

Abstract
Endogenous extracellular adenosine level fluctuates in an activity-dependent manner and with sleep–wake cycle, modulating synaptic transmission and short-term plasticity. Hebbian-type long-term plasticity introduces intrinsic positive feedback on synaptic weight changes, making them prone to runaway dynamics. We previously demonstrated that co-occurring, weight-dependent heterosynaptic plasticity can robustly prevent runaway dynamics. Here we show that at neocortical synapses in slices from rat visual cortex, adenosine modulates the weight dependence of heterosynaptic plasticity: blockade of adenosine A1 receptors abolished weight dependence, while increased adenosine level strengthened it. Using model simulations, we found that the strength of weight dependence determines the ability of heterosynaptic plasticity to prevent runaway dynamics of synaptic weights imposed by Hebbian-type learning. Changing the weight dependence of heterosynaptic plasticity within an experimentally observed range gradually shifted the operating point of neurons between an unbalancing regime dominated by associative plasticity and a homeostatic regime of tightly constrained synaptic changes. Because adenosine tone is a natural correlate of activity level (activity increases adenosine tone) and brain state (elevated adenosine tone increases sleep pressure), modulation of heterosynaptic plasticity by adenosine represents an endogenous mechanism that translates changes of the brain state into a shift of the regime of synaptic plasticity and learning. We speculate that adenosine modulation may provide a mechanism for fine-tuning of plasticity and learning according to brain state and activity.
SIGNIFICANCE STATEMENT Associative learning depends on brain state and is impaired when the subject is sleepy or tired. However, the link between changes of brain state and modulation of synaptic plasticity and learning remains elusive. Here we show that adenosine regulates weight dependence of heterosynaptic plasticity: adenosine strengthened weight dependence of heterosynaptic plasticity; blockade of adenosine A1 receptors abolished it. In model neurons, such changes of the weight dependence of heterosynaptic plasticity shifted their operating point between regimes dominated by associative plasticity or by synaptic homeostasis. Because adenosine tone is a natural correlate of activity level and brain state, modulation of plasticity by adenosine represents an endogenous mechanism for translation of brain state changes into a shift of the regime of synaptic plasticity and learning.
Keywords: adenosine, heterosynaptic plasticity, learning rules, neuron models, synaptic plasticity, visual cortex
Introduction
Adenosine, a metabolite of ATP and a ubiquitous neuromodulator in the brain, is released from neurons and astrocytes in an activity-dependent manner. During high-frequency neuronal firing, adenosine and ATP can be coreleased with neurotransmitter into the extracellular space, where ATP is broken down to adenosine by ectonucleotidases (Wall and Dale, 2008; Lovatt et al., 2012; Pajski and Venton, 2013). More dramatic rises in adenosine level are observed during pathological elevations of activity, such as epileptic seizures (Van Gompel et al., 2014). In physiological conditions, adenosine levels rise (specifically in the basal forebrain and neocortex) throughout waking, and decrease during subsequent sleep (Porkka-Heiskanen et al., 2000; Bjorness and Greene, 2009). Increased adenosine tone is one of the factors mediating use-dependent increases in sleep pressure as indicated by increased slow-wave activity (0.5–4 Hz) during non-rapid eye movement (non-REM) sleep (Rétey et al., 2005; Bjorness and Greene, 2009; Halassa et al., 2009; Porkka-Heiskanen and Kalinchuk, 2011). Rises of adenosine level during waking contribute to somnogenesis through actions in multiple brain areas, including inhibition of basal forebrain neurons (Strecker et al., 2000).
In the neocortex, the most widespread adenosine receptor is the G-protein-coupled A1 receptor (A1R; Dunwiddie and Masino, 2001). Its activation suppresses synaptic transmission by reducing presynaptic release probability and hyperpolarizing neurons (Kerr et al., 2013; Bannon et al., 2014; Zhang et al., 2015). Thus, activity-dependent rise of extracellular adenosine concentration provides a negative feedback on synaptic transmission and neural activity.
Apart from these immediate effects on synaptic transmission, adenosine, by changing presynaptic release, may also modulate the induction of long-term synaptic plasticity. Evidence has suggested that adenosine can modulate plasticity in the hippocampus (Dias et al., 2013; Sebastião and Ribeiro, 2015) and at thalamocortical inputs to the auditory cortex (Blundon et al., 2011; Blundon and Zakharenko, 2013). However, the role of adenosine in long-term plasticity at the intracortical connections is unexplored. In the neocortex, long-term plastic changes are weight dependent (Volgushev et al., 2000; Hardingham et al., 2007; Lee et al., 2012; Chistiakova et al., 2014). Both homosynaptic changes at synapses, which were active during plasticity induction, and changes at heterosynaptic sites, which were not activated presynaptically during the induction, correlate with the initial state of the release mechanisms assessed by paired-pulse ratio (PPR) or with release probability (Volgushev et al., 2000; Hardingham et al., 2007; Lee et al., 2012). Initial state of a synapse is thus one of the factors determining the outcome of the plastic changes. Weight-dependent heterosynaptic plasticity can robustly prevent runaway dynamics imposed on synaptic weight changes by Hebbian-type learning rules, and thus is a strong candidate mechanism for homeostatic regulation of synaptic weight changes (Chen et al., 2013).
Because adenosine plays a role in both modulation of transmitter release at the synaptic level and regulation of sleep pressure and sleep–wake cycle at the systems level, it may provide a link between changes of the brain state and state-dependent modulation of synaptic plasticity. To address this possibility, we ask: how do changes of the activation of adenosine receptors affect long-term synaptic plasticity? In slices from the visual cortex of rats, we studied how properties of homosynaptic and heterosynaptic plasticity of excitatory responses in layer 2/3 pyramidal neurons are modified during blockade or activation of adenosine receptors. The most prominent result was the abolition of weight dependence of heterosynaptic plasticity during blockade of A1Rs, and strengthening of the weight dependence by adenosine. Using computer simulations, we show that these experimentally observed changes alter the homeostatic effect of heterosynaptic plasticity on synaptic weights. Without weight dependence, heterosynaptic plasticity cannot prevent runaway potentiation or depression of synaptic weights and thus loses its homeostatic role. Heterosynaptic plasticity with even weak weight dependence effectively prevents runaway dynamics of synaptic weights, and this homeostatic effect increases with further strengthening of the weight dependence. We conclude that modulation of heterosynaptic plasticity by adenosine may represent a novel mechanism of regulation of synaptic changes and learning in different brain states.
Materials and Methods
All experimental procedures comply with the US National Institutes of Health regulations and were approved by the Institutional Animal Care and Use Committee of the University of Connecticut. Details of slice preparation and recording are similar to those used in previous studies (Volgushev et al., 2000; Lee et al., 2012). Slices were prepared from male Wistar rats [median age, 24 d; mean ± SEM, 24.6 ± 0.17 d; range, 15–34 d; 97% of the distribution between postnatal day (P) 21 and P29] obtained from Charles River or Harlan. Rats were anesthetized with isoflurane and decapitated. Then, brains were quickly removed and placed into an ice-cold oxygenated artificial CSF solution (ACSF) containing (in mm) the following: 125 NaCl, 25 NaHCO3, 25 glucose, 3 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, bubbled with 95% O2/5% CO2, pH 7.4. Coronal slices (350 μm thick) containing the visual cortex were prepared from the right hemisphere. After ≥1 h recovery at room temperature, individual slices were transferred to a recording chamber mounted on an Olympus BX-50WI microscope equipped with infrared differential interference contrast optics. Recordings were made at 28–32°C.
Adenosine (Sigma-Aldrich, catalog #A4036; CAS #58-61-7) was dissolved in ACSF to a 1 mm stock. Eight-cyclopentyl-1,3-dipropylxanthine (DPCPX; Sigma-Aldrich, catalog #C101, CAS #102146-07-6) was dissolved in >99.9% DMSO (Sigma-Aldrich, catalog #D8418, CAS #67-68-5) to a 1 mm stock. Final concentration of DMSO in bath was <0.05%. DPCPX (30 nm) or adenosine (20 μm) was present in the bath throughout the respective experiments for ≥30 min before beginning of recordings.
Intracellular recording and synaptic stimulation.
