n = 10) | .35 | ||||
Abbreviations: BMI, body mass index; HbA1c, hemoglobin A1c (<42 mmol/mol indicates normoglycemia; 42‐47 mmol/mol indicates prediabetes and >48 mmol/mol indicates diabetes); N/A, not applicable.
Mean, standard deviation (SD) and range are presented in the table. Group differences were tested by independent two‐sample t‐test and Chi square tests, and significant group differences are indicated by asterisks (*).
Patients were treated with various antipsychotics in both groups (both monotherapy and polypharmacy), including first generation antipsychotics (perphenazine, zuclopenthixol and chlorprothixene) and second generation antipsychotics (clozapine, olanzapine, aripiprazole, risperidone, paliperidone, quetiapine, ziprasidone, amisulpride and sertindole) with no differences between groups (P values > .35) (Table 1).
3.1. Primary outcome
At baseline, mean body weight in the exenatide group was 118.3 ± 16.0 kg vs 111.7 ± 18.0 kg in the placebo group, with no differences between groups (P = .23). During the intervention, the exenatide and placebo groups experienced similar reductions in body weight (2.2 ± 3.3 and 2.2 ± 4.4 kg, respectively) (Figure 2). This effect on body weight was significant (P = .004), without effect of Group (P = .25) or a Time × Group interaction (P = .98) (Table 2). Accordingly, the mean BMI decreased significantly (P = .004) in both groups from 39.5 ± 3.5 to 38.7 ± 3.7 kg/m2 in the exenatide group and from 38.6 ± 6.3 to 37.8 ± 6.7 kg/m2 in the placebo group, with no Group differences (P = .64) and no Time × Group interaction (P = .97). Intention‐to‐treat analysis on primary outcome showed similar results; the effect of Time on body weight was significant (P = .004), and no effect of Group (P = .22) or Time × Group interaction were found (P = .98). Post hoc correction for smoking status did not significantly change our result concerning the primary outcome. Post hoc analysis concerning change in body weight scores, with baseline body weight as covariate, was also insignificant (P = .87).
Figure 2.
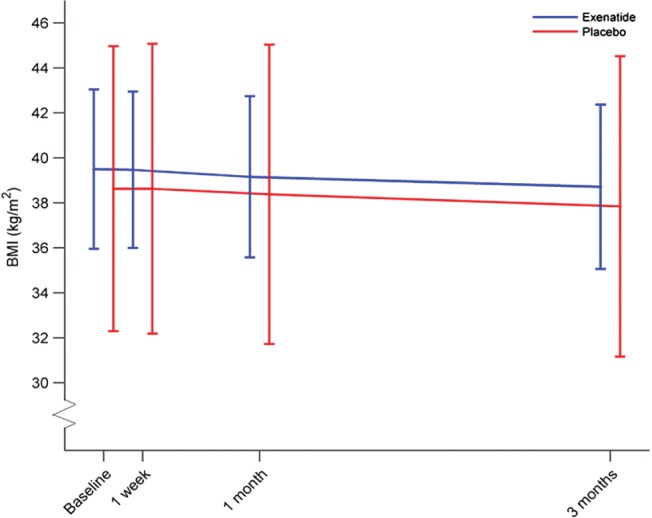
Time lines of mean body mass index (BMI) over a 3‐month period after glucagon‐like peptide‐1 receptor agonist (GLP‐1RA) treatment in obese patients with schizophrenia. Blue line: exenatide group; red line: placebo group. Repeated measures analysis of variance showed no treatment effect of exenatide treatment, Time × Group interaction (P = .98). The x‐axis shows time points for body weight measurements. Horizontal lines on these time points are BMI standard deviations within the groups.
Table 2.
