

Abstract
The first transmembrane (TM1) helix in the red cell anion exchanger (AE1, Band 3, or SLC4A1) acts as an internal signal anchor that binds the signal recognition particle and directs the nascent polypeptide chain to the endoplasmic reticulum (ER) membrane where it moves from the translocon laterally into the lipid bilayer. The sequence N-terminal to TM1 forms an amphipathic helix that lies at the membrane interface and is connected to TM1 by a bend at Pro403. Southeast Asian ovalocytosis (SAO) is a red cell abnormality caused by a nine-amino acid deletion (Ala400–Ala408) at the N-terminus of TM1. Here we demonstrate, by extensive (∼4.5 μs) molecular dynamics simulations of TM1 in a model 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine membrane, that the isolated TM1 peptide is highly dynamic and samples the structure of TM1 seen in the crystal structure of the membrane domain of AE1. The SAO deletion not only removes the proline-induced bend but also causes a “pulling in” of the part of the amphipathic helix into the hydrophobic phase of the bilayer, as well as the C-terminal of the peptide. The dynamics of the SAO peptide very infrequently resembles the structure of TM1 in AE1, demonstrating the disruptive effect the SAO deletion has on AE1 folding. These results provide a precise molecular view of the disposition and dynamics of wild-type and SAO TM1 in a lipid bilayer, an important early biosynthetic intermediate in the insertion of AE1 into the ER membrane, and extend earlier results of cell-free translation experiments.
The anion exchanger AE1 (also called Band 3 or SLC4A1) is an abundant glycoprotein in the plasma membrane of the red cell where it mediates the electro-neutral exchange of chloride and bicarbonate ions.1−3 Human protein contains 911 amino acids with a single N-glycosylation site at Asn642. The protein consists of two domains: the amino-terminal cytosolic domain that provides a link to the underlying cytoskeleton4,5 and the carboxyl-terminal membrane domain that spans the lipid bilayer 14 times6−11 and is responsible for the anion transport function.12 A structure of the membrane domain of human AE1 has been recently elucidated by X-ray crystallography (Figure 1A) that confirms the 14-TM model.13
Figure 1.

Role of TM1 in the synthesis of the transmembrane domain of AE1. (A) Structure of the transmembrane region of AE1 or Band 3. (B) Sequence alignment between the sequences of the wild type and the SAO mutant in the region of TM1. The SAO AE1 mutant is missing nine residues (Ala400–Ala408) from the N-terminus of transmembrane segment 1 (TM1). The numbering refers to the wild-type sequence. The three proline residues are highlighted, as are two key aspartate residues. (C) Scanning N-glycosylation mutagenesis experiments35 suggest TM1 acts as a signal-anchor sequence during AE1 biosynthesis. TM1 moves laterally into the lipid bilayer after synthesis of TM2 or after synthesis of TM2 and -3, which are transiently in the endoplasmic reticulum lumen followed by TM4 that acts as a stop-transfer sequence. (D) Structure of this sequence in the AE1 crystal structure, consisting of a cytosolic helix H1 connected by a sharp bend to the TM1 helix that begins at P403 and terminates at T431 with a slight kink at Pro419. (E) The ensemble of 21 wild-type structures of residues 389−430 resolved by nuclear magnetic resonance (labeled WT-NMR) has a high degree of structural variability, especially in their N- and C-terminal regions. For the sake of clarity, they have been fitted onto the structure of the wild-type transmembrane region using the Cα atoms of residues 403–418. (F) The nuclear magnetic resonance ensemble of 21 structures of SAO mutant residues 389−430 (SAO-NMR) has less structural variability, consisting predominantly of a single helical segment. For the sake of clarity, they have been fitted onto the structure of the wild-type transmembrane region using the Cα atoms of residues 409–417. Model helices of the (G) Arg384–Lys430 wild-type sequence (WT-HEL1) and (H) the SAO deletion (SAO-HEL1) as constructed by PyMol. The kinks are due to the proline residues at positions 403 and 419.
Southeast Asian ovalocytosis (SAO) is an inherited condition caused by a nine-amino acid deletion [residues Ala400–Ala408 (Figure 1B)] in the boundary between the cytosolic domain and the first transmembrane segment (TM1) of AE1.14−16 SAO Band 3 is incapable of mediating anion transport14,17 and is unable to bind anion transport inhibitors such as stilbene disulfonates.18,19 Individuals with SAO are heterozygotes, and their red cells contain both SAO (45%) and normal (55%) AE1. Their anion transport activity is severely reduced (∼50% of normal20) because of the presence of nonfunctional SAO AE1. AE1 is a dimer, and the presence of SAO AE1 in the heterodimer affects the structure, transport, and inhibitor binding properties of the normal subunit.20−22 SAO AE1 exhibits a circular dichroism spectrum similar to that of normal AE1, suggesting that there is little difference in secondary structure.18,19 Interestingly, differential thermal calorimetry studies of SAO membranes have shown that SAO AE1 does not undergo the thermal transition exhibited by normal AE1.18 These studies indicate that although the TM helices of SAO AE1 are formed they are not packed together properly. As convincingly shown by the recent crystal structure,13 the dimer interface consists of a four-helix bundle consisting of TM5 and -6 from the gate domain and does not involve TM1 directly, which is located in the core domain. It is therefore plausible that the gate domain is folded correctly, allowing dimer formation, while the core domain is disordered, and is akin to a molten globule.
The level of expression of SAO AE1 in transfected HEK and MDCK cells is lower than that of normal AE1, and the misfolded protein is retained in the endoplasmic reticulum (ER) where it is subjected to more rapid degradation.23 SAO AE1 can, however, form heterodimers with normal AE1, facilitating its trafficking to the cell surface.23,24 SAO AE1 can also be transported to the cell surface in K562 cells that express glycophorin A.25 Glycophorin A interacts with AE1 in the ER26 and is known to facilitate the trafficking of AE1 to the cell surface.27−30 Arg61 in glycophorin A is proposed to interact directly with Glu658 in AE1 to create the Wright (Wr) blood group antigen.31 The abundant expression of SAO AE1 in red cells indicates that trafficking of the protein to the plasma membrane in red cell precursors is not severely impaired likely because of its interaction with glycophorin A and heterodimer formation.