Whole-cell recordings were made from layer 2/3 pyramidal cells from the visual cortex. Monosynaptic EPSPs were evoked using two pairs of stimulating electrodes (S1 and S2) placed in L4, below the L2/3 recording site (as in Bannon et al., 2014). Paired-pulse stimuli (50 ms interpulse interval) were applied to S1 and S2 in alternating sequence, so that each input was stimulated with paired pulses every 15 s. To test for the possible contribution of inhibition, evoked PSPs were recorded at depolarized potentials between −50 and −40 mV. Only those PSPs that were still depolarizing at this membrane potential were considered excitatory and included in the analysis. To verify that two stimulated inputs were independent, we studied correlations of their properties, such as latency or induced plastic changes. If two stimulation electrodes would activate overlapping sets of presynaptic fibers, these properties in the two inputs would be correlated. For the subset of cells in which both inputs fulfilled stability criteria (see below), we did not find such correlations (r = 0.097, p = 0.43 for latency; r = 0.11, p = 0.38 for plastic changes). Therefore, we conclude that S1 and S2 activated nonoverlapping inputs to recorded neurons. Membrane potential and input resistance were monitored throughout the experiments; cells in which either parameter changed by >15% by the end of recording were discarded.
Plasticity induction.
After recording control EPSPs (12 ± 0.1 min), synaptic stimulation was stopped and an induction protocol was applied. Homosynaptic plasticity was induced via a spike-timing-dependent plasticity (STDP) pairing protocol. Pairing procedure consisted of three trains (1/min) of 10 bursts (1 Hz) of five depolarizing pulses (5 ms, 100 Hz, 0.4–1.5 nA; current intensity adjusted to evoke 4–5 spikes per burst) through the recording electrode, with an EPSP evoked at one of the two independent inputs preceding each burst of spikes by 10 ms (Fig. 1A). The remaining unpaired input experienced all effects of postsynaptic activation but without any presynaptic stimulation. Intracellular tetanization consisted of the same pattern of postsynaptic activation: three trains (1/min) of 10 bursts (1 Hz) of five depolarizing pulses (5 ms, 100 Hz, 0.4–1.5 nA) through the recording electrode, but without any concurrent presynaptic stimulation. Following induction, synaptic stimulation was resumed, and test EPSPs were recorded for another 40–60 min. Note that unpaired inputs in experiments with pairing and all inputs in intracellular tetanization experiments experienced the exact same pattern of postsynaptic firing during plasticity induction. This facilitated comparison of heterosynaptic changes in these two groups of inputs.
Figure 1.

Induction of long-term plasticity by pairing procedure. A, Scheme of pairing protocol. Two independent inputs to a layer 2/3 pyramidal neuron are stimulated by bipolar electrodes. Pairing consisted of three trains (1/min) of 10 bursts (1 Hz) of five pulses (5 ms, 100 Hz, 0.4–1.5 nA) through the recording electrode, with an EPSP evoked at one input 10 ms before each burst of spikes (paired input). Unpaired inputs were not stimulated during the induction. B, C, Example time course of EPSP amplitudes evoked by the first pulse in a paired-pulse paradigm. Vertical gray bars show timing of plasticity induction. Color-coded insets show averaged responses to paired-pulse stimuli before and after plasticity induction. Examples of potentiation at paired inputs, and depression, no change, and potentiation at unpaired inputs in control solution. These examples are also representative of plasticity induced under DPCPX or adenosine.
Data analysis.
Data analysis was made using custom-written programs in Matlab (MathWorks). All inputs included in the analysis fulfilled the following criteria: (1) stability of EPSP amplitudes during the control period, (2) stability of the membrane potential and input resistance throughout the recording, and (3) stability of the onset latency and kinetics of the rising slope of the EPSP. EPSP amplitudes are measured as the difference between the mean membrane potential during two time windows, the first time window placed before the EPSP onset and the second window placed on the rising slope of the EPSP, just before the peak. Amplitude of the second EPSP in the paired-pulse stimulation paradigm was measured using windows of the same duration and separation, but shifted by the length of the interpulse interval (50 ms). The PPR was calculated as the amplitude of the second EPSP divided by the amplitude of the first. Significance is denoted in the figures corresponding to p values of 0.05 (*), 0.01 (**), and 0.001 (***). Homogeneity of variance was assessed using a Brown–Forsythe or nonparametric Levine's test. One-way ANOVAs with either Tukey's or Tamhane's T2 post hoc are used. For correlations, Pearson's r was used.
Model of pyramidal neuron.
To investigate how observed changes of heterosynaptic plasticity affect its ability to counteract runaway dynamics of synaptic weights imposed by Hebbian-type learning, we used model simulations. For all simulations, we used an established reduced model of a cortical pyramidal cell (Bazhenov et al., 2002; Chen et al., 2012, 2013; Lemieux et al., 2014). This model was first proposed as a reduction of a multicompartmental pyramidal cell model, and consists of two electrically coupled compartments, dendritic and axosomatic (Mainen and Sejnowski, 1996). The current balance equations for the two compartments of the model are as follows (Eqs. 1 and 2): Cm (dVS/dt) = −g(VS − VD) − ISint; and Cm (dVD/dt) = gleak(VD − Eleak) − g (VD − VS) − ISint − Isyn, where Cm is the membrane capacitance, VS and VD is membrane potential, and ISint and IDint is the sum of all active intrinsic currents in axosomatic and dendritic compartments respectively; g is conductance between the two compartments. Isyn is the sum of synaptic currents. Since the kinetics of all currents are much faster in the axosomatic than in the dendritic compartment, the membrane potential in axosomatic compartment VS can be set at equilibrium state. Using singular perturbations analysis, it can be shown that the VS quickly reaches the manifold of slow motion defined by equation dVs/dt = F(VS) = 0, thus Equation 4 (below) can be substituted by the following (Eq. 3): g(VS − VD) = − ISint.
This reduced model has been successfully used in previous cortical network studies (Chen et al., 2012, 2013; Lemieux et al., 2014). For all currents (Na+, K+, Ca2+, Cl−) all conductances and their kinetics have been previously listed (Chen et al., 2012, 2013).
AMPA-type synapses were located at the dendritic compartment (see Fig. 6A). The synaptic current at each synapse was simulated by first-order activation kinetics expressed as follows (Eq. 4): Isyn= Wsyn[O](V − Esyn), where Wsyn is synaptic weight (defined in the range between 0 and 0.0303 mS/cm2), and Esyn is the reversal potential (Esyn = 0 mV). The fraction of open channels [O] is calculated as follows (Eqs. 5 and 6): d[O]/dt = α(1 − [O]) [T] − β[O]; and [T] = AH(t0 +tmax − t) H(t − t0), where H is Heaviside (step−) function, t0 is the time of receptor activation, t is simulation time, A = 0.5, and tmax = 0.3 ms. The rate constants α and β are 1.1 and 0.19 ms−1 respectively.
Figure 6.

A neuron model with homosynaptic and heterosynaptic plasticity. A–C, A model neuron with 100 input synapses expressing homosynaptic and heterosynaptic plasticity. STDP rules (B) were symmetrical at 50 synapses (orange), and depression-biased at the other 50 synapses (violet). Heterosynaptic plasticity rules were the same at all synapses, and included weight dependence of the probability and magnitude of change (C, left and middle; Eqs. 10 and 11). Rightmost plot in C shows example relation between initial weights and weight changes with a random component (Eq. 11, σ) calculated for initial synaptic weights from 0 to 0.03 mS/cm2 (0.0005 mS/cm2 increment), and regression line through these points. D, E, Dynamics of synaptic weights (D) and distributions of synaptic weights in the beginning and at the end of simulation (E) for two groups of synapses, with symmetrical (top) and depression-biased STDP (bottom) and heterosynaptic plasticity. Each synapse was driven by individual spike trains with Poisson-distributed interspike intervals (averaged frequency, 3 Hz; averaged correlation between spike trains, 0.5).
Short-term plasticity was implemented in all synapses using a simple phenomenological model of synaptic depression. The depression variable D represents the amount of available synaptic resources expressed as follows (Eq. 7): D = 1 − [1 − Di (1 − U)] exp[−(t − ti)/τ], where U = 0.07 is the fraction of resources used per action potential, τ = 700 ms is the time constant of recovery of the synaptic resources, Di is the value of D immediately before the ith event, and (t − ti) is the time after ith event.
STDP was implemented as in previous modeling studies (Song et al., 2000; Kempter et al., 2001; Rubin et al., 2001; Song and Abbott, 2001): every synaptic pairing event could potentially trigger a change of synaptic weight, and the direction and magnitude of synaptic modification are determined by the time difference between presynaptic and postsynaptic spikes as expressed in the following equations (Eqs. 8 and 9): dWsyn+ = a+ (exp[−(tpost − tpre)/τ+]); and dWsyn− = −a− (exp[(tpost − tpre)/τ−]).