Results on primary and secondary outcomes after 3 months of treatment
| Exenatide (n = 20) | Placebo (n = 20) | Time | Group | Time × Group | |||
|---|---|---|---|---|---|---|---|
| Baseline | End‐of‐trial | Baseline | End‐of‐trial | P value | P value | P value | |
| Body weight (kg) | 118.3 ± 16.0 | 116.0 ± 16.9 | 111.7 ± 18.0 | 109.1 ± 19 | .004* | .25 | .98 |
| [84.4‐150.7] | [82.2‐151.9] | [88.2‐149.7] | [82.6‐145.9] | ||||
| BMI (kg/m2) | 39.5 ± 3.5 | 38.7 ± 3.7 | 38.6 ± 6.3 | 37.8 ± 6.7 | .004* | .64 | .97 |
| [31‐48] | [30.2‐47.9] | [30.1‐55.1] | [28.6‐55.11] | ||||
| Waist circumference (cm) | 128.4 ± 11.1 | 127.7 ± 12.1 | 125.3 ± 13.5 | 124.4 ± 14.9 | .33 | .56 | .48 |
| [106‐150] | [101‐149] | [105‐150] | [100‐149] | ||||
| Hip circumference (cm) | 124.0 ± 8.6 | 122 ± 9.3 | 118.5 ± 13.4 | 118.1 ± 14.8 | .21 | .23 | .42 |
| [110‐147] | [110‐146] | [98‐152] | [96‐153] | ||||
| Central 24 h systolic blood pressure (mm Hg) | 122.4 ± 14.6 | 115.6 ± 13.0 | 110.5 ± 8.9 | 111.9 ± 10.1 | .048* | .06 | .004* |
| [100‐143] | [95‐140] | [97‐141] | [94‐132] | ||||
| n = 15 | n = 15 | n = 16 | n = 16 | ||||
| Central 24 h diastolic blood pressure (mm Hg) | 83.7 ± 12.4 | 81.6 ± 11.4 | 78.1 ± 9.5 | 78.6 ± 8.9 | .48 | .24 | .26 |
| [65‐107] | [63‐101] | [67‐96] | [63‐96] | ||||
| n = 15 | n = 15 | n = 16 | n = 16 | ||||
| 24 h heart rate (beats/min) | 85.0 ± 11.4 | 87.9 ± 10.0 | 88.6 ± 12.9 | 85.4 ± 12.3 | .28 | .72 | .051 |
| [71‐111] | [69‐105] | [63‐122] | [66‐110] | ||||
| n = 19 | n = 19 | n = 20 | n = 18 | ||||
| Pulse wave velocity (m/s) | 6.1 ± 0.8 | 5.9 ± 0.8 | 5.7 ± 0.7 | 5.7 ± 1.0 | .10 | .25 | .007* |
| [4.9‐7.3] | [4.9‐7.3] | [4.7‐7.4] | [4.7‐7.9] | ||||
| n = 18 | n = 18 | n = 17 | n = 17 | ||||
| 24 h systolic blood pressure (mm Hg) | 131.6 ± 13.7 | 126.6 ± 12.4 | 123.4 ± 10.6 | 123.5 ± 10.5 | .034* | .07 | .08 |
| [111‐155] | [105‐153] | [109‐154] | [105‐143] | ||||
| n = 16 | n = 16 | n = 17 | n = 17 | ||||
| 24 h diastolic blood pressure (mm Hg) | 81.4 ± 11.1 | 78.73 ± 10.1 | 76.5 ± 9.0 | 76.9 ± 8.6 | .26 | .31 | .13 |
| [64‐102] | [62‐97] | [64‐93] | [63‐96] | ||||
| n = 16 | n = 16 | n = 17 | n = 17 | ||||
| Visceral adipose tissue (kg) | 2.2 ± 1.1 | 2.1 ± 1.0 | 2.0 ± 1.2 | 1.9 ± 1.1 | .29 | .56 | .71 |
| [1.1‐4.4] | [1.0‐4.5] | [0.6‐4.6] | [0.5‐4.1] | ||||
| n = 15 | n = 15 | n = 15 | n = 15 | ||||
| Body fat mass (%) | 47.0 ± 5.5 | 47.0 ± 5.4 | 46.9 ± 6.8 | 45.7 ± 7.2 | .30 | .62 | .36 |
| [37.6‐55.6] | [37.7‐55.3] | [34.2‐56.9] | [32.9‐55.1] | ||||
| n = 19 | n = 19 | ||||||
| Muscle‐mass (kg) | 59.9 ± 11.7 | 59.2 ± 12.5 | 57.4 ± 7.5 | 56.4 ± 8.2 | .08 | .42 | .81 |
| [42.3‐83] | [41.4‐83] | [44.2‐70.4] | [45.8‐72.1] | ||||
| n = 20 | n = 19 | ||||||
| Android‐to‐gynoid fat mass ratio | 1.21 ± 0.16 | 1.21 ± 0.18 | 1.23 ± 0.23 | 1.22 ± 0.26 | .70 | .82 | .58 |
| [0.99‐1.61] | [0.95‐1.58] | [0.92‐1.64] | [0.9‐1.81] | ||||
| n = 19 | n = 19 | n = 18 | n = 18 | ||||
Abbreviation: BMI, body mass index.