TM1 in AE1 acts as a signal-anchor sequence to target the nascent chain to the ER and mediates cotranslational insertion of the growing polypeptide into the membrane.32−34 During AE1 biosynthesis, TM1 can move laterally into the lipid bilayer in its proper orientation only after the synthesis of TM2 and the short loop connecting it to TM1 (Figure 1C). Scanning N-glycosylation mutagenesis and cell-free translation experiments35 have localized the C-terminal end of the hydrophobic span of TM1 to Phe423 during AE1 biosynthesis.9 Stable integration of TM2 into the membrane to act as a stop-transfer sequence requires the signal sequence properties of TM1; however, an overly long intervening sequence allowed translocation of TM2 into the ER lumen.36,37 N-Glycosylation sites engineered into TM2 could be N-glycosylated in the cell-free translation system, leading to the suggestion that TM2 and TM3 form a re-entrant loop that is transiently exposed to the ER lumen during biosynthesis but folds into the protein at a later stage.9 Thus, TM1 may not insert into the lipid bilayer until after the synthesis of TM4 (Figure 1C), which acts as an efficient stop-transfer sequence.36 Regardless of the point at which TM1 moves out of the translocon, it is located within the lipid bilayer before the majority of AE1 synthesis is completed. Indeed, TM1 expressed alone can act as a signal anchor, target the ER, and insert into the membrane in its proper orientation.36,38
Hydrophobicity plot analyses suggest that TM1 begins at Val405 and extends to Phe423, creating a 19-amino acid hydrophobic segment long enough to span the hydrophobic core of a lipid bilayer as an α-helix.8 In the crystal structure of AE1, the TM1 helix begins at Pro403 and ends at Thr431.13 Ensembles of structures of both wild-type TM1 [residues 389–430, labeled WT-NMR (Figure 1E)] and the SAO deletion mutant [SAO-NMR (Figure 1F)] in a 1:1 (v/v) chloroform/methanol detergent have been resolved by nuclear magnetic resonance (NMR).39 These structures are missing the RDIRR sequence at the N-terminus that, because of its charged nature, is likely to lie at the interface between the lipids and water.
The hydrophobic length of TM1 was found to be critical for efficient membrane insertion of AE1, and the SAO deletion compromised this function.38 The same region of SAO AE1 was poorly N-glycosylated, showing that TM1 was impaired in its ability to function as a signal-anchor sequence.9,40 Scanning N-glycosylation of AE1 and SAO AE1 containing an insertion in EC loop 2 to allow efficient N-glycosylation showed that the positions of the C-terminal ends of TM1 in SAO AE1 in the ER membrane were the same as in wild-type AE1.40 This suggests that TM1 in SAO AE1 assumes a transmembrane disposition by pulling in polar residues from the cytosolic side into the membrane.
In this paper, we studied the effect of the SAO deletion on the structure and dynamics of TM1 of AE1 in a model membrane using molecular dynamics (MD) simulations. Our approach was to simulate the dynamics of a peptide corresponding to residues 384–430, which includes the hydrophobic core of TM1, in a phospholipid bilayer, and thereby to study the behavior of TM1 in the ER membrane during the biosynthesis of AE1. All simulations were repeated with the SAO mutant to determine the effect of this deletion on the interaction of the shortened TM1 of AE1 with the lipid bilayer. Our results provide the first view of the segmental dynamics of signal-anchor TM1 in a lipid bilayer and the effect of a deletion mutation linked to a red cell shape change that disrupts the proper folding of AE1.
Methods
Model helices of sequences Arg384–Lys430 and Arg389–Lys430 were constructed using PyMol. The ensembles of the NMR structures39 for the sequence Arg383–Lys430 were downloaded from the Protein Data Bank (PDB entries 1BNK and 1BNX, respectively). The relevant structure was first converted to a MARTINI coarse-grained representation41 by version 2.4 of the martinize Python script42 with an elastic network applied between all beads that were within 0.5 and 0.9 nm of one another. The force constant was 500 kJ mol–1 nm–2. The resulting coarse-grained peptide was then placed at the center of a box with dimensions of 0.95 nm × 0.95 nm × 0.70 nm. The energy of the system was then minimized for 10 steps using version 4.6.x of the GROMACS molecular dynamics package,43 and 180 coarse-grained 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipids were randomly added, ensuring no clashes. A simple POPC lipid bilayer was chosen because it is a major component of erythrocyte membranes. No additional lipid species were added because the lateral diffusion of lipids is a slow process and hence would not be resolved by our molecular dynamics simulations performed here. The z dimension of the box was increased to 1.1 nm, and all the coordinates were moved up by 0.2 nm, thereby leaving a 0.2 nm gap above and below the lipids. A large number of water beads (5000) were then added, again randomly. The energy of the system was again minimized using the steepest descent algorithm until machine precision was reached. Then the dynamics of the system was simulated for 10 ns using an integration time step of 20 fs. This was found to be sufficient to allow the lipid bilayer to form. The gaps at the top and bottom of the box introduced a bias ensuring the bilayer always formed in the x–y plane, simplifying subsequent analysis. Standard parameters for a MARTINI MD simulation were used. A Verlet cutoff scheme was employed, while van der Waals interactions were cut off at 1.2 nm with a switching function applied from 0.9 nm. Electrostatic forces were calculated using the reaction-field method with a cutoff of 1.5 nm and a relative dielectric constant of 15. The dielectric constant beyond the cutoff was set to infinity. A Berendsen thermostat applied separately to the lipids, protein, and solvent with a relaxation time of 1.0 ps was used to maintain the temperature at 310 K. The pressure was held at 1.0 bar using a Berendsen barostat applied semi-isotropically with a relaxation time of 2.0 ps and a compressibility of 3 × 10–4 bar–1.
The final frame from this self-assembly simulation44 was then converted back to an atomistic representation,45 with the protein having neutral termini and protonating Asp396 and Asp399 as required. This conversion procedure occasionally failed because of steric clashes between the protein and lipids (Table 1). The GROMOS53a6 atomistic force field was used.46 A short 0.1 ns molecular dynamics simulation with the position of the protein restrained was run before a 10 ns unrestrained molecular dynamics simulation. Both simulations used an integration time step of 2 fs with the lengths of all bonds involving a hydrogen restrained using the LINCS algorithm. A Verlet cutoff scheme was used, and electrostatic forces were calculated using the particle mesh Ewald method using a real space cutoff of 1.2 nm. van der Waals forces were cut off at 1.2 nm. The temperature was maintained at 310 K using a Langevin thermostat with a relaxation time of 2 ps. Finally, the pressure was held at 1 bar by a Berendsen barostat applied semi-isotropically with a relaxation time of 1 ps and a compressibility of 4.46 × 10–5 bar–1.
Table 1. Details of the MD Simulationsa.
| sequence | ID | initial structure(s) | D396 and D399 protonated | no. of repeats | no. of completed MD | no. inserted |
|---|---|---|---|---|---|---|
| wild-type | WT-HEL1 | helix with RDIRR | no | 50 | 39 | 22 |
| WT-HEL2 | helix | no | 50 | 41 | 23 | |
| WT-NMR | NMR ensemble | no | 63 | 60 | 43 | |
| SAO deletion | SAO-HEL1 | helix with RDIRR | yes | 50 | 48 | 40 |
| SAO-HEL2 | helix | yes | 50 | 46 | 36 | |
| SAO-NMR | NMR ensemble | yes | 63 | 58 | 40 | |
| SAO-HEL1* | helix with RDIRR | no | 50 | 48 | 34 | |
| SAO-HEL2* | helix | no | 50 | 48 | 34 | |
| SAO-NMR* | NMR ensemble | no | 63 | 58 | 39 | |
| total | 489 | 446 | 311 |
The alphanumeric ID is used to refer to the different sets of simulations. Three different sets of initial structures are used to seed the MD simulations: a model helix covering Arg384–Lys430 (HEL1), an ensemble of all 21 structures covering Arg389–Lys430 determined by NMR (NMR), and, to allow comparison, a second model helix covering Arg389–Lys430 (HEL2). Fifty simulations of each of the model helices were run, while each of the structures in the NMR ensembles was repeated three times, making a total of 63 simulations. A proportion of simulations fail to reach completion. Of these, a further subset does not have the protein in a transmembrane orientation and is therefore discarded from subsequent analysis. Because it appears that the SAO deletion mutation causes Asp396 and Asp399 to enter the lipid bilayer, we assume that these residues are protonated and therefore neutral. To check the effect of this assumption, all the SAO deletion mutation simulations were repeated with Asp396 and Asp399 in their default charged state. These simulations are marked with asterisks.