In equations 8 and 9, dWsyn is the change of synaptic weight, a+ and a− are the maximal amplitude of potentiation and depression that could be induced by a single pair of presynaptic and postsynaptic spikes, tpost and tpre are the timing of postsynaptic and presynaptic spikes, and τ+ and τ− are the time constants of synaptic potentiation and synaptic depression.
Heterosynaptic plasticity was implemented according to rules derived from the results of in vitro experiments presented in this paper and from results of our prior studies (Volgushev et al., 2000; Chistiakova and Volgushev, 2009; Lee et al., 2012; Chen et al., 2013). Weight dependence of heterosynaptic plasticity was implemented using Equations 10–12 (Results).
Further details of the rules for STDP and heterosynaptic plasticity used in the model are described in Results, Figures 6–8, and related text.
Figure 7.

Prevention of runaway dynamics in the model neuron depends on the weight-dependent component to heterosynaptic plasticity. A1–A3, Example relations between initial weights and heterosynaptic weight changes with contribution of the weight-dependent component (Eq. 12, F) of 0, 0.3, and 1; and initial and final distributions of weights of two groups of synapses in models implementing these relations. Two groups of synapses had symmetrical or depression-biased STDP rules as shown in the insets in B. B, Dependence of the final weights (mean ± SD) of two groups of synapses on the contribution of the weight-dependent component of heterosynaptic plasticity. Black are mean ± SD of initial synaptic weights. Arrows (top) show R2 of correlations between initial PPR and amplitude changes at heterosynaptic inputs measured in electrophysiological experiments with DPCPX, control, and adenosine. Gradient areas show mean ± SD of R2 for each group obtained with bootstrapping (1000 resamplings; 0.0172 ± 0.019; 0.165 ± 0.091; 0.362 ± 0.104; for the three groups).
Figure 8.

Prevention of runaway dynamics in the model depends on the slope of weight dependence of heterosynaptic plasticity. A1–A3, Example relations between heterosynaptic weight changes and initial weights for slope factors (Sf) 10, 40, and 70, as indicated, and initial and final distributions of weights of two groups of synapses in models implementing these relations. B, Dependence of the final weights (mean ± SD) of two groups of synapses on the slope of weight dependence of heterosynaptic plasticity, as shown on the right (Eq. 11, SlopeFactor). Black are mean ± SD of initial synaptic weights. Two groups of synapses had STDP rules as in Figure 6 model.
Results
Homosynaptic and heterosynaptic plasticity in layer 2/3 pyramidal neurons
We made whole-cell recordings from layer 2/3 pyramidal neurons in slices from the visual cortex of adolescent Wistar rats. Small-amplitude monosynaptic EPSPs were evoked by electric pulses applied through two pairs of stimulating electrodes using a paired-pulse stimulation protocol with a 50 ms interpulse interval. The PPR is defined as the averaged amplitude of the EPSP evoked by the second pulse in a pair divided by the averaged amplitude of the EPSP evoked by the first pulse.
Figure 1 shows examples of long-term plasticity induced by a pairing procedure in an STDP paradigm. The pairing procedure consisted of three trains (1/min) of 10 bursts (1 Hz) of five depolarizing pulses (5 ms, 100 Hz, 0.4–1.5 nA) applied through the recording electrode, with an EPSP evoked at one of the two independent inputs 10 ms before each burst of spikes (Fig. 1A). Typically, this pre-before-post pairing protocol led to a long-term potentiation (LTP) of EPSP amplitude (Fig. 1B), though in some experiments long-term depression (LTD) or no changes of synaptic transmission were observed. On average, paired inputs expressed a significant potentiation to 128.1 ± 7.7% of baseline (paired t test, p < 0.05, n = 31). Notably, unpaired inputs could also express LTP or LTD (Fig. 1C). Because unpaired inputs were not presynaptically activated during the induction, these changes represent heterosynaptic plasticity.
The situation experienced by unpaired inputs, postsynaptic spiking without concurrent presynaptic activation, is recapitulated via a purely postsynaptic induction protocol of intracellular tetanization (Fig. 2A). Intracellular tetanization consisted of the same pattern of postsynaptic activation as the pairing procedure: three trains (1/min) of 10 bursts (1 Hz) of five pulses (5 ms, 100 Hz, 0.4–1.5 nA; Fig. 2A) through the recording electrode, but without any concurrent presynaptic stimulation. Thus, all plastic changes after intracellular tetanization can be considered heterosynaptic. Following intracellular tetanization, we have observed heterosynaptic LTP, LTD, or no change (Fig. 2B).
Figure 2.

Induction of long-term plasticity by intracellular tetanization. A, Scheme of intracellular tetanization protocol. Intracellular tetanization consisted of the same postsynaptic firing as pairing: three trains (1/min) of 10 bursts (1 Hz) of five pulses (5 ms, 100 Hz, 0.4–1.5 nA) through the recording electrode. However, no inputs were stimulated during the intracellular tetanization. B, Examples of potentiation, no changes, and depression induced by intracellular tetanization. Same conventions as in Figure 1.
Weight dependence of homosynaptic and heterosynaptic plasticity
Previous research has established that both homosynaptic and heterosynaptic plasticity are weight dependent. Following induction of homosynaptic plasticity in paired recordings (Hardingham et al., 2007), or induction of heterosynaptic plasticity by intracellular tetanization (Volgushev et al., 1997, 2000; Chen et al., 2013), inputs with a low PPR (and thus a higher release probability) tend to undergo LTD, while inputs with a high PPR (lower release probability) tend to undergo potentiation. Furthermore, the outcome of plasticity is determined individually at each input onto a neuron: intracellular tetanization may lead to potentiation of one input, but depression of the other input to the same neuron (Volgushev et al., 2000; Chen et al., 2013). In neurons recorded in control extracellular solution, EPSP amplitude changes correlated with initial PPR in all three groups of inputs (Fig. 3, blue triangles): paired inputs (Pearson's r = 0.427, n = 31, p < 0.05) and unpaired inputs (r = 0.338, n = 25, p = 0.1) in experiments with STDP, and in the inputs from experiments with intracellular tetanization (r = 0.391, n = 41, p < 0.01). These results confirm that both Hebbian-type associative plasticity and heterosynaptic plasticity are weight dependent.
Figure 3.

Homosynaptic and heterosynaptic plasticity are weight dependent. A–D, Correlation between initial PPR and changes of EPSP amplitude following induction protocol at paired inputs (A), at unpaired inputs (B), following intracellular tetanization (C), and at all heterosynaptic sites (D, pooled data from B and C). Recordings were conducted in either control ACSF (CTRL, blue triangles), or on the background of 30 nm selective A1R antagonist DPCPX (green circles) or 20 μm adenosine (red squares). A, At paired inputs (homosynaptic sites), changes in EPSP amplitudes were positively correlated with the initial PPR in all experimental series, regardless of background adenosine receptor ligands. B, C, At unpaired inputs and inputs undergoing intracellular tetanization (both of which may be considered heterosynaptic), changes of EPSP amplitudes were positively correlated with the initial PPR in control solution and on the background of 20 μm adenosine. On the background of A1R blockade with DPCPX, there were still cases of potentiation and depression, but the relationship of EPSP change to initial synaptic strength (initial PPR) was abolished. D, Pooled data (from B and C) representing all heterosynaptic sites. *p < 0.05; **p < 0.01; ***p < 0.001.
Because paired inputs most often expressed LTP and were potentiated on average (Fig. 4A, right, blue bars), their weight dependence was shifted upward toward potentiation (Fig. 3A, blue symbols and blue regression line). At heterosynaptic sites (unpaired or after intracellular tetanization), potentiation and depression were balanced such that despite significant potentiation or depression of individual inputs, net changes were centered around 100% of baseline values (Fig. 4D, right). Regression lines for these inputs (in isolation; Fig. 3B,C, blue) or pooled together (Fig. 3D, blue) crossed 100% of baseline amplitude at PPR values of ∼1.
Figure 4.