Mean, standard deviation (SD) and range (in square brackets) are presented as mean ± SD (min‐max) in the table. “n” is provided when the number of validated and/or obtained data points of a variable was <20. P values were analysed using repeated measures analysis of variance and significant group differences are indicated by asterisks (*). Columns represent effects of Time, Group and Time × Group interaction (treatment).
3.2. Secondary outcomes
Plasma exenatide significantly increased in the exenatide group compared to the placebo group (P = .002) (Table 3). Exenatide treatment compared to placebo (Time × Group interaction) significantly reduced central 24‐hour systolic blood pressure (P = .004) and pulse wave velocity (P = .007).
Significant effects of Time were found on central 24‐hour systolic blood pressure (P = .05), peripheral 24‐hour systolic blood pressure (P = .03), HbA1c (P = .001), fasting plasma glucose (P = .002), plasma exenatide (P = .002), triglyceride (P < .001), total cholesterol (P < .001), low‐density lipoprotein (P < .001), very low‐density lipoprotein (P < .001) and high‐density lipoprotein cholesterol (P < .001). Regarding plasma exenatide, we found an effect of Group (P < .001), but no effect of Group was found for any other secondary outcomes (P > .06). Post hoc correction for smoking status did not significantly change results concerning any secondary outcomes.
Among 20 patients treated with exenatide, 13 developed significant binding of labeled exendin, ie anti‐exenatide antibodies, compared to none in the placebo group (P = .004). Post hoc analyses excluding these 13 patients did not significantly alter conclusions pertaining to the primary outcome (P = .47) or any of the secondary outcomes. Plasma concentrations of exenatide are reported only for subjects without concomitant measurable antibodies (4 weeks, n = 13 and end‐of‐trial, n = 7). Two to five Mobil‐O‐Graph 24‐hour PWA Monitor parameter measurements were of poor quality and were excluded from statistical analyses (Table 2). Correction for age, mean arterial pressure (MAP) at baseline, and delta MAP (MAP end‐of‐ trial minus MAP baseline) did not remove the significant Time × Group interaction for pulse wave velocity (P = .02). Because of invalid or missing measurements from DEXA scans, the numbers of analysed measurements are less than 20. Mean values for secondary outcome measures are provided in Tables 2 and 3.
3.3. Adverse events
The exenatide group reported more diarrhea (n = 5, 21.7%; P = .02) and fatigue (n = 4, 17.4%; P = .04) compared to 0% in the placebo group (Table 4). No other differences in adverse events were observed (P values > 0.16).
Table 4.
Incidences of adverse events reported in the 2 groups during 3 months of treatment
| Exenatide (n = 23) | Placebo (n = 22) | P value | |
|---|---|---|---|
| Nausea | 30.4% (n = 7) | 18.2% (n = 4) | .34 |
| Diarrhea | 21.7% (n = 5) | 0% (n = 0) | .02* |
| Vomiting | 4.3% (n = 1) | 4.5% (n = 1) | .97 |
| Injection site erythema | 0% (n = 0) | 4.5% (n = 1) | .30 |
| Fatigue | 17.4% (n = 4) | 0% (n = 0) | .04* |
| Pneumonia | 4.3% (n = 1) | 9.1% (n = 2) | .52 |
| Debut of type 2 diabetes | 0% (n = 0) | 4.5% (n = 1) | .30 |
| Hospital admission | 8.6% (n = 2) | 13.6% (n = 4) | .58 |
| Psychiatric symptoms | 17.4% (n = 4) | 4.5 % (n = 1) | .17 |
| Psychiatric admission | 13% (n = 3) | 0 % (n = 0) | .16 |
Columns show frequencies of reported adverse events in percentage and the number (n) of events for all enrolled patients (intention‐to‐treat principle) for both groups. Pearson's Chi2 analyses were performed and significant group differences are indicated by asterisks (*).