Table 1 describes how many simulations were run. Fifty repeats of each of the model helices were tried, and three repeats of each of the 21 structures in the NMR ensemble, making a total of 63, were also run. Simulations were not included in the final analysis either because they failed to complete the pipeline, usually because the conversion back to atomistic coordinates was not successful, or because the sequence did not adopt a transmembrane orientation. This was defined as the sequence having Cα atoms 1.4 nm above and below the midplane of the bilayer at the end of the self-assembly process.
Between 44 and 88% of simulations satisfied the criteria described above (Table 1). These were then analyzed as follows. First the sequence was divided into segments, as defined in Figures 2–4. For each frame of the trajectory, the upper and lower leaflets and the midplane of the membrane were defined using the phosphate atoms of the lipids. Then the helicity of each segment was determined using the STRIDE algorithm.47 The helical axis of the segment was calculated by finding the first eigenvector of the backbone heavy atoms. It is defined as pointing toward the C-terminus. The tilt angle can then be calculated using linear algebra. Next the depth of the segment is calculated by subtracting the membrane midplane from the center of mass of the segment. All atoms within 0.6 nm of each residue were examined to determine the local environment, such as the accessibility to water. The depth of each residue, relative to the membrane midplane, was also calculated. All this analysis was performed in Python using the MDAnalysis48 module. Graphs were plotted using gnuplot, and all images were rendered using VMD.
Figure 2.

The wild-type TM1 peptide is highly dynamic and samples an ensemble of conformations. (A) The peptide remains mostly helical during the ensemble of 50 filtered simulations. As expected, the proline residues initiate regions of high helicity. We therefore defined three segments that each started from one of the three proline residues: an interfacial amphipathic helical segment (H1) and two transmembrane helical segments (TM1a and TM1b). (B) Illustrative snapshot taken from the end of the one of the molecular dynamics simulations. The peptide is colored according to panel A. The positions of the phosphate groups of the lipid bilayer are denoted with empty circles. Note the distortion of the inner leaflet of the bilayer induced by the peptide. (C) To-scale schematic representation of the average conformation with segment tilts, lengths, and helicity annotated. The average extent of the lipid bilayer as defined by the positions of the phosphate atoms is shaded gray. (D) Density plot showing the variation in the depth and tilt angle of the three segments of the peptide. The center of mass of TM1a (colored red) remains close to the center of the lipid bilayer at Phe411, and the segment explores tilt angles between 0° and 50°, centered around ∼20°.
Figure 4.

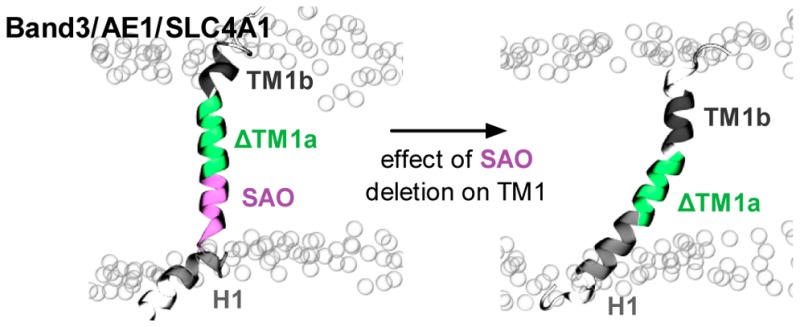
Effect of deletion of residues Ala400–Ala408 in the SAO mutant on the position and dynamics of the TM1 peptide in the lipid bilayer. (A) To facilitate comparison, the wild-type sequence was divided into four sequences: an interfacial amphipathic segment (H1) and a short transmembrane segment (TM1b) that are also present in the SAO mutant and two central transmembrane segments (SAO and ΔTM1a). The SAO segment consists of the nine residues that are deleted in the mutant, and the ΔTM1a segment is the TM1 segment without the six residues at the N-terminus. The colors defined here are used throughout this figure. Illustrative snapshots, to-scale schematics of the average conformations, with the lengths, tilts, and helicity of each segment labeled, and density plots showing the variation in the depth and tilt angle of all segments for (B) the wild-type sequence and (C) the SAO deletion mutant. Note that the H1 segment is pulled into the lipid bilayer in the SAO peptide, as well as the TM1b segment, to compensate for the nine-amino acid deletion.
Results
A single transmembrane helix, such as TM1 of AE1, is likely to be more dynamic on its own in a lipid bilayer, as will be the case during biosynthesis, than when confined within the full transmembrane protein. This variation in conformation sampled by the sequence Arg384–Lys430, which includes TM1, makes it well-suited to being studied by molecular dynamics simulation. Although there are two ensembles of NMR structures for the wild-type sequence [Arg389–Lys430, labeled WT-NMR (Figure 1E and Table 1)] and the corresponding SAO deletion [SAO-NMR (Figure 1F)] that we used in our simulations, it is likely that at least some members of these ensembles are not representative of native conformational states due to the nonphysiological solvent mixture of chloroform and methanol [1:1 (v/v)].39 We therefore also constructed two model α-helices, one with the extended RDIRR sequence at the N-terminus [WT-HEL1 (Figure 1G)] and one without the extension (WT-HEL2). The latter has the same sequence as the NMR structure [WT-NMR (Figure 1E)], facilitating direct comparison. The slight kinks in the model helices are introduced by the three proline residues that cannot participate in backbone hydrogen bonding.
Together with the ensembles of NMR structures (21 each for the WT-NMR and SAO-NMR peptides), these structures form a set of putative initial conformations for the wild-type sequence. SAO deletion variants of the longer [SAO-HEL1 (Figure 1H)] and shorter (SAO-HEL2) helices as well as the NMR ensemble (SAO-NMR) of structures were also considered. Because the deletion of residues Ala400–Ala408 is likely to result in Asp396 and Asp399 entering the hydrophobic core of the lipid bilayer, we have assumed that these residues are protonated in the SAO mutant. To check the effect of this assumption, we repeated all the SAO deletion simulations with both these residues in their default, charged state; these are labeled SAO-HEL1*, SAO-HEL2*, and SAO-NMR*.
In the remainder of the paper, we shall focus on the behavior of the sequence Arg389–Lys430 assuming it adopts an initial helical conformation [WT-HEL1 (Table 1)] and the corresponding SAO deletion with both Asp396 and Asp399 protonated (SAO-HEL1). Where appropriate, we compared to the results for the wild-type sequence and SAO deletion mutant that were modeled as a α-helix without the N-terminal RDIRR motif (WT-HEL2 and SAO-HEL2) or the ensemble of NMR structures (WT-NMR and SAO-NMR). Because it is likely that an individual simulation could become trapped in a metastable conformation, we ran a large number of simulations and analyzed their statistical behavior. Fifty simulations of either α-helix were run (Table 1). Because each NMR ensemble contains 21 structures, three repeats of each were run, making 63 simulations for each ensemble. Overall, therefore, 489 simulations were run, each 10 ns long, making in total 4.46 μs of dynamics.