Effects of adenosine on PPR and net plastic outcome at homosynaptic and heterosynaptic inputs. A, Initial (preinduction) PPR (left) and mean EPSP amplitude change (right) induced at paired inputs in three drug conditions. Error bars show SEM; gray dash marks denote individual cases. B, Percentage of inputs that expressed significant potentiation or depression, or did not change (t test comparing baseline period to after induction period for each input). Data for paired inputs, for three drug groups. Inset in each bar shows number of cases. A χ2 test revealed no significant relationship between adenosine receptor manipulation and the frequency of potentiation, depression, or no change. Note that potentiation is generally a more common outcome than depression. C, Mean changes of EPSP amplitude in inputs that underwent potentiation, that underwent depression, or that did not change. Paired inputs in three drug conditions. The mean magnitude of potentiation was larger under adenosine than in the DPCPX group. D, Initial PPR (left) and mean EPSP amplitude change (right) induced at heterosynaptic inputs in three drug conditions. Conventions as in A. E, Percentage of heterosynaptic inputs that expressed significant potentiation or depression, or did not change, in three drug groups. Blocking tonic A1R activation (DPCPX group) results in more cases of depression and fewer cases of potentiation than expected (χ2 test). The adenosine group exhibited fewer cases of depression and more cases of potentiation than expected. Standardized residuals of ≥1.4 are denoted with #, indicating that these counts deviated the most from expected observations. F, Mean changes of EPSP amplitude in heterosynaptic inputs that underwent potentiation, that underwent depression, or that did not change. No significant effects of adenosine receptor manipulation on magnitude of EPSP change.
Adenosine-modulation of STDP at paired inputs
Adenosine is an endogenous neuromodulator known to fluctuate in a use-dependent manner and during the sleep–wake cycle. Our prior research in the same preparation has demonstrated that the predominant effect of adenosine on excitatory transmission to layer 2/3 pyramidal neurons is a reduction in presynaptic release probability (and thus an increase in the PPR) via activation of presynaptic A1Rs (Bannon et al., 2014; Zhang et al., 2015). It also revealed a background level of adenosine receptor activation in slices during recordings in control extracellular solution (Bannon et al., 2014). To study effects of adenosine on homosynaptic and heterosynaptic plasticity, we performed experiments in the presence of either the selective antagonist of A1Rs, DPCPX (30 nm), or adenosine (Ado, 20 μm) in the extracellular solution. This resulted in either a blockade of endogenous adenosine tone (and thus reduced level of activation of adenosine receptors) or an elevated level of receptor activation.
The PPR was reduced in the DPCPX group compared with the adenosine group (Fig. 4A, left; F(2,93) = 3.591, p < 0.05, Tukey's post hoc test). These results corroborate our previous findings of adenosine effects on short-term plasticity and are consistent with presynaptic action of adenosine on A1Rs, leading to reduction of the average release probability (Bannon et al., 2014; Zhang et al., 2015).
Next, we asked how adenosine affects the outcome of plasticity. As described above, in control solution pairing induced a net potentiation (Fig. 4A). On the background of A1R antagonism by DPCPX, averaged responses after the pairing procedure were not different from baseline (105.9 ± 7.1% of baseline, n = 30, p = 0.89). The relationship between initial PPR and EPSP change was preserved in this group (Fig. 3A, green circles), and thus the tendency of DPCPX to reduce PPR is consistent with the observation of less potentiation but more depression in these conditions. Indeed, a PPR of >2 was observed in 5 of 31 paired inputs studied in control solution, but only 1 of 30 paired inputs studied under DPCPX. There was a tendency for the relative proportion of the number of potentiating and depressing inputs to shift toward depression in DPCPX (Fig. 4B). In control solution, the ratio of potentiated to depressed inputs was 16:5. On the background of DPCPX, the ratio was 11:10 (Fig. 4B).
Surprisingly, there was also no significant net increase of averaged EPSP amplitude in n = 35 paired inputs recorded on the background of adenosine (111.5 ± 8.5% of baseline, n = 35, p = 0.37). Individual cases of robust potentiation and depression were still observed, though their average was centered on 100% of baseline. This result is unexpected because the correlation between initial PPR and EPSP change was preserved (Fig. 3A, red squares), and the PPR was increased in adenosine (Fig. 4A, left). Moreover, the magnitude of EPSP amplitude increase averaged over inputs that expressed significant potentiation was higher in the adenosine group compared with the DPCPX group (Fig. 4C; F(2,34) = 4.060, p < 0.05, Tukey's post hoc test). A partial explanation for these results may be that adenosine strengthened the weight-dependent relationship (indicated by the highest r value among three groups, r = 0.534; Fig. 3A), and that stronger weight dependence became a dominating factor, which overrides the potentiation bias imposed by STDP rules, thus leading to more balanced synaptic changes.
Adenosine modulation of heterosynaptic plasticity
Effects of activation or blockade of adenosine receptors on heterosynaptic plasticity were more pronounced than at homosynaptic sites. The most dramatic effect was an abolishment of weight dependence of heterosynaptic plasticity in the DPCPX group: no significant correlation between initial PPR and EPSP amplitude changes was found at unpaired inputs in STDP experiments (Fig. 3B, green circles; r = −0.106, n = 24, p = 0.6) or after intracellular tetanization (Fig. 3C, green circles; r = −0.142, n = 40, p = 0.4). In contrast, on the background of adenosine, the relationship between initial PPR and EPSP change was strengthened in both groups (Fig. 3B,C, red squares; r = 0.502, n = 23, p < 0.05 for unpaired inputs; r = 0.689, n = 43, p < 0.001, after intracellular tetanization). Because unpaired inputs in STDP experiments and inputs studied in intracellular tetanization experiments (1) experienced the same pattern of postsynaptic spiking without concurrent presynaptic activation during plasticity induction and (2) demonstrated the same modulation of weight dependence of plasticity by adenosine, the two groups were pooled and subsequently considered together as “heterosynaptic inputs.”
In the pooled group of heterosynaptic inputs (Fig. 3D), correlation between initial PPR and plastic changes was not significant in DPCPX (r = −0.093, N = 64, p = 0.47), was significant in control solution (r = 0.394, n = 66, p < 0.001), and was strongest in adenosine (r = 0.60, N = 66, p < 0.001). A multiple linear regression for heterosynaptic inputs including PPR, drug condition, and their interaction as predictors revealed significant contribution of the interaction term (F(2,190) = 5.857, p < 0.01), demonstrating that the slopes of the regression lines were different between groups. The abolition of weight dependence in the DPCPX group demonstrates that endogenous adenosine tone plays a mandatory role in the establishment of weight dependence of heterosynaptic plasticity. Moreover, the ability of adenosine to strengthen the weight dependence indicates that adenosine receptor activation fundamentally modulates the weight-dependent property of heterosynaptic plasticity.
In the DPCPX group, the average EPSP amplitude following induction of heterosynaptic plasticity was significantly depressed to 89.92 ± 4.9% of baseline (paired t test, n = 64, p < 0.05, Fig. 4D, right). In the adenosine group, net EPSP change was biased toward potentiation (117.80 ± 7.24%), but was not significantly different from 100% of baseline (paired t test, n = 66, p = 0.11). Net EPSP change in the adenosine group was significantly higher than net change in the DPCPX group (Fig. 4D, right; F(2,193) = 4.702, p = 0.01, Tukey's post hoc). A χ2 test revealed a significant relationship between adenosine receptor manipulation and the frequency of potentiation, depression, or no change (X2 = 9.647, n = 196, p < 0.05). In the DPCPX group, more cases of depression and fewer cases of potentiation were observed. In the adenosine group, fewer cases of depression and more cases of potentiation were observed than expected (Fig. 4E; Pearson's χ2 test, χ2 = 9.65, df = 4, p = 0.047). The magnitude of potentiation or depression was not different between the three groups (Fig. 4F; depression, F(2,67) = 0.263 p = 0.77; potentiation F(2,67) = 0.811 p = 0.45), suggesting that the differences observed between drug groups in the mean EPSP change were attributable to differences in the frequency of occurrence, but not magnitude of plasticity at individual synapses.
In summary, A1R antagonism abolished weight dependence of heterosynaptic plasticity and biased plastic changes toward depression. In contrast, exogenous adenosine resulted in stronger weight dependence of plasticity, increased the initial PPR, and biased plastic outcome toward potentiation.