A total of 6 serious adverse events were registered during the trial, but none were regarded as related to trial medication. In the exenatide group, 2 patients were admitted to a somatic hospital during participation. One patient had known intermittent suicidal behavior and was admitted following a suicide attempt with acetaminophen. One patient was admitted because of exacerbation of known irritable bowel syndrome resulting in dehydration and an unintended lithium‐intoxication. In the placebo group, 4 patients were admitted to somatic hospital during participation, 2 patients because of pneumonia, 1 patient because of acute dizziness (assumed to represent benign paroxysmal positional vertigo) and 1 patient because of the onset of type 2 diabetes (this patient was hospitalized for 4 days, received insulin and metformin and completed the trial).
4. DISCUSSION
This is the first report on GLP‐1RA effects on body weight and other metabolic and physiological outcomes in obese schizophrenia patients. With reductions in body weight of approximately 2.3 kg in both the exenatide and placebo‐treated groups, a body weight‐lowering effect of exenatide once‐weekly could not be identified after 3 months of treatment. This finding was surprising, as previous placebo‐controlled trials with GLP‐1RAs in obese diabetic, as well as obese non‐diabetic, non‐psychiatric patients, have consistently reported body weight losses.29 Specifically, 2 mg exenatide once‐weekly for 3 months resulted in weight loss of approximately 3 kg compared to placebo in several clinical studies in obese type 2 diabetic individuals.30, 31
In diabetic and non‐diabetic populations with obesity, treatment duration and dose of GLP‐1RA are positively correlated to a body weight‐lowering effect.29 Both the dose of exenatide and duration of treatment may have affected the outcome of our trial; however, as the most prominent weight loss normally is achieved within the first 16 weeks of treatment, we consider duration of treatment here to be adequate.32 For saxenda (the GLP‐1RA liraglutide for obesity) the liraglutide dose (3 mg once‐daily) is almost 2‐fold the antidiabetic dose of liraglutide in victoza (up to 1.8 mg once‐daily), and discontinuation of saxenda is recommended if body weight is not reduced by 5% within the first 12 weeks.33 This suggests that exenatide once‐weekly might also be more effective in higher doses in treating obesity; however, if treatment is effective, weight loss would be expected in the early phase of treatment. Here we used exenatide off‐label; thus, regulatory and ethical considerations prevented us from using higher doses. Our results do not support use of exenatide once‐weekly in cases of antipsychotic‐associated obesity; however, we cannot exclude that weight loss might have been induced with more than 2 mg exenatide per week, or if treatment exceeded 3 months. Variability in the groups of 3.3 kg (exenatide) vs 4.4 kg (placebo) observed on primary endpoint was larger than our a priori expectation of ±2.5 kg. This larger variability may reflect a bias of different degrees of motivation to achieve weight loss through diet and/or physical exercise in the 2 groups. As we did not systematically assess diet and physical exercise, theoretical between‐group differences regarding these factors could have influenced our findings.
In contrast to prior studies of GLP‐1RAs, all patients in this study were treated with antipsychotics. Although antipsychotics represent complex pharmacology affecting multiple receptor systems (eg serotonergic, histaminergic and cholinergic receptors) all antipsychotics share affinity for the dopamine D2 receptor.8 Therefore, the absence of effect of exenatide on body weight in our patients could suggest that the body weight‐lowering effect of GLP‐1RAs may interact with dopaminergic signaling. Because of the known influence of dopaminergic signaling on appetite regulation in the brain reward system, we speculate that the observed GLP‐1RA resistance (ie no weight loss) in our antipsychotic‐treated patients is associated with the central (cerebral) effects of GLP‐1.12 Supporting this notion, a recent study showed that GLP‐1R activation increased dopamine‐turnover in the amygdala, and that food‐related reward, as well as the anorexic effect of GLP‐1, were associated with dopamine D2 receptor signaling.34 However, we cannot rule out that blockade of other receptor systems (eg cholinergic,35 histaminergic36 and serotonergic37) also interferes with the body weight‐lowering effect of GLP‐1RAs.