We started by assuming that the sequence spans the membrane once, and therefore, the first step was to embed the different structures in our set of putative conformations into a membrane, in this case a simple POPC lipid bilayer as described in Methods. To allow each conformation to relax, the first half of each trajectory was then discarded and the resulting data set analyzed. Our rationale is that repeating and analyzing many, short simulations of the sequence Arg384–Lys430 is likely to better sample the dynamics than running a few, much longer simulations.49
The Sequence R384–K430 Is, on Average, Helical in the Membrane but Is Highly Dynamic
Twenty-two of the initial 50 WT-HEL1 simulations were successfully embedded and simulated for 10 ns in a transmembrane orientation in a POPC lipid bilayer. Examining the average helicity of this ensemble (Figure 2A) shows that, as one might expect, the sequence is mainly helical with the termini being less helical, in agreement with the NMR studies. The three prolines (Pro391, Pro403, and Pro419) all approximately mark the start of a local region of increased helicity. To simplify the task of analyzing the ensembles of simulations, we defined three segments (Figure 2A) that are mainly helical and start at each of these proline residues (H1, TM1a, and TM1b). The center of mass (COM) of the first segment (H1) tends, on average, to be found at the interface of the lipid bilayer and the cytoplasm; we define the interface by the position of the phosphate atoms in the POPC lipids. It adopts an angle of 66° relative to the bilayer normal and is 81% helical, on average, and therefore can be described as an interfacial helix, which is not surprising considering its amphipathic nature. The next two segments form a kinked helix spanning the bilayer and are therefore labeled TM1a and TM1b. TM1a is longer and more helical, and its COM is approximately at the center of the bilayer. It is slightly tilted, making an angle of 27°, on average, with the bilayer normal. TM1b is much shorter and less helical, and its COM is close to the extracellular side of the membrane. Like H1, it is tilted away from the membrane normal, making an angle of 60° on average. TM1a and TM1b therefore form a kinked transmembrane helix.
As one might expect for an isolated transmembrane sequence, this average conformation hides a considerable degree of dynamics. If we characterize each segment by a coordinate consisting of its tilt and COM depth, then the resulting density plot (Figure 2D) suggests that the depth of the COM of all three segments varies by ∼1 nm and H1, TM1a, and TM1b explore a wide range of tilt angles (0–100°, 0–50°, and 45–100°, respectively). Taking the average and dynamical descriptions together gives us a more complete picture of the behavior of the sequence Arg384–Lys430 in a lipid bilayer. A similar image emerges if we examine the simulations of the shorter model helix [WT-HEL2 (Figure S1)] or the ensemble of NMR conformations [WT-NMR (Figure S2)]. There are, however, some notable differences. Removing the RDIRR sequence from the model helix appears to allow the H1 segment to explore a wider range of conformations (Figure S1) and reduces its helicity. This is consistent with the charged RDIRR sequence interacting with the lipids and thereby restricting the dynamics of the H1 segment. The behavior of the flanking segments, H1 and TM1b, is different when the simulations of the ensemble of wild-type NMR structures are analyzed (Figure S2). The helicity of both segments is reduced, and they explore a far wider range of tilt angles, resulting in an average tilt angle of 108° for the H1 segment. These differences are likely due to bias introduced by some of the conformations present in the NMR ensemble: in several of these, the N- and C-termini are bent around sufficiently that they will be initially embedded within the lipid bilayer. Although this conformation is probably unstable, it is highly likely that the simulations are not long enough to allow the termini to escape the bilayer (see, for example, Figure S2B), and hence, the overall behavior is biased. We attribute this behavior of the peptide in the NMR experiment to the nonphysiological solvent mixture used.
The SAO Deletion Results in a Helical, Dynamical Transmembrane Peptide
Now let us consider how the SAO-HEL1 ensemble behaves. Like the wild-type sequence, it is predominantly helical (Figure 3A) with fraying at both termini. Crucially, one of the prolines (Pro403) is missing in the mutant, leading to two rather than three segments, starting at Pro391 and Pro419. The first segment, which we call TM1a′, is composed of the residues that make up H1 in the wild-type sequence and the second half of the TM1a sequence. It retains a helicity of 94% (Figure 3C), and its COM is 0.8 nm below the midplane of the lipid bilayer, on average. TM1a′ is more tilted than TM1a in the wild-type sequence, making an angle of 34°, on average, with the bilayer normal. The second segment, TM1b, comprises the same residues as the wild-type sequence and behaves similarly with one exception: while it has an average helicity of 76% and an average tilt angle of 60%, the COM is only 0.9 nm, on average, above the bilayer midplane, a shift of 0.4 nm closer to the center of the bilayer. Like that of the wild-type sequence, this average behavior hides a high degree of dynamics. The vertical position of the COMs of both segments varies by up to 1.5 nm, while the TM1a′ segment explores tilt angles in the range of 10–55° and the TM1b segment, like the wild type, a wider range of tilt angles (10–130°). A similar picture is observed when we analyze either the ensemble of simulations started from the shorter model helix [SAO-HEL2 (Figure S3)] or the ensemble of simulations started from the NMR structures [SAO-NMR (Figure S4)]. Interestingly, the TM1b segment is both more helical and less dynamic when the RDIRR sequence is absent (Figure S3D), suggesting that the latter binds more strongly to the bilayer, as one might expect; therefore, when it is present, it anchors the sequence, leaving the TM1b segment more free, whereas when it is absent, the TM1b segment instead interacts more closely with the bilayer. This analysis assumes that Asp396 and Asp399 in TM1a′ are protonated and therefore neutral. If they are deprotonated, the overall effect is to reduce the helicity in the vicinity of these two residues (Figures S5–S7) and pull both the TM1a′ and TM1b segments toward the cytoplasmic side of the membrane.
Figure 3.

The SAO deletion mutant peptide also samples an ensemble of conformations. (A) The peptide remains mostly helical during the ensemble of 50 filtered simulations. As expected, proline and glycine residues initiate and terminate regions of high helicity. Two putative transmembrane segments were defined, each starting from one of the two proline residues, a central segment (TM1a′) and a shorter segment identical to that found in the wild-type sequence (TM1b). (B) Illustrative snapshot taken from the end of one of the molecular dynamics simulations. The peptide is colored according to panel A. The positions of the phosphate groups of the lipid bilayer are shown as empty circles. (C) To-scale schematic representation of the average conformation with segment tilts, lengths, and helicity annotated. The average extent of the lipid bilayer as defined by the phosphate atoms is shaded gray. (D) Density plot showing the variation in the depth and tilt angle of both segments. The center of mass of the larger transmembrane segment (TM1a′, colored dark blue) remains on average 0.8 nm below the center of the lipid bilayer, and the segment explores tilt angles between 10° and 60°, centered around ∼35°.