Adenosine strengthens the involvement of presynaptic mechanisms in expression of plasticity
To evaluate the relative contribution of presynaptic changes to the expression of plasticity, we plotted changes in EPSP amplitude against changes in the PPR following plasticity induction (Fig. 5). Changes in EPSP amplitude were negatively correlated with changes in the PPR. Potentiation was associated with reductions in the PPR, indicative of an increase of release probability. Depression was associated with increases in the PPR, indicative of a decrease in release probability. Thus, expression of plastic changes at both homosynaptic and heterosynaptic sites was at least partially presynaptic. In control solution, Pearson's r values were r = −0.522 (n = 31, p < 0.01) at homosynaptic sites (Fig. 5A, paired inputs, blue triangles) and r = −0.311 (n = 66, p < 0.01) at heterosynaptic sites (Fig. 5B, blue triangles). Blockade of A1Rs by DPCPX diminished the strength of this relationship (r = −0.212, n = 30, p = 0.26 at paired inputs; r = −0.279, n = 64, p < 0.05 at heterosynaptic sites), while adenosine strengthened it (r = −0.704, n = 35, p < 0.001 at paired inputs; r = −0.582, n = 66, p < 0.001 at heterosynaptic sites). A strengthening of this relationship (and higher R2 value) indicates that more of the variance in plastic outcome could be explained by changes in the PPR, suggesting an increased contribution of presynaptic component in expression of plasticity.
Figure 5.

Homosynaptic and heterosynaptic plasticity are expressed partially via presynaptic mechanisms. A, B, Correlation of changes in the PPR with changes in EPSP amplitude at paired inputs (A) and heterosynaptic inputs (B; unpaired inputs and after intracellular tetanization). Data for three drug groups: control ACSF (blue triangles); 30 nm A1R antagonist DPCPX (green circles); 20 μm adenosine (red squares). Changes in the PPR were negatively correlated with EPSP amplitude changes. This relationship was generally strengthened as a function of adenosine receptor activation. Compared with control, r values were higher in adenosine, but lower in DPCPX groups.
To summarize, our experimental results show that adenosine modulates synaptic plasticity (1) by changing the initial PPR, which is an index used for assessing properties of presynaptic release; (2) by changing the strength of the dependence of the outcome of plasticity on initial PPR and the slope of this dependence; and (3) by modulating the contribution of presynaptic mechanisms in the expression of plasticity. This multitude of actions establishes adenosine as a central player in the regulation of synaptic plasticity. The most dramatic effects of adenosine were on heterosynaptic plasticity: blockade of A1Rs eliminated the dependence of heterosynaptic changes on initial PPR, while their increased activation strengthened this dependence. We chose these most prominent effects of adenosine on heterosynaptic plasticity to investigate their influence on dynamics of synaptic weights in a model neuron with plastic synapses, which allows us to extend analysis of the effects of heterosynaptic plasticity beyond what is possible in the slice.
Homosynaptic and heterosynaptic plasticity in a neuron model
Weight-dependent and balanced potentiation and depression are defining properties of the heterosynaptic plasticity described above and in our prior work (Volgushev et al., 2000; Lee et al., 2012; Chen et al., 2013; Chistiakova et al., 2015). Heterosynaptic plasticity endowed with these features represents a strong candidate homeostatic mechanism for preventing runaway dynamics of synaptic weight changes incurred by Hebbian-type learning rules (Chen et al., 2013; Chistiakova et al., 2015). We hypothesized that, by modulating the weight dependence of heterosynaptic changes, adenosine modulates the homeostatic role of heterosynaptic plasticity. To test this hypothesis and reveal computational consequences of the modulation of weight dependence of heterosynaptic plasticity by adenosine, we built a conductance-based neuron model with plastic synapses, based on previous work (Chen et al., 2013). The model neuron received inputs from 100 synapses expressing calcium-dependent homosynaptic and heterosynaptic plasticity (Fig. 6A). Because for the purposes of this study we needed a model in which synaptic weights express runaway dynamics in either direction (as typical for models with Hebbian-type learning rules alone), we introduced different STDP rules in two groups of synapses. Homosynaptic changes were governed by symmetrical STDP rules in 50 synapses and, by a depression-biased STDP, at the other 50 synapses (Fig. 6A,B). Without heterosynaptic plasticity, this model expressed robust runaway dynamics of synaptic weights in either direction over a broad range of input patterns, or differing proportions of synapses with symmetrical or depression-biased STDP rules (data not shown). Therefore, to facilitate comparison of the effects of heterosynaptic plasticity on runaway dynamics to the maximum and to the minimum weight, we opted for a 50:50 split of synapses. Rules for heterosynaptic plasticity were the same at all synapses (Fig. 6C). Heterosynaptic changes occurred when intracellular calcium level exceeded the threshold level, with a probability P expressed as follows (Eq. 10): P = 3000 × (Wsyn − 0.5 × Wmax)2 + 0.1, where Wsyn represents synaptic weight ranging from 0 to Wmax (0.03 mS/cm2).
The magnitude of synaptic change dWsyn was calculated as follows (Eq. 11):
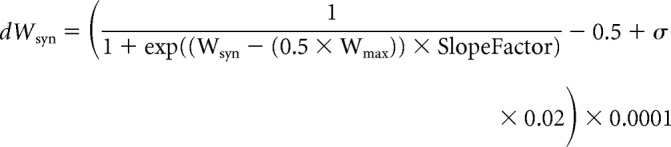 |
where σ is a random variable derived from the standard normal distribution with zero mean and SD of 3. Variable SlopeFactor (Sf = 100 in the base model) determines the slope of the weight dependence of heterosynaptic changes. Stimulation of this model with irregular activity at the inputs (e.g., spike trains with Poisson-distributed interspike intervals; averaged frequency, 3 Hz; mean correlation between pairs of spike trains, 0.5) leads to a robust segregation of the weights of synapses from the two groups, but without runaway dynamics and saturation of the weights of synapses (Fig. 6D,E).
Changing contribution of weight dependence alters homeostatic effect of heterosynaptic plasticity
In the model described above, the correlation between the initial weight and weight change is very high, with average R2 = 0.95 ± 0.01 (N = 50 realizations as in Fig. 6C, right). This means that initial weight predicted ∼95% of the variance in the outcome of heterosynaptic plasticity. In real synapses, however, R2 values for the correlations between initial PPR and synaptic change at heterosynaptic sites were <0.4. Thus, the initial state of a synapse is only one of multiple factors determining the outcome (direction and magnitude) of plastic change in real synapses. We found that the strength of the correlation between initial PPR and EPSP change, and thus percentage of variance of heterosynaptic changes explained by initial state of synapse, systematically changes as a function of adenosine receptor activation. In DPCPX, control, and adenosine groups, R2 values at heterosynaptic sites were 0.009, 0.155, and 0.360, respectively.
We also accounted for the contribution of other factors (unrelated to the initial state of synapse) to the outcome of heterosynaptic plasticity in the model neuron. We introduced an additional random term in the calculation of synaptic weight change dW′syn (Eq. 12): dW′syn = dWsyn × F + Rand × (1 − F), where dWsyn is calculated as in Equation 11; Rand is a normally distributed random variable with zero mean and variance of 0.003, and F is a fraction of the weight-dependent changes contributing to the weight change. Varying F would then modify the amount of variance of plastic changes attributable to the weight dependence and thus be analogous to R2 measured in experiments.
In the model with F = 0 contribution of weight dependence, heterosynaptic changes are random and cannot prevent runaway dynamics of synaptic weights: synapses with symmetrical STDP rules potentiate to the maximal value, and weights of synapses with depression-biased rules bunch around zero (Fig. 7A1). Heterosynaptic plasticity with F = 0.3 contribution of weight-dependent component robustly prevents saturation of synaptic weights (Fig. 7A2). Importantly, the weights of two groups of synapses still could be separated in this model and even in the model with F = 1 contribution of weight dependence to heterosynaptic plasticity, corresponding to R2 ∼0.95 (Fig. 7A3). Summary results of this series of simulations show that heterosynaptic plasticity with even a mild contribution of weight dependence (∼10%) can prevent runaway potentiation and depression (Fig. 7B). The homeostatic effect of heterosynaptic plasticity rapidly increases with the increase of the contribution of weight-dependent component up to F = ∼0.5, but changes less with further strengthening of the weight dependence (Fig. 7B). Notably, adenosine modulates the weight dependence exactly within the range of values that bear the strongest impact on the homeostatic effect of heterosynaptic plasticity. During blockade of A1Rs with the selective antagonist DPCPX, R2 values for the relationship between initial PPR and heterosynaptic plastic changes were 0.017 ± 0.02 (mean ± SD, estimated using bootstrapping). These values (Fig. 7B, green region) correspond to absent or negligible contribution of weight dependence and negligible homeostatic effect of heterosynaptic plasticity, thus representing permissive conditions for runaway synaptic changes. In control experiments with background level of adenosine receptor activation, R2 values were 0.165 ± 0.09 (Fig. 7B, blue region). This range covers the transition from a mode of operation that allows strong segregation of synaptic weights, to a regime in which the magnitude of segregation decreases, and runaway dynamics are robustly prevented. In experiments with strong activation of adenosine receptors (20 μm adenosine in the bath) R2 values were 0.362 ± 0.10 (Fig. 7B, red region). In that range, heterosynaptic plasticity has a strong homeostatic effect, the tendency for runaway dynamics is strongly counteracted, and postsynaptic activity would have a stabilizing effect on synaptic weights. Thus, changing the level of activation of adenosine receptors changes the weight dependence and the homeostatic effect of heterosynaptic plasticity over a range from completely absent to extremely strong.