Corresponding to the modest reductions in body weight, we found no effect of exenatide on waist and hip circumference, body composition or muscle and fat tissue distribution. In contrast to a GLP‐1RA study in non‐diabetic obese patients,38 we found unexpected significant increases in HbA1c, fasting plasma glucose and lipids after 3 months; however, parallel increases in these parameters were also observed in the placebo group. This could indicate that these metabolic parameters in antipsychotic‐treated patients may not be as responsive to weight loss as would be expected in the background population.39 In contrast, a study using exenatide twice‐daily and once‐weekly in patients with type 2 diabetes found a significant lowering of HbA1c, regardless of weight loss.30
In accordance with previous observations in non‐psychiatric populations, we found that exenatide significantly lowered systolic blood pressure (Table 2).30 Moreover, exenatide significantly lowered pulse wave velocity indicative of reduced arterial stiffness, which may be part of the physiological mechanisms behind GLP‐1RA‐induced blood pressure reduction. The values of PWV observed in this study are numerically lower than reported reference values corrected for age and blood pressure.40, 41 However, Mobil‐O‐Graph appears to obtain PWV values, which are slightly lower than the SphygmoCor measurements.26
The significant changes in systolic blood pressure after exenatide treatment were similar to those observed in non‐psychiatric patients, suggesting that the patients had received adequate doses of the drug, and that the “peripheral” effect of exenatide was retained. Post hoc correction for smoking status did not significantly change results on primary outcome or secondary outcomes and, to our knowledge, smoking has not been shown to counteract the effect of GLP‐1RA treatment.
At end‐of‐trial, exenatide was detected in plasma in the exenatide group, confirming that trial medication was active and was administered according to protocol. According to previous pharmacokinetic studies on exenatide once‐weekly, steady‐state levels are reached within 6‐7 weeks of treatment.42 Also, the absolute mean levels (84.9 pmol/L) are similar to those reported in previous studies of exenatide once‐weekly in non‐psychiatric patients.43 As could be expected from a previous study,42 13 out of 20 patients in the exenatide group developed anti‐exenatide antibodies at end‐of‐trial compared to none in the placebo group, but the presence of anti‐exenatide antibody did not alter our results.42
Methodological strengths of the current study include the randomized, placebo‐controlled, double‐blinded trial design, with weight loss as the primary outcome. We used a standard antidiabetic, fixed dose of 2 mg exenatide once‐weekly, previously shown to result in weight loss in obese patients with type 2 diabetes, as well as in obese non‐diabetic subjects. The once‐weekly administration by trial personnel ensured full medication compliance, which was further verified by plasma measurements of exenatide, and the home visits contributed to optimizing trial adherence as reflected in a low attrition rate of 11%30 Exenatide was well‐tolerated and only diarrhea (P = .02) and fatigue (P = .04) were more commonly reported in the exenatide group (Table 4). In comparison with GLP‐1RA studies in diabetic and obese non‐psychiatric patients, the exenatide group in the present study reported fewer gastrointestinal side effects overall. It is known that dopamine D2 receptor antagonism promotes an anti‐emetic effect,44 (eg olanzapine has proven efficacy to reduce chemotherapy‐induced nausea).45 Possibly, the anti‐emesis of antipsychotics resulted in fewer gastrointestinal side effects than would be expected.
We aimed to conduct a proof‐of‐concept trial, and we intentionally included a naturalistic trial population, as reflected in the included patients’ broad medication profiles. We cannot exclude that subgroups of obese antipsychotic‐treated patients, eg patients treated with monotherapy using aripiprazole, a partial dopamine D2 agonist, or with antipsychotics with relatively low dopaminergic affinity (eg quetiapine, olanzapine or clozapine) may benefit from GLP‐1RA treatment. Moreover, we cannot exclude that other GLP‐1RAs (eg liraglutide) or studies of longer treatment duration will show positive effects.
5. CONCLUSIONS
Three months of treatment with the GLP‐1RA, exenatide once‐weekly, had no placebo‐corrected effect on body weight in our obese, antipsychotic‐treated patients with schizophrenia. Because antipsychotics have dopamine D2‐antagonism in common, our results could suggest that the body weight‐lowering effect of GLP‐1RAs may involve dopaminergic signaling. Obesity and cardiovascular health remain critical public healthcare challenges in patients treated with antipsychotics, and the lack of efficient, long‐term treatment regimens, encourage future attempts to prevent weight‐gain when initiating antipsychotic treatment.
ACKNOWLEDGMENTS
The authors would like to thank Gitte S. Andersen (Mental Health Centre Glostrup, Glostrup, Denmark) for administering trial medication, and Lis Larsen (Department of Clinical Physiology, Nuclear Medicine & PET, Rigshospitalet, Glostrup, Denmark) for technical assistance. Also, we would like to express our gratitude to the patients who participated in this trial.