The SAO Deletion Causes the N- and C-Termini To Be Pulled into the Membrane by Five and Four Residues, Respectively
Overall, a picture is emerging from the simulations of the SAO deletion causing the C-terminus to be pulled into the lipid bilayer. To gain a more detailed view, we need to make a direct comparison between the wild-type sequence and the SAO deletion mutant. To achieve this, let us define segments that are the same in both sequences (Figure 4A). The wild-type sequence is then described by the behavior of four segments: H1 as before, then the SAO sequence, the remainder of TM1a, which we call ΔTM1a, and TM1b, also as described above. The SAO deletion mutant is therefore identical, except the nine-residue SAO segment is missing. Repeating exactly the same analysis as before and considering first the average and then the dynamical behavior show that the average effect of the deletion is for the H1 segment to be pulled into the lipid bilayer from the cytoplasmic side by 0.6 nm on average (Figure 4B,C) and for the ΔTM1a and TM1b segments to be pulled into the lipid bilayer from the extracellular side by 0.5 and 0.4 nm, respectively. The average conformations of the ΔTM1a and TM1b segments are not significantly altered; however, the H1 segment becomes slightly more helical, and the tilt angle decreases from 66° to 40°. All segments remain highly dynamic, with those firmly embedded in the lipid bilayer (SAO and ΔTM1a) displaying a variation in tilt angles smaller than those of the other segments (H1 and TM1b). Similar trends are seen when the simulations starting from the shorter model helix or the ensembles of NMR structures are analyzed (Figures S8 and S9). Our overall picture is now more nuanced: the SAO deletion causes both ends of the protein to be pulled into the lipid bilayer by approximately the same amount, causing the N-terminus to be become more helical, thereby forming an approximately continuous helical region with the ΔTM1a segment, as shown in Figure 3.
To more precisely determine how much each end of the sequence moves in response to the SAO deletion, we have calculated the average distance of each residue relative to the midplane of the lipid bilayer over all the simulations in each ensemble (Figure 5A). This shows that, on average, the N-terminus is pulled in by five residues and the C-terminus is pulled in by four residues. Repeating this analysis for the simulations seeded by either the shorter model helix or the ensemble of NMR structures yields similar but not identical patterns; the former suggests the N- and C-termini are pulled in by six and three residues, respectively, while the latter suggests that the eight residues of the N-terminus are pulled into the bilayer (Figure S10). We treat the latter result with caution because of the previously noted problems with trapped structures and equilibration. Taken together, these results suggest that while both termini are pulled into the bilayer, the effect is stronger at the N-terminus than at the C-terminus. This conclusion assumes both Asp396 and Asp399 are protonated; repeating the analysis with both residues charged alters the behavior. For either model helix, the N-terminus is now only pulled into the bilayer by three residues on average (Figure S11), while the C-terminus is pulled in by six residues. As described above, the effect is different for the ensemble of NMR structures where the N- and C-termini are pulled in by six and three residues, respectively.
Figure 5.

Structure of the transmembrane segment of the SAO deletion mutant that is different from the structure of wild-type TM1. (A) Average distance from the central plane of the lipid bilayer for each residue in both the wild-type and SAO deletion mutant sequences. This analysis suggests that deleting the nine residues causes the N-terminus to shift toward the extracellular side by five residues and the C-terminus to shift toward the intracellular side by four residues. (B) Shifting the sequence by these amounts allows us to find the sequence in the SAO mutant equivalent to the TM1 segment in the wild type. (C) Comparing the ensemble of structures generated by the simulations with the structure of TM1 found in the full AE1 structure shows that the equivalent sequence in the SAO mutant both is more different on average, with higher root-mean-square deviation values, and rarely samples conformations similar to that seen in the experimental structure of the transmembrane part of AE1.
Unlike That of the Wild-Type Sequence, the Transmembrane Helix of the SAO Deletion Mutant Rarely Resembles TM1 in the Structure of AE1
Using this result, we can align residues in both the wild-type and SAO deletion mutant sequences based on their depth in the membrane (Figure 5B). This shows that, for example, Tyr403 in the SAO mutant is found at the same depth as Val409 in the wild-type sequence, allowing us to identify the residues in the SAO deletion mutant equivalent in terms of their depth in the bilayer to those of the TM1a segment of the wild type. If we consider the biosynthesis of AE1 for a moment, then TM1 will be the first transmembrane helix ejected from the translocon into the membrane (Figure 1C). As our simulations suggest, it is likely that TM1 on its own is highly dynamic once it moves into the lipid bilayer. It is, however, reasonable to assume that TM1 samples, perhaps only occasionally, the conformation it ultimately adopts in the folded structure of the whole AE1 transmembrane protein. If this were not true, it would imply either that the structure adopted by TM1 in the whole protein has a high energy, and is therefore not favored, or that the local environment around TM1 in the folded protein is significantly different from what it experiences immediately after synthesis. Consistent with this idea, we find that a few (8%) of the structures sampled by TM1 in our wild-type simulations are very similar to the conformation of the same residues in the full AE1 structure, as defined by having a Cα root-mean-square deviation of <0.05 nm. The conformations adopted by the equivalent residues in the SAO deletion mutant are on average more different and also very rarely (0.2%) sample conformations similar to that adopted by TM1 in the full AE1 structure. This hints at the disruptive effect of deleting residues Ala400–Ala408 on the biosynthesis and proper folding of AE1.
Discussion
The red blood cells of heterozygotes with SAO have an abnormal shape and exhibit an ∼50% decrease in their level of anion transport because of the presence of nonfunctional SAO AE1. SAO results from a nine-residue deletion at the N-terminal end of TM1 of AE1, which removes a bend in the protein chain resulting in a misfolded protein (Figure 1B). TM1 acts as a signal-anchor sequence targeting the nascent chain to the ER, and therefore, it is important to consider the effect of this mutation on the biosynthesis of AE1 where TM1 is initially isolated in the membrane before the distal transmembrane segments are ejected from the translocon and inserted into the membrane. Indeed, cell-free translation experiments9,36 have indicated that TM2 and -3 translocate into the ER lumen with TM4 acting as a stop-transfer sequence. Here we have shown by extensive MD simulations that the wild-type sequence Arg384–Lys430, which includes TM1, is predominantly helical in conformation and can be characterized by three helical segments: an amphipathic helix (H1) that lies at the interface between the lipid bilayer and the cytoplasm and a transmembrane helix composed of two helical segments that begins at Pro403 and is kinked at a conserved proline residue (Pro419). This average view hides, however, considerable dynamics, as expected for a single helix in a lipid bilayer. The same overall picture is recovered if we seed the simulations with structures resolved by NMR experiments39 or start with a model of the sequence as a classical α-helix (Figures 2, S1, and S2).
Removing the nine residues that cause SAO, Ala400–Ala408, from near the N-terminal end of TM1 not only alters its average conformation but also causes the N- and C-termini to be pulled into the lipid bilayer to accommodate this deletion (Figures 4, S8, and S9). We estimate this effect is more pronounced at the N-terminus with at least five residues entering the membrane (Figures 5 and S10). As a consequence, the sequence adopts a single, kinked transmembrane helix, but with no interfacial H1 helix (Figures 3, S3, and S4). Starting the simulations from either a model of the sequence as a helix or structures resolved by NMR experiments39 leads to similar conclusions. This result assumes that both aspartic acid residues in the H1 helix that are pulled into the bilayer become protonated. Leaving both these N-terminal residues in their default charged state still results in the sequence folding into a single, kinked transmembrane helix (Figures S5–S7); however, as expected, only three residues are pulled into the bilayer from the N-terminus (Figure S11), while six residues are pulled in from the C-terminus.