Homeostatic action of heterosynaptic plasticity depends on the slope of weight dependence
In the simulations described above, we have investigated a scenario in which the weight dependence of heterosynaptic plasticity was altered due to the changing contribution of factors unrelated to the initial weight, and thus could be modeled by adding a random variable. This changed both the slope of the weight dependence and the spread of data points around the regression line (Fig. 7A1–A3, compare top plots). In an alternative scenario, only the slope of the weight dependence may change, while the spread of points around the regression line remains the same. In experiments, we did observe markedly different slopes of the dependence of heterosynaptic plasticity on the initial PPR (Fig. 3). Note that because of large intrinsic variability of plastic changes in electrophysiological experiments, it is not possible to discriminate between changes of slope and changes of the variance around the slope, even for our large samples (N = 64, 66, 66 for three groups of heterosynaptic inputs). To isolate the effect of changing slope of weight dependence of heterosynaptic plasticity on dynamics of synaptic changes, we systematically changed the SlopeFactor in Eq. 2, while keeping the same all other parameters of simulation.
In the model with heterosynaptic changes independent of the initial weight (SlopeFactor = 0), synaptic weights of the inputs with symmetrical STDP windows were potentiated to the maximal value, and weights of the inputs with depression-biased STDP windows were depressed to zero (data not shown, but summarized in Fig. 8B). Introducing a minimal weight dependence (SlopeFactor = 10; Fig. 8A1) to heterosynaptic changes prevented runaway depression and significantly limited runaway potentiation of synaptic weights. Increasing the slope of weight dependence increased the homeostatic force of heterosynaptic plasticity (Fig. 8A2,A3). Overall, influence of the slope of weight dependence of heterosynaptic changes on dynamics of synaptic weights and their final distributions (Fig. 8) was similar to the influence of changing the relative contribution of weight-dependent changes (Fig. 7).
To summarize, our model simulations show that weight dependence is a necessary feature for heterosynaptic plasticity to counteract runaway dynamics of synaptic weights imposed by Hebbian-type learning rules. Heterosynaptic plasticity without weight dependence did not prevent runaway dynamics. However, even a weak weight dependence effectively prevented runaway dynamics of synaptic weights and their saturation. Thus, results of model simulation demonstrate that modulation of weight dependence of heterosynaptic plasticity, e.g., by adenosine as observed in our slice experiments, can be a powerful tool for regulation of the final outcome of synaptic changes.
Discussion
Previous studies have found that heterosynaptic plasticity with weight-dependent properties can maintain homeostasis of synaptic weights and prevent runaway potentiation or depression of the weights of individual synapses imposed by the Hebbian-type plasticity rules (Chen et al., 2013; Chistiakova et al., 2014, 2015). Here we demonstrate that adenosine receptor activation modulates the weight dependence of synaptic changes. Antagonism of tonically activated adenosine A1Rs abolished weight dependence of plasticity at heterosynaptic sites and weakened it at homosynaptic sites. Increased activation of adenosine receptors strengthened the weight dependence at both homosynaptic and heterosynaptic sites. Our model simulations show that changing the weight dependence within the experimentally observed range can strengthen or abolish the homeostatic effect of heterosynaptic plasticity and thus fundamentally change the dynamics of synaptic weights and their final equilibrium state. Thus, our study identified a novel mechanism of regulation of synaptic changes by neuromodulatory control of weight dependence of heterosynaptic plasticity.
Weight dependence is essential for the homeostatic role of heterosynaptic plasticity
Homosynaptic plasticity with Hebbian-type learning rules introduces positive feedback on synaptic weight changes, making them prone to runaway dynamics. Concurrent heterosynaptic plasticity can robustly prevent runaway potentiation or depression and avoid saturation of synaptic weights at maximal or minimal values (Chen et al., 2013; Volgushev et al., 2016). Results of our present study show that the weight-dependent property of heterosynaptic plasticity is essential to this homeostatic role. In a model that lacks weight dependence of the heterosynaptic changes, STDP drove synaptic weights to their boundary values. Introducing a weight-dependent component to heterosynaptic plasticity counteracted the runaway dynamics. Increasing the contribution of weight dependence shifted final distributions of synaptic weights further away from the maxima and minima, keeping synaptic weights well within their dynamic range. The percentage of the contribution of weight dependence to heterosynaptic plasticity in the model (Eq. 12, F) is analogous to percentage of variance in the experimentally observed EPSP changes accounted for by knowledge of the initial PPR (R2 value of the EPSP change vs PPR correlation). Thus, under A1R antagonism, the R2 value was virtually 0, corresponding to F = 0 in the model, a setting that could not constrain runaway dynamics imposed by Hebbian plasticity. Increasing adenosine receptor activation increased the strength of the relationship (higher R2 values), and in the model with corresponding parameters (increasing F > 0), synaptic weight changes were constrained and their runaway dynamics effectively prevented.
Factors determining synaptic changes
It is common practice to describe STDP protocols with specific timing windows to be “potentiating” or “depressing” by design. Indeed, such protocols create the desired plastic outcome on average over many inputs, corroborating these labels. The well documented ability of associative plasticity to induce changes in opposite directions after the same STDP procedure is often overlooked (for recent reviews, see Chistiakova et al., 2014, 2015). This diversity of plastic outcomes implies the involvement of additional factors, other than basic STDP rules. One of these factors is weight dependence of plasticity. Prior research revealed weight dependence of homosynaptic plasticity in a variety of synapses (van Rossum et al., 2000; Sjöström et al., 2001; Hardingham et al., 2007; Volgushev et al., 2016). Our results confirmed that in all drug conditions, homosynaptic plasticity was weight dependent. Adenosine, by modulating the strength of the weight-dependent relationship, changed the relative predictive power of the weight-dependent rules among other possible determinants of homosynaptic plasticity. For example, in conditions of increased adenosine, the strength of weight dependence and thus the contribution of weight-dependent factors might increase relative to “protocol-specific” rules determined by STDP windows, and plastic outcome would be more representative of weight-dependent rules than STDP rules. This is corroborated by both a balancing of net plastic outcome around 100% and an increased R2 of the PPR-by-EPSP relationship in the adenosine group. For heterosynaptic plasticity, effects of adenosine on weight dependence are even stronger, and contribution of the weight-dependent factor to heterosynaptic changes can range from nonsignificant to strong. The difference between homosynaptic versus heterosynaptic plasticity in susceptibility to adenosine modulation indicates that diverse sets of mechanisms may mediate communication between postsynapses and presynapses, and coordinate presynaptic and postsynaptic plastic changes. Candidate mechanisms include the nitric oxide (NO) pathway activated either in an NMDA-dependent manner (O'Dell et al., 1991; Nugent et al., 2007) or by purely postsynaptic protocols (Volgushev et al., 2000; Phillips et al., 2008; Lee et al., 2012), as well as pathways via metabotropic glutamate receptors and endocannabinoids (Maejima et al., 2001; Chevaleyre and Castillo, 2003; Sjöström et al., 2004; Chevaleyre et al., 2006; Duguid and Sjöström, 2006). Results of the present study add adenosine signaling to this list—either as a new separate pathway or as a modulator. These signaling pathways differ by activation requirements (whether or not coincidence of presynaptic and postsynaptic activity is necessary) and by the spatial domain of their action: extremely limited for glutamate spillover, modest for NO and endocannabinoids, and quite large for astrocytic adenosine release. The difference in the sensitivity of weight dependence of homosynaptic versus heterosynaptic plasticity to the level of activation of adenosine receptors may reflect the interplay between the activation requirements and the spatial range of action. Revealing details of this interaction requires further work.