ClinicalTrials.gov identifier: NCT01794429
Conflict of interest
P. L. I. reports no competing interests. F. K. K. has received lecture fees from, is part of Advisory Boards of and/or has consulted for AstraZeneca, Boehringer Ingelheim Pharmaceuticals, Bristol‐Myers Squibb, Eli Lilly and Company, Merck Sharp & Dohme, Novartis, Novo Nordisk, Sanofi and Zealand Pharma. B. V. B., N. B., U. B. A. and N. R. J. report no financial relationships with commercial interests. J. J. H. has received lecture fees from, is part of Advisory Boards of and/or has consulted for AstraZeneca, Boehringer Ingelheim Pharmaceuticals, Bristol‐Myers Squibb, Eli Lilly and Company, Merck Sharp & Dohme, Novartis, Novo Nordisk, Sanofi and Zealand Pharma. B. Y. G. is the leader of a Lundbeck Foundation Center of Excellence for CINS, which is partially financed by an independent grant from the Lundbeck Foundation based on international review and partially financed by the Mental Health Services in the Capital Region of Denmark, the University of Copenhagen, and other foundations. All grants are the property of the Mental Health Services in the Capital Region of Denmark and administrated by them. She has nothing else to declare. B. H. E. has received lecture fees from and/or is part of Advisory Boards of Bristol‐Myers Squibb, Eli Lilly and Company, Janssen‐Cilag, Otsuka Pharma Scandinavia and Takeda Pharmaceutical Company.
Author contributions
All authors fulfill authorship criteria of the ICMJE by substantial contribution to the conception and design, to acquisition of data, or to the analysis and interpretation of the data. All authors have made a substantial contribution to drafting the article or reviewing it critically. The trial was investigator‐initiated and data analysis was conducted without influence from the pharmaceutical industry. We also affirm that there was no editorial direction or censorship from any pharmaceutical company. The content of the manuscript has not been previously published in whole or substantial part and is not under consideration for publication by any other journal. All authors have given final approval of this version of the article to be published.
Ishøy PL, Knop FK, Broberg BV, Bak N, Andersen UB, Jørgensen NR, Holst JJ, Glenthøj BY and Ebdrup BH. Effect of GLP‐1 receptor agonist treatment on body weight in obese antipsychotic‐treated patients with schizophrenia: A randomized, placebo‐controlled trial Diabetes Obes Metab, 2017;19(2):162–171.
This work was generously supported by grants from the University of Copenhagen to Dr. Ishøy (211‐0649/11‐3012), and from the University of Copenhagen/Mental Health Services, Capital Region of Denmark to Dr. Ebdrup. A Lundbeck Foundation grant supported Centre for Clinical Intervention and Neuropsychiatric Schizophrenia Research, CINS (R25‐A2701). The TAO study is investigator‐initiated and not sponsored by pharmaceutical industry.
REFERENCES
- 1. McGrath J, Saha S, Chant D, Welham J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev. 2008;30(1):67‐76. [DOI] [PubMed] [Google Scholar]
- 2. Dieset I, Andreassen OA, Haukvik UK. Somatic comorbidity in schizophrenia: some possible biological mechanisms across the life span. Schizophr Bull. 2016;42(6):1316‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS One. 2013;8(1):e55176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saha S, Chant D, Mcgrath J. A systematic review of mortality in schizophrenia. Arch Gen Psychiatry. 2007;64(10):1123‐1131. [DOI] [PubMed] [Google Scholar]
- 5. Baskin ML, Ard J, Franklin F, Allison DB. Prevalence of obesity in the United States. Obes Rev. 2005;6(1):5‐7. [DOI] [PubMed] [Google Scholar]
- 6. Allison DB, Newcomer JW, Dunn AL, et al. Obesity among those with mental disorders: a National Institute of Mental Health meeting report. Am J Prev Med. 2009;36(4):341‐350. [DOI] [PubMed] [Google Scholar]
- 7. Megna JL, Schwartz TL, Siddiqui U, Herrera Rojas M. Obesity in adults with serious and persistent mental illness: a review of postulated mechanisms and current interventions. Ann Clin Psychiatry. 2011;23(2):131‐140. [PubMed] [Google Scholar]
- 8. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III ‐ the final common pathway. Schizophr Bull. 2009;35(3):549‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol. 1989;40:191‐225. [DOI] [PubMed] [Google Scholar]
- 10. Stice E, Spoor S, Ng J, Zald DH. Relation of obesity to consummatory and anticipatory food reward. Physiol Behav. 2009;97(5):551‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caravaggio F, Hahn M, Nakajima S, Gerretsen P, Remington G, Graff‐Guerrero A. Reduced insulin‐receptor mediated modulation of striatal dopamine release by basal insulin as a possible contributing factor to hyperdopaminergia in schizophrenia. Med Hypotheses. 2015;85(4):391‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nielsen MØ, Nielsen MØ, Rostrup E, Wulff S, Glenthøj B, Ebdrup BH. Striatal reward activity and antipsychotic‐associated weight change in patients with schizophrenia undergoing initial treatment. JAMA Psychiatry. 2016;73(2):1‐8. [DOI] [PubMed] [Google Scholar]