In all cases, the simulations produce a transmembrane helix that is highly dynamic. This helix ultimately becomes TM1 in the transmembrane region of AE1.13 By comparing the ensemble of transmembrane structures seen in our simulations and the recent structure of the transmembrane region of AE1,13 we have shown that, although dynamic, the wild-type sequence occasionally samples conformations very similar to that seen in the folded structure of AE1. It is possible, therefore, to see how this sequence can become incorporated into the fully folded structure of AE1. The SAO deletion, however, leads to an ensemble of transmembrane structures that samples conformations similar to TM1 in AE1 less frequently, and consequently, one can see how this mutation could lead to packing defects in AE1, leading to its physiological effects. We have noted that in the crystal structure of the transmembrane region of AE1, the helical portion of TM1 finishes around two helical turns later than we have seen in our simulations at Thr431. This may be because the local environment around TM1 in the full structure causes that region to adopt a helical conformation.
To validate these results, let us compare the simulations to the results of N-glycosylation scanning mutagenesis experiments by considering the average water accessibility of each residue (Figure 6). If we consider the wild-type sequence, then, as expected, the transmembrane SAO and ΔTM1a segments are only marginally accessible to water. Overall, as we move farther from the central transmembrane region, the water accessibility increases, as expected. The “sawtooth” pattern with peaks every three or four residues seen in the N-terminus is consistent with the formation of amphipathic helices lying at the interface between the lipid bilayer and the solvent, as seen in the behavior of the H1 segment (Figure 2). The fact that a similar pattern with a smaller magnitude is seen in the TM1b segment is consistent both with this sequence forming a helix less often and with it being more buried in the bilayer (Figure 2).
Figure 6.
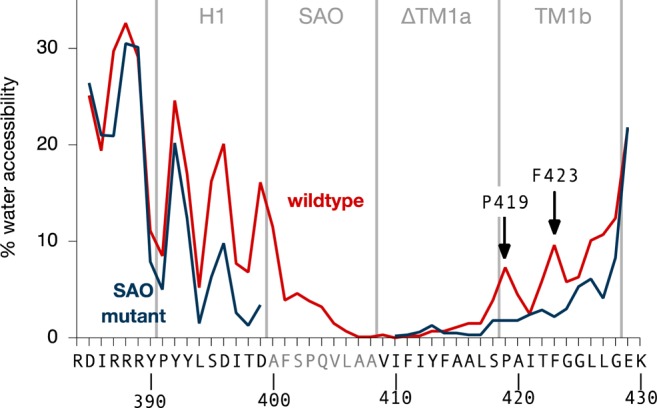
Water accessibility of the wild-type and SAO mutant peptide. A residue is defined as being accessible to water if at least one water oxygen atom is found within 0.6 nm of the residue, averaged over all the simulations. The sawtooth pattern is due to the amphipathic nature of the H1 helix in the flanking segments, which lie approximately perpendicular to the membrane normal with one side facing water and the other facing the lipid bilayer. This positions polar and/or charged residues Arg387, Arg388, Tyr392, Asp396, and Asp399 facing water and nonpolar Ile386, Pro391, Leu394, and Ile 397 facing the lipid bilayer. Considering the C-terminal end of TM1, our simulations predict that the first residues that are accessible to water are Pro419 and Phe423.
N-Glycosylation is a cotranslational event that occurs on the luminal side of the ER membrane while the nascent polypeptide chain is located within the translocon. N-Glycosylation acceptor sites must be located a minimum of 12 and 14 residues (12 + 14 rule) from the proximal and distal ends, respectively, of the hydrophobic regions of transmembrane segments to be efficiently N-glycosylated in the lumen of the ER.8 Scanning N-glycosylation mutagenesis can therefore be used to predict the position of the lumen ends of transmembrane segments during biosynthesis. N-Glycosylation experiments40 using cell-free translation and transfected HEK cells with an insertion in the short loop between TM1 and -2 in AE1 to facilitate N-glycosylation identified Pro419 as the end of the hydrophobic region of TM1 in both wild-type and SAO AE1. This indicates that the position of TM1 in the translocon is the same in wild-type and SAO AE1. The efficiency of N-glycosylation of the SAO constructs was always lower than that of the wild-type protein, suggesting that the SAO deletion impairs the signal-anchor function of TM1.
Subsequent N-glycosylation scanning experiments using transfected HEK cells and constructs without the insertion revealed that introduced N-glycosylation sites within TM2 and -3 in AE1 could be N-glycosylated. This led to the suggestions that TM2 and -3 are translocated into the ER lumen during biosynthesis and folded into the protein as re-entrant loops. These experiments found that the luminal end of TM1 was located at Phe423, one helical turn more distal than Pro419, the estimate made with the insertion constructs. Interestingly, residues Pro419 and Phe423 are the positions of the first two peaks in water accessibility on the extracellular side of the protein, regardless of the initial structure(s) used in the simulation (Figures 6 and S12). This is evidence that our simulations are accurately modeling the dynamics of this peptide in a simple lipid bilayer, placing these residues on the same side of a helix at the membrane interface region. In our simulations of the sequence Arg384–Lys430 in AE1 SAO, because TM1b is pulled into the lipid bilayer (Figure 4B) by the deletion, the sawtooth pattern is missing and it is not until Gly428 that the water accessibility is >10%. We note that in the crystal structure of AE1 the helical portion of TM1 begins at Pro403 and ends at Thr431,13 around two helical turns later than where we have assumed TM1b finishes, well into the aqueous phase. In addition, TM1 is buried within the protein structure with little exposure to the lipid bilayer. Thus, it is not surprising that the shortened TM1 segment in AE1 SAO cannot assume a native conformation, resulting in disruption of the packing of TM segments.
It is notable but not surprising that the three proline residues in the sequence we have considered are important in determining the secondary structure. Because of its inability to form backbone hydrogen bonds with the residue four positions toward the N-terminus, proline is acknowledged as “breaking” α-helices. We observed that local increases in the helicity tended to coincide with a proline residue, consistent with this suggestion. These three prolines are conserved,1 and it has been suggested for a long time that proline residues not only are enriched in the transmembrane helices of transport proteins50 but also can play important roles in the functioning of membrane proteins. Like their soluble cousins, proline residues in membrane proteins can also induce helix initiation. This notion is supported by our simulations in which the sequences distal to proline residues are highly helical. For example, it has been suggested that straightening the kinks introduced into a transmembrane helix by a proline is a way of storing energy (as strain) in membrane proteins, such as transporters, that can be unwound later in the functional cycle.51
We have made several assumptions throughout that it is important to clarify. The first is that the structure of the polypeptide, including the first transmembrane segment TM1, of AE1 can be described as a helix, and the second is that it is preferable to run a large number of short simulations rather than a few long simulations. Given we obtain similar results when the simulations are seeded with structures determined by NMR experiments,39 these assumptions appear to be reasonable. We have also assumed that this polypeptide is inserted into a transmembrane orientation by the translocon; again this is realistic given the experimental data.32−34 Finally, we have assumed that a simple POPC lipid bilayer is a good mimic for a single helix that has been just ejected from the translocon into the ER membrane.