Thus, plastic outcome at a synapse can be thought of as a function of at least two factors. First is the timing, number, and pattern of presynaptic and postsynaptic spikes in an STDP protocol, which at homosynaptic sites sets the direction and magnitude of plastic changes, and at heterosynaptic sites determines the profile of calcium rise that may lead to plasticity. Second is a weight-dependent rule, which operates at both homosynaptic and heterosynaptic sites, fine-tunes the outcome, and can be modulated by adenosine.
Shifting regimes of learning
Because of adenosine's ability to modulate the weight dependence of synaptic plasticity, and because the effects of this modulation are so pronounced, adenosine can play a unique role in modulation of regimes of learning. Blockade of adenosine receptors eliminates weight dependence and permits runaway-like dynamics of synaptic weights and their maximal segregation under unconstrained STDP rules. Higher levels of adenosine receptor activation strengthen weight dependence of heterosynaptic plasticity, enhancing its homeostatic role while still permitting a segregation of synaptic weights. Thus, via modulation of weight dependence, adenosine can shift the operating point of the cell between two regimes. One regime is dominated by associative learning rules allowing more profound segregation of weights. The other regime is dominated by weight-dependent plasticity, with a strong homeostatic effect on synaptic weights. Notably, in this regime, any strong postsynaptic activity will have a homeostatic effect on synaptic weights. The ability to shift between these operating modes allows the brain to exploit conceivable advantages of either regime. It is likely that a dynamic regulation of the relative balance of associative and homeostatic rules is fundamental to permitting new learning and maintaining homeostasis.
Outlook: adenosine as a link between changes of brain state and modulation of plasticity?
Sleep pressure is the homeostatic force involved in regulation of the sleep–wake cycle and slow-wave activity during non-REM sleep (Borbély and Achermann, 1999; Steriade, 2001; Vyazovskiy et al., 2011; Tononi and Cirelli, 2006; Landolt, 2008). The accumulation of sleep pressure and corresponding increases in slow-wave activity are use dependent (Huber et al., 2004; Tononi and Cirelli, 2006; Vyazovskiy et al., 2011). A wealth of evidence links adenosine with modulation of slow-wave activity (Benington et al., 1995; Rétey et al., 2005; Bjorness and Greene, 2009; Bjorness et al., 2009; Porkka-Heiskanen and Kalinchuk, 2011; Urry and Landolt, 2015) and implicates rises in extracellular adenosine concentration in mediating sleep pressure and compensatory increase of slow-wave activity following prolonged waking or use (Fellin et al., 2009; Halassa et al., 2009). An influential hypothesis relating sleep and plasticity submits that imbalances in synaptic weight changes induced during waking are met with synaptic homeostasis during subsequent sleep (Tononi and Cirelli, 2006, 2012, 2014). Our results establishing the role of adenosine in modulating the homeostatic component of heterosynaptic plasticity identify a possible mechanism of state-dependent modulation of plasticity regimes. We propose that increases in sleep pressure, which co-occur with adenosine increases (such as following increased episodes of spiking during associative learning or long waking), would shift synaptic plasticity toward a regime dominated by normalizing, homeostatic rules. Dissipation of sleep pressure and lower adenosine levels would permit associative learning rules to dominate and change synapses toward the ends of their dynamic range. A constellation of plasticity rules whose relative importance can be changed in an analog fashion by neuromodulation adds a fine-tuning mechanism for synaptic learning and homeostasis. In this scenario, the balance between synaptic learning and homeostasis is not restricted to discrete states dictated by phases of sleep or waking, but would be continuous as a function of sleep pressure. This gradual engagement of a homeostatic regime might also be relevant at smaller temporal and spatial scales, such as when excessive firing of a neuronal ensemble, either produced by normal sensory input or by pathological seizure activity, causes adenosine release. These stabilizing regimes may also be particularly useful in pathological situations in which the adenosine level is known to rise dramatically, such as during ischemic events or seizures (Van Gompel et al., 2014).
Adenosine therefore alters the homeostatic force on synaptic weight changes by shifting regimes of plasticity. When adenosine receptor activation is absent or low, the weight-dependent relationship is weak and unbalancing regimes of learning defined by STDP rules dominate, pushing synaptic weights toward extreme values. Increasing adenosine levels increases the contribution of weight dependence to heterosynaptic plastic changes and shifts plasticity toward a homeostatic regime where homosynaptic STDP is tightly constrained. These shifts of plasticity regimes mediated by changes of adenosine concentration may take place on both a slow and global scale (sleep pressure) as well as on a fast and local scale (episodes of high neuronal activity). Dynamic regulation of the balance between STDP and weight-dependent plasticity is likely a significant feature of cortical processing controlled by neuromodulation. It is imaginable that state-dependent alterations in neuromodulation shift regimes of plasticity and learning toward those dominated by associative rules or toward those dominated by homeostatic forces to suit the needs of the organism and according to the brain state.
Footnotes
This work was supported by National Institutes of Health Grant R01 MH087631 to M.V. and M.B. M.V. was partially supported by a Humboldt Research Award from the Alexander von Humboldt Foundation.
The authors declare no competing financial interests.
References
- Bannon NM, Zhang P, Ilin V, Chistiakova M, Volgushev M (2014) Modulation of synaptic transmission by adenosine in layer 2/3 of the rat visual cortex in vitro. Neuroscience 260:171–184. 10.1016/j.neuroscience.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ (2002) Model of thalamocortical slow-wave sleep oscillations and transitions to activated states. J Neurosci 22:8691–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benington JH, Kodali SK, Heller HC (1995) Stimulation of A1 adenosine receptors mimics the electroencephalographic effects of sleep deprivation. Brain Res 692:79–85. 10.1016/0006-8993(95)00590-M [DOI] [PubMed] [Google Scholar]
- Bjorness TE, Greene RW (2009) Adenosine and sleep. Curr Neuropharmacol 7:238–245. 10.2174/157015909789152182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorness TE, Kelly CL, Gao T, Poffenberger V, Greene RW (2009) Control and function of the homeostatic sleep response by adenosine A1 receptors. J Neurosci 29:1267–1276. 10.1523/JNEUROSCI.2942-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundon JA, Zakharenko SS (2013) Presynaptic gating of postsynaptic synaptic plasticity A plasticity filter in the adult auditory cortex. Neuroscientist 19:465–478. 10.1177/1073858413482983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundon JA, Bayazitov IT, Zakharenko SS (2011) Presynaptic gating of postsynaptically expressed plasticity at mature thalamocortical synapses. J Neurosci 31:16012–16025. 10.1523/JNEUROSCI.3281-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbély AA, Achermann P (1999) Sleep homeostasis and models of sleep regulation. J Biol Rhythms 14:559–570. 10.1177/074873099129000894 [DOI] [PubMed] [Google Scholar]
- Chen JY, Chauvette S, Skorheim S, Timofeev I, Bazhenov M (2012) Interneuron-mediated inhibition synchronizes neuronal activity during slow oscillation. J Physiol 590:3987–4010. 10.1113/jphysiol.2012.227462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Lonjers P, Lee C, Chistiakova M, Volgushev M, Bazhenov M (2013) Heterosynaptic plasticity prevents runaway synaptic dynamics. J Neurosci 33:15915–15929. 10.1523/JNEUROSCI.5088-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38:461–472. 10.1016/S0896-6273(03)00235-6 [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE (2006) Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci 29:37–76. 10.1146/annurev.neuro.29.051605.112834 [DOI] [PubMed] [Google Scholar]