- 13. Buchanan RW, Kreyenbuhl J, Kelly DL, et al. The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mukundan A, Faulkner G, Cohn T, Remington G. Antipsychotic switching for people with schizophrenia who have neuroleptic‐induced weight or metabolic problems. Cochrane Database Syst Rev. 2010;(2):1‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gabriele JM, Dubbert PM, Reeves RR. Efficacy of behavioural interventions in managing atypical antipsychotic weight gain. Obes Rev. 2009;10(4):442‐455. [DOI] [PubMed] [Google Scholar]
- 16. Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medications used to attenuate antipsychotic‐related weight gain and metabolic abnormalities: a systematic review and meta‐analysis. Neuropsychopharmacology. 2010;35(7):1520‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Faulkner G, Cohn T, Remington G. Interventions to reduce weight gain in schizophrenia. Schizophr Bull. 2007;33(3):654‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jarskog LF, Hamer RM, Catellier DJ, et al. Metformin for weight loss and metabolic control in overweight outpatients with schizophrenia and schizoaffective disorder. Am J Psychiatry. 2013;170(9):1032‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holst JJ. The physiology of glucagon‐like peptide 1. Physiol Rev. 2007;87(4):1409‐1439. [DOI] [PubMed] [Google Scholar]
- 20. Ebdrup BH, Knop FK, Ishøy PL, et al. Glucagon‐like peptide‐1 analogs against antipsychotic‐induced weight gain: potential physiological benefits. BMC Med. 2012;10(1):92‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ishøy PL, Knop FK, Vilsbøll T, Glenthøj BY, Ebdrup BH. Sustained weight loss after treatment with a glucagon‐like Peptide‐1 receptor agonist in an obese patient with schizophrenia and type 2 diabetes. Am J Psychiatry. 2013;170(6):681‐682. [DOI] [PubMed] [Google Scholar]
- 22. Ishøy PL, Knop FK, Broberg BV, et al. Treatment of antipsychotic‐associated obesity with a GLP‐1 receptor agonist — protocol for an investigator‐ initiated prospective, randomised, intervention study: the TAO study protocol. BMJ Open. 2014;4:e004158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moher D, Hopewell S, Schulz KF, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340:c869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gøtzsche PC. Blinding during data analysis and writing of manuscripts. Control Clin Trials. 1996;17(4):285‐290; discussion 290–293. [DOI] [PubMed] [Google Scholar]
- 25. Weiss W, Gohlisch C, Harsch‐Gladisch C, Tölle M, Zidek W, van der Giet M. Oscillometric estimation of central blood pressure: validation of the Mobil‐O‐Graph in comparison with the SphygmoCor device. Blood Press Monit. 2012;17(3):128‐131. [DOI] [PubMed] [Google Scholar]
- 26. Luzardo L, Lujambio I, Sottolano M, et al. 24‐h ambulatory recording of aortic pulse wave velocity and central systolic augmentation: a feasibility study. Hypertens Res. 2012;35(10):980‐987. [DOI] [PubMed] [Google Scholar]
- 27. Kielgast U, Holst JJ, Madsbad S. Antidiabetic actions of endogenous and exogenous GLP‐1 in type 1 diabetic patients with and without residual β‐cell function. Diabetes. 2011;60(5):1599‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wewer Albrechtsen NJ, Hartmann B, Veedfald S, et al. Hyperglucagonaemia analysed by glucagon sandwich ELISA: nonspecific interference or truly elevated evels? Diabetologia. 2014;57(9):1919‐1926. [DOI] [PubMed] [Google Scholar]
- 29. Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon‐like peptide‐1 receptor agonists on weight loss: systematic review and meta‐analyses of randomised controlled trials. BMJ. 2012;344:d7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buse JB, Drucker DJ, Taylor KL, et al. DURATION‐1: exenatide once weekly produces sustained glycemic control and weight loss over 52 weeks. Diabetes Care. 2010;33(6):1255‐1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buse JB, Nauck M, Forst T, et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION‐6): a randomised, open‐label study. Lancet. 2013;381(9861):117‐124. [DOI] [PubMed] [Google Scholar]
- 32. Davies MJ, Bergenstal R, Bode B, et al. Efficacy of liraglutide for weight loss among patients with type 2 diabetes. JAMA. 2015;314(7):687. [DOI] [PubMed] [Google Scholar]
- 33. European Medicines Agency. Annex I: summary of product characteristics for Saxenda;URL: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/003780/WC500185786.pdf. Accessed April 16, 2015.