Acknowledgments
We thank Joanne Bright for her early interest in this project.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00966.
Twelve additional figures (PDF)
P.W.F. and M.S.P.S. are grateful to the Wellcome Trust for funding (092970/788Z/10/Z). This work was initiated by R.A.F.R. during a sabbatical leave in 2002 supported by the University of Toronto.
The authors declare no competing financial interest.
Supplementary Material
References
- Reithmeier R. A. F.; Casey J. R.; Kalli A. C.; Sansom M. S. P.; Alguel Y.; Iwata S. (2016) Band 3, the human red cell chloride/bicarbonate anion exchanger (AE1, SLC4A1), in a structural context. Biochim. Biophys. Acta, Biomembr. 1858, 1507–1532. 10.1016/j.bbamem.2016.03.030. [DOI] [PubMed] [Google Scholar]
- Alper S. L. (2006) Molecular physiology of SLC4 anion exchangers. Exp Physiol 91, 153–161. 10.1113/expphysiol.2005.031765. [DOI] [PubMed] [Google Scholar]
- Tanner M. J. (1997) The structure and function of band 3 (AE1): recent developments. Mol. Membr. Biol. 14, 155–65. 10.3109/09687689709048178. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Kiyatkin A.; Bolin J. T.; Low P. S. (2008) Crystallographic structure and functional interpretation of the cytoplasmic domain of erythrocyte membrane band 3 Crystallographic structure and functional interpretation of the cytoplasmic domain of erythrocyte membrane band 3. Blood 96, 2925–2933. [PubMed] [Google Scholar]
- Low P. S. (1986) Structure and function of the cytoplasmic domain of band 3: center of erythrocyte membrane-peripheral protein interactions. Biochim. Biophys. Acta, Rev. Biomembr. 864, 145–167. 10.1016/0304-4157(86)90009-2. [DOI] [PubMed] [Google Scholar]
- Fujinaga J.; Tang X. B.; Casey J. R. (1999) Topology of the membrane domain of human erythrocyte anion exchange protein, AE1. J. Biol. Chem. 274, 6626–33. 10.1074/jbc.274.10.6626. [DOI] [PubMed] [Google Scholar]
- Bonar P.; Schneider H. P.; Becker H. M.; Deitmer J. W.; Casey J. R. (2013) Three-dimensional model for the human Cl-/HCO3 - exchanger, AE1, by homology to the E. coli ClC protein. J. Mol. Biol. 425, 2591–2608. 10.1016/j.jmb.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Popov M.; Tam L. Y.; Li J.; Reithmeier R. A. F. (1997) Mapping the ends of transmembrane segments in a polytopic membrane protein. Scanning N-glycosylation mutagenesis of extracytosolic loops in the anion exchanger, band 3. J. Biol. Chem. 272, 18325–18332. 10.1074/jbc.272.29.18325. [DOI] [PubMed] [Google Scholar]
- Cheung J. C.; Li J.; Reithmeier R. A. F. (2005) Topology of transmembrane segments 1–4 in the human chloride/bicarbonate anion exchanger 1 (AE1) by scanning N-glycosylation mutagenesis. Biochem. J. 390, 137–144. 10.1042/BJ20050315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Casey J. R. (2004) The substrate anion selectivity filter in the human erythrocyte Cl -/HCO3\n- exchange protein, AE1. J. Biol. Chem. 279, 23565–23573. 10.1074/jbc.M401380200. [DOI] [PubMed] [Google Scholar]
- Barneaud-Rocca D.; Etchebest C.; Guizouarn H. (2013) Structural model of the anion exchanger 1 (SLC4A1) and identification of transmembrane segments forming the transport site. J. Biol. Chem. 288, 26372–26384. 10.1074/jbc.M113.465989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinstein S.; Ship S.; Rothstein A. (1978) Anion transport in relation to proteolytic dissection of band 3 protein. Biochim. Biophys. Acta, Biomembr. 507, 294–304. 10.1016/0005-2736(78)90424-8. [DOI] [PubMed] [Google Scholar]
- Arakawa T.; Kobayashi-Yurugi T.; Alguel Y.; Iwanari H.; Hatae H.; Iwata M.; Abe Y.; Hino T.; Ikeda-Suno C.; Kuma H.; Kang D.; Murata T.; Hamakubo T.; Cameron A. D.; Kobayashi T.; Hamasaki N.; Iwata S. (2015) Crystal structure of the anion exchanger domain of human erythrocyte band 3. Science (Washington, DC, U. S.) 350, 680–684. 10.1126/science.aaa4335. [DOI] [PubMed] [Google Scholar]
- Schofield A. E.; Reardon D. M.; Tanner M. J. A. (1992) Defective anion transport activity of the abnormal band 3 in hereditary ovalocytic red blood cells. Nature 355, 836–838. 10.1038/355836a0. [DOI] [PubMed] [Google Scholar]
- Jarolim P.; Palek J.; Amato D.; Hassan K.; Sapak P.; Nurse G. T.; Rubin H. L.; Zhai S.; Sahr K. E.; Liu S. C. (1991) Deletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc. Natl. Acad. Sci. U. S. A. 88, 11022–6. 10.1073/pnas.88.24.11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohandas N.; Winardi R.; Knowles D.; Leung A.; Parra M.; George E.; Conboy J.; Chasis J. (1992) Rapid Publication Molecular Basis for Membrane Rigidity of Hereditary Ovalocytosis. J. Clin. Invest. 89, 686–692. 10.1172/JCI115636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. D.; Ring S. M.; Schofield A. E.; Tanner M. J. (1993) The expression of the abnormal human red cell anion transporter from South-East Asian ovalocytes (band 3 SAO) in Xenopus oocytes. FEBS Lett. 330, 186–190. 10.1016/0014-5793(93)80270-5. [DOI] [PubMed] [Google Scholar]
- Moriyama R.; Ideguchi H.; Lombardo C. R.; Van Dort H. M.; Low P. S. (1992) Structural and functional characterization of band 3 from Southeast Asian ovalocytes. J. Biol. Chem. 267, 25792–25797. [PubMed] [Google Scholar]
- Sarabia V. E.; Casey J. R.; Reithmeier R. A. F. (1993) Molecular characterization of the band 3 protein from Southeast Asian ovalocytes. J. Biol. Chem. 268, 10676–10680. [PubMed] [Google Scholar]
- Jennings M. L.; Gosselink P. G. (1995) Anion exchange protein in Southeast Asian ovalocytes: heterodimer formation between normal and variant subunits. Biochemistry 34, 3588–3595. 10.1021/bi00011a013. [DOI] [PubMed] [Google Scholar]
- Kuma H.; Abe Y.; Askin D.; Bruce L. J.; Hamasaki T.; Tanner M. J. A.; Hamasaki N. (2002) Molecular Basis and Functional Consequences of the Dominant Effects of the Mutant Band 3 on the Structure of Normal Band 3 in Southeast Asian Ovalocytosis †. Biochemistry 41, 3311–3320. 10.1021/bi011678+. [DOI] [PubMed] [Google Scholar]
- Salhany J. M.; Schopfer L. M. (1996) Interactions between mutant and wild-type band 3 subunits in hereditary Southeast Asian ovalocytic red blood cell membranes. Biochemistry 35, 251–257. 10.1021/bi952411b. [DOI] [PubMed] [Google Scholar]