- Chistiakova M, Bannon NM, Bazhenov M, Volgushev M (2014) Heterosynaptic plasticity: multiple mechanisms and multiple roles. Neuroscientist 20:483–498. 10.1177/1073858414529829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakova M, Bannon NM, Chen JY, Bazhenov M, Volgushev M (2015) Homeostatic role of heterosynaptic plasticity: models and experiments. Front Comput Neurosci 9:89. 10.3389/fncom.2015.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakova M, Volgushev M (2009) Heterosynaptic plasticity in the neocortex. Exp Brain Res 199:377–390. 10.1007/s00221-009-1859-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias RB, Rombo DM, Ribeiro JA, Henley JM, Sebastião AM (2013) Adenosine: setting the stage for plasticity. Trends Neurosci 36:248–257. 10.1016/j.tins.2012.12.003 [DOI] [PubMed] [Google Scholar]
- Duguid I, Sjöström PJ (2006) Novel presynaptic mechanisms for coincidence detection in synaptic plasticity. Curr Opin Neurobiol 16:312–322. 10.1016/j.conb.2006.05.008 [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24:31–55. 10.1146/annurev.neuro.24.1.31 [DOI] [PubMed] [Google Scholar]
- Fellin T, Halassa MM, Terunuma M, Succol F, Takano H, Frank M, Moss SJ, Haydon PG (2009) Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc Natl Acad Sci U S A 106:15037–15042. 10.1073/pnas.0906419106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG (2009) Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61:213–219. 10.1016/j.neuron.2008.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham NR, Hardingham GE, Fox KD, Jack JJ (2007) Presynaptic efficacy directs normalization of synaptic strength in layer 2/3 rat neocortex after paired activity. J Neurophysiol 97:2965–2975. 10.1152/jn.01352.2006 [DOI] [PubMed] [Google Scholar]
- Huber R, Ghilardi MF, Massimini M, Tononi G (2004) Local sleep and learning. Nature 430:78–81. 10.1038/nature02663 [DOI] [PubMed] [Google Scholar]
- Kempter R, Gerstner W, van Hemmen JL (2001) Intrinsic stabilization of output rates by spike-based Hebbian learning. Neural Comput 13:2709–2741. 10.1162/089976601317098501 [DOI] [PubMed] [Google Scholar]
- Kerr MI, Wall MJ, Richardson MJ (2013) Adenosine A1 receptor activation mediates the developmental shift at layer 5 pyramidal cell synapses and is a determinant of mature synaptic strength. J Physiol 591:3371–3380. 10.1113/jphysiol.2012.244392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolt HP. (2008) Sleep homeostasis: a role for adenosine in humans? Biochem Pharmacol 75:2070–2079. 10.1016/j.bcp.2008.02.024 [DOI] [PubMed] [Google Scholar]
- Lee CM, Stoelzel C, Chistiakova M, Volgushev M (2012) Heterosynaptic plasticity induced by intracellular tetanization in layer 2/3 pyramidal neurons in rat auditory cortex. J Physiol 590:2253–2271. 10.1113/jphysiol.2012.228247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux M, Chen JY, Lonjers P, Bazhenov M, Timofeev I (2014) The impact of cortical deafferentation on the neocortical slow oscillation. J Neurosci 34:5689–5703. 10.1523/JNEUROSCI.1156-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt D, Xu Q, Liu W, Takano T, Smith NA, Schnermann J, Tieu K, Nedergaard M (2012) Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc Natl Acad Sci U S A 109:6265–6270. 10.1073/pnas.1120997109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31:463–475. 10.1016/S0896-6273(01)00375-0 [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Sejnowski TJ (1996) Influence of dendritic structure on firing pattern in model neocortical neurons. Nature 382:363–366. 10.1038/382363a0 [DOI] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA (2007) Opioids block long-term potentiation of inhibitory synapses. Nature 446:1086–1090. 10.1038/nature05726 [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Hawkins RD, Kandel ER, Arancio O (1991) Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci U S A 88:11285–11289. 10.1073/pnas.88.24.11285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajski ML, Venton BJ (2013) The mechanism of electrically stimulated adenosine release varies by brain region. Purinergic Signal 9:167–174. 10.1007/s11302-012-9343-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KG, Hardingham NR, Fox K (2008) Postsynaptic action potentials are required for nitric-oxide-dependent long-term potentiation in CA1 neurons of adult GluR1 knock-out and wild-type mice. J Neurosci 28:14031–14041. 10.1523/JNEUROSCI.3984-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Kalinchuk AV (2011) Adenosine, energy metabolism and sleep homeostasis. Sleep Med Rev 15:123–135. 10.1016/j.smrv.2010.06.005 [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, McCarley RW (2000) Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience 99:507–517. 10.1016/S0306-4522(00)00220-7 [DOI] [PubMed] [Google Scholar]
- Rétey JV, Adam M, Honegger E, Khatami R, Luhmann UF, Jung HH, Berger W, Landolt HP (2005) A functional genetic variation of adenosine deaminase affects the duration and intensity of deep sleep in humans. Proc Natl Acad Sci U S A 102:15676–15681. 10.1073/pnas.0505414102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin J, Lee DD, Sompolinsky H (2001) Equilibrium properties of temporally asymmetric Hebbian plasticity. Phys Rev Lett 86:364–367. 10.1103/PhysRevLett.86.364 [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA (2015) Neuromodulation and metamodulation by adenosine: impact and subtleties upon synaptic plasticity regulation. Brain Res 1621:102–113. 10.1016/j.brainres.2014.11.008 [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB (2004) Endocannabinoid-dependent neocortical layer-5 LTD in the absence of postsynaptic spiking. J Neurophysiol 92:3338–3343. 10.1152/jn.00376.2004 [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB (2001) Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron 32:1149–1164. 10.1016/S0896-6273(01)00542-6 [DOI] [PubMed] [Google Scholar]
- Song S, Abbott LF (2001) Cortical development and remapping through spike timing-dependent plasticity. Neuron 32:339–350. 10.1016/S0896-6273(01)00451-2 [DOI] [PubMed] [Google Scholar]
- Song S, Miller KD, Abbott LF (2000) Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat Neurosci 3:919–926. 10.1038/78829 [DOI] [PubMed] [Google Scholar]
- Steriade M. (2001) Active neocortical processes during quiescent sleep. Arch Ital Biol 139:37–51. [PubMed] [Google Scholar]
- Strecker RE, Morairty S, Thakkar MM, Porkka-Heiskanen T, Basheer R, Dauphin LJ, Rainnie DG, Portas CM, Greene RW, McCarley RW (2000) Adenosinergic modulation of basal forebrain and preoptic/anterior hypothalamic neuronal activity in the control of behavioral state. Behav Brain Res 115:183–204. 10.1016/S0166-4328(00)00258-8 [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C (2006) Sleep function and synaptic homeostasis. Sleep Med Rev 10:49–62. 10.1016/j.smrv.2005.05.002 [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C (2012) Time to be SHY? Some comments on sleep and synaptic homeostasis. Neural Plast 2012:415250. 10.1155/2012/415250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tononi G, Cirelli C (2014) Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81:12–34. 10.1016/j.neuron.2013.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urry E, Landolt HP (2015) Adenosine, caffeine, and performance: from cognitive neuroscience of sleep to sleep pharmacogenetics. Curr Top Behav Neurosci 25:331–366. 10.1007/7854_2014_274 [DOI] [PubMed] [Google Scholar]
- Van Gompel JJ, Bower MR, Worrell GA, Stead M, Chang SY, Goerss SJ, Kim I, Bennet KE, Meyer FB, Marsh WR, Blaha CD, Lee KH (2014) Increased cortical extracellular adenosine correlates with seizure termination. Epilepsia 55:233–244. 10.1111/epi.12511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rossum MC, Bi GQ, Turrigiano GG (2000) Stable Hebbian learning from spike timing-dependent plasticity. J Neurosci 20:8812–8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgushev M, Voronin LL, Chistiakova M, Singer W (1997) Relations between long-term synaptic modifications and paired-pulse interactions in the rat neocortex. Eur J Neurosci 9:1656–1665. 10.1111/j.1460-9568.1997.tb01523.x [DOI] [PubMed] [Google Scholar]
- Volgushev M, Balaban P, Chistiakova M, Eysel UT (2000) Retrograde signaling with nitric oxide at neocortical synapses. Eur J Neurosci 12:4255–4267. 10.1046/j.0953-816X.2000.01322.x [DOI] [PubMed] [Google Scholar]
- Volgushev M, Chen JY, Ilin V, Goz R, Chistiakova M, Bazhenov M (2016) Partial breakdown of input specificity of STDP at individual synapses promotes new learning. J Neurosci 36:8842–8855. 10.1523/JNEUROSCI.0552-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G (2011) Local sleep in awake rats. Nature 472:443–447. 10.1038/nature10009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall M, Dale N (2008) Activity-dependent release of adenosine: a critical re-evaluation of mechanism. Curr Neuropharmacol 6:329–337. 10.2174/157015908787386087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Bannon NM, Ilin V, Volgushev M, Chistiakova M, (2015) Adenosine effects on inhibitory synaptic transmission and excitation-inhibition balance in the rat neocortex. J Physiol 593:825–841. 10.1113/jphysiol.2014.279901 [DOI] [PMC free article] [PubMed] [Google Scholar]