- 34. Anderberg RH, Anefors C, Bergquist F, Nissbrandt H, Skibicka KP. Dopamine signaling in the amygdala, increased by food ingestion and GLP‐1, regulates feeding behavior. Physiol Behav. 2014;136:135‐144. [DOI] [PubMed] [Google Scholar]
- 35. Weston‐Green K, Huang X‐F, Lian J, Deng C. Effects of olanzapine on muscarinic M3 receptor binding density in the brain relates to weight gain, plasma insulin and metabolic hormone levels. Eur Neuropsychopharmacol. 2012;22(5):364‐373. [DOI] [PubMed] [Google Scholar]
- 36. He M, Deng C, Huang X‐F. The role of hypothalamic H1 receptor antagonism in antipsychotic‐induced weight gain. CNS Drugs. 2013;27(6):423‐434. [DOI] [PubMed] [Google Scholar]
- 37. Rasmussen H, Ebdrup BH, Oranje B, Pinborg LH, Knudsen GM, Glenthøj B. Neocortical serotonin2A receptor binding predicts quetiapine associated weight gain in antipsychotic‐naive first‐episode schizophrenia patients. Int J Neuropsychopharmacol. 2014:1‐8. [DOI] [PubMed] [Google Scholar]
- 38. Astrup A, Carraro R, Finer N, et al. Safety, tolerability and sustained weight loss over 2 years with the once‐daily human GLP‐1 analog, liraglutide. Int . Obes (Lond). 2012;36(6):843‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ebdrup BH, Knop FK, Madsen A, et al. Glucometabolic hormones and cardiovascular risk markers in antipsychotic‐treated patients. J Clin Psychiatry. 2014;75(9):e899‐e905. [DOI] [PubMed] [Google Scholar]
- 40. Díaz A, Galli C, Tringler M, Ramírez A, Cabrera Fischer EI. Reference values of pulse wave velocity in healthy people from an urban and rural argentinean population. Int J Hypertens. 2014;2014:653239:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reference Values for Arterial Stiffness’ Collaboration . Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: “establishing normal and reference values”. Eur Heart J. 2016;31(19):2338‐2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fineman M, Flanagan S, Taylor K, et al. Pharmacokinetics and pharmacodynamics of exenatide extended‐release after single and multiple dosing. Clin Pharmacokinet. 2011;50(1):65‐74. [DOI] [PubMed] [Google Scholar]
- 43. Drucker DJ, Buse JB, Taylor K, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open‐label, non‐inferiority study. Lancet. 2008;372(9645):1240‐1250. [DOI] [PubMed] [Google Scholar]
- 44. Herrstedt J. Risk‐benefit of antiemetics in prevention and treatment of chemotherapy‐induced nausea and vomiting. Expert Opin Drug Saf. 2004;3(3):231‐248. [DOI] [PubMed] [Google Scholar]
- 45. Chiu L, Chow R, Popovic M, et al. Efficacy of olanzapine for the prophylaxis and rescue of chemotherapy‐induced nausea and vomiting: a systematic review and meta‐analysis. Support Care Cancer. 2016;24(5):2381‐2392. [DOI] [PubMed] [Google Scholar]