- Cheung J. C.; Cordat E.; Reithmeier R. A. F. (2005) Trafficking defects of the Southeast Asian ovalocytosis deletion mutant of anion exchanger 1 membrane proteins. Biochem. J. 392, 425–434. 10.1042/BJ20051076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernova M. N.; Jarolim P.; Palek J.; Alper S. L. (1995) Overexpression of AE1 Prague, but not of AE1 SAO, inhibits wild-type AE1 trafficking in Xenopus oocytes. J. Membr. Biol. 148, 203–210. 10.1007/BF00207276. [DOI] [PubMed] [Google Scholar]
- Patterson S. T.; Li J.; Kang J. A.; Wickrema A.; Williams D. B.; Reithmeier R. A. F. (2009) Loss of specific chaperones involved in membrane glycoprotein biosynthesis during the maturation of human erythroid progenitor cells. J. Biol. Chem. 284, 14547–14557. 10.1074/jbc.M809076200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang A. J.; Reithmeier R. A. F. (2009) Interaction of anion exchanger 1 and glycophorin A in human erythroleukaemic K562 cells. Biochem. J. 421, 345–356. 10.1042/BJ20090345. [DOI] [PubMed] [Google Scholar]
- Young M. T.; Beckmann R.; Toye A. M.; Tanner M. J. (2000) Red-cell glycophorin A-band 3 interactions associated with the movement of band 3 to the cell surface. Biochem. J. 350 (Part 1), 53–60. 10.1042/bj3500053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. D.; Tanner M. J. (1992) Glycophorin A facilitates the expression of human band 3-mediated anion transport in Xenopus oocytes. J. Biol. Chem. 267, 22163–70. [PubMed] [Google Scholar]
- Groves J. D.; Tanner M. J. (1994) The effects of glycophorin A on the expression of the human red cell anion transporter (band 3) in Xenopus oocytes. J. Membr. Biol. 140, 81–88. 10.1007/BF00234488. [DOI] [PubMed] [Google Scholar]
- Auffray I. (2001) Glycophorin A dimerization and band 3 interaction during erythroid membrane biogenesis: in vivo studies in human glycophorin A transgenic mice. Blood 97, 2872–2878. 10.1182/blood.V97.9.2872. [DOI] [PubMed] [Google Scholar]
- Bruce L. J.; Ring S. M.; Anstee D. J.; Reid M. E.; Wilkinson S.; Tanner M. J. (1995) Changes in the blood group Wright antigens are associated with a mutation at amino acid 658 in human erythrocyte band 3: a site of interaction between band 3 and glycophorin A under certain conditions. Blood 85, 541–547. [PubMed] [Google Scholar]
- Braell W. A.; Lodish H. F. (1982) Cotranslationally Anion Transport Protein Is Inserted into Microsomes. Cell 28, 23–31. 10.1016/0092-8674(82)90371-3. [DOI] [PubMed] [Google Scholar]
- Kopito R. R.; Lodish H. F. (1985) Primary structure and transmembrane orientation of the murine anion exchange protein. Nature 316, 234–238. 10.1038/316234a0. [DOI] [PubMed] [Google Scholar]
- Braeil W. A.; Lodish H. F. (1981) Biosynthesis of the Erythrocyte Anion. J. Biol. Chem. 256, 11337–11344. [PubMed] [Google Scholar]
- Cheung J. C.; Reithmeier R. A. F. (2007) Scanning N-glycosylation mutagenesis of membrane proteins. Methods 41, 451–459. 10.1016/j.ymeth.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Ota K.; Sakaguchi M.; Hamasaki N.; Mihara K. (1998) Assessment of topogenic functions of anticipated transmembrane segments of human band 3. J. Biol. Chem. 273, 28286–91. 10.1074/jbc.273.43.28286. [DOI] [PubMed] [Google Scholar]
- Ota K.; Sakaguchi M.; Hamasaki N.; Mihara K. (2000) Membrane integration of the second transmembrane segment of band 3 requires a closely apposed preceding signal-anchor sequence. J. Biol. Chem. 275, 29743–29748. 10.1074/jbc.M002468200. [DOI] [PubMed] [Google Scholar]
- Kanki T.; Young M. T.; Sakaguchi M.; Hamasaki N.; Tanner M. J. A. (2003) The N-terminal Region of the Transmembrane Domain of Human Erythrocyte Band 3. J. Biol. Chem. 278, 5564–5573. 10.1074/jbc.M211662200. [DOI] [PubMed] [Google Scholar]
- Chambers E. J.; Bloomberg G. B.; Ring S. M.; Tanner M. J. (1999) Structural studies on the effects of the deletion in the red cell anion exchanger (band 3, AE1) associated with South East Asian ovalocytosis. J. Mol. Biol. 285, 1289–307. 10.1006/jmbi.1998.2392. [DOI] [PubMed] [Google Scholar]
- Cheung J. C.; Reithmeier R. A. F. (2005) Membrane integration and topology of the first transmembrane segment in normal and Southeast Asian ovalocytosis human erythrocyte anion exchanger 1. Mol. Membr. Biol. 22, 203–214. 10.1080/09687860500093115. [DOI] [PubMed] [Google Scholar]
- Periole X., and Marrink S.-J. (2013) The Martini Coarse-Grained Force Field. In Methods in Molecular Biology (Monticelli L., and Salonen E., Eds.) pp 533–565, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- de Jong D. H.; Lopez C. A.; Marrink S. J. (2013) Molecular view on protein sorting into liquid-ordered membrane domains mediated by gangliosides and lipid anchors. Faraday Discuss. 161, 347. 10.1039/C2FD20086D. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. (2008) GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Scott K. A.; Bond P. J.; Ivetac A.; Chetwynd A. P.; Khalid S.; Sansom M. S. P. (2008) Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure 16, 621–630. 10.1016/j.str.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Stansfeld P. J.; Sansom M. S. P. (2011) From Coarse Grained to Atomistic: A Serial Multiscale Approach to Membrane Protein Simulations. J. Chem. Theory Comput. 7, 1157–1166. 10.1021/ct100569y. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C.; Villa A.; Mark A. E.; van Gunsteren W. F. (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–76. 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- Frishman D.; Argos P. (1995) Knowledge-based protein secondary structure assignment. Proteins: Struct., Funct., Genet. 23, 566–79. 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. (2011) MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coveney P. V.; Wan S. (2016) On the calculation of equilibrium thermodynamic properties from molecular dynamics. Phys. Chem. Chem. Phys. 18, 30236–30240. 10.1039/C6CP02349E. [DOI] [PubMed] [Google Scholar]
- Brandl C. J.; Deber C. M. (1986) Hypothesis about the function of membrane-buried proline residues in transport proteins. Proc. Natl. Acad. Sci. U. S. A. 83, 917–21. 10.1073/pnas.83.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler P. W.; Sansom M. S. P. (2013) The pore of voltage-gated potassium ion channels is strained when closed. Nat. Commun. 4, 1872. 10.1038/ncomms2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.