

Abstract
In screening a library of plant extracts from ~1000 species native to the Southeastern United States, Lobelia cardinalis was identified as containing nicotinic acetylcholine receptor (nicAchR) binding activity which was relatively non-selective for the α4β2- and α7-nicAchR subtypes. This nicAchR binding profile is atypical for plant-derived nicAchR ligands, the majority of which are highly selective for α4β2-nicAchRs. Its potential therapeutic relevance is noteworthy since agonism of α4β2- and α7-nicAchRs is associated with anti-inflammatory and neuroprotective properties. Bioassay-guided fractionation of L. cardinalis extracts led to the identification of lobinaline, a complex binitrogenous alkaloid, as the main source of the unique nicAchR binding profile. Purified lobinaline was a potent free radical scavenger, displayed similar binding affinity at α4β2- and α7-nicAchRs, exhibited agonist activity at nicAchRs in SH-SY5Y cells, and inhibited [3H]-dopamine (DA) uptake in rat striatal synaptosomes. Lobinaline significantly increased fractional [3H] release from superfused rat striatal slices preloaded with [3H]-DA, an effect that was inhibited by the non-selective nicAchR antagonist mecamylamine. In vivo electrochemical studies in urethane-anesthetized rats demonstrated that lobinaline locally applied in the striatum significantly prolonged clearance of exogenous DA by the dopamine transporter (DAT). In contrast, lobeline, the most thoroughly investigated Lobelia alkaloid, is an α4β2-nicAchR antagonist, a poor free radical scavenger, and is a less potent DAT inhibitor. These previously unreported multifunctional effects of lobi-naline make it of interest as a lead to develop therapeutics for neuropathological disorders that involve free radical generation, cholinergic, and dopaminergic neurotransmission. These include neurodegenerative conditions, such as Parkinson’s disease, and drug abuse.
Keywords: Lobelia cardinalis, Multifunctional alkaloid, Nicotinic acetylcholine receptors, Dopamine transporter, Free radical scavenger, Dopaminergic neurodegeneration
Graphical Abstract
1. Introduction
Plants are a rich source of nicotinic acetylcholine receptor (nicAchR) ligands used as medicines, drug leads, and/or pharmacological probes [1.]. In plants, metabolites active at nicAchRs are believed to function as chemical defenses against herbivorous insects [2–4]. Nicotine (NIC) is a well-known example of a naturally occurring insecticide present in Nicotiana tabacum (tobacco) that, upon ingestion, targets and activates nicAchRs present in the insect central nervous system (CNS) producing aversive stimuli and/or death [2, 4]. NIC also activates nicAchRs present in the human CNS, which underlies its rewarding and neuroprotective properties [4, 5]. The latter effect has generated interest in the development of nicAchR agonists as neuroprotective agents [6–13]. NIC itself is undergoing evaluation to assess its therapeutic efficacy in early stage of Parkinson’s disease (PD) patients (https://www.michaeljfox.org/) [6].
The neuroprotective effects of nicAchR ligands are primarily a function of agonist activity at α4β2- and α7-nicAchR subtypes [6, 7, 9–14]. Considering plants are known to synthesize nicAchR ligands of astonishing complexity and diversity, the screening of plant extracts to discover novel nicAchRs ligands with potential as neuroprotective drug leads seems logical [1, 15]. However, the majority of nicAchR ligands that have been isolated from plant sources are either α4β2-nicAchRs selective agonists (e.g. cytisine), or selective antagonists at α7-nicAchRs (e.g. methyllycaconitine) [1, 16–18]. Agonists selective for α4β2-nicAchRs are likely to induce dependence, whereas some α7-nicAchR selective antagonists are associated with toxicity [15, 16, 19–21]. Therefore, development of a screen that enables rapid identification of plant extracts which contain metabolites with appropriate nicAchR selectivity is necessary to efficiently discover novel neuroprotective drug leads from plant sources [15].
In the present study, high-throughput pharmacological screening (HTPS) was performed on a library of aqueous plant extracts prepared from ~1,000 species in an effort to discover novel nicAchR ligands with greater therapeutic potential as neuroprotective agents, as compared to previously investigated ligands. The “differential smart screen” (DSS), as previously described by Littleton et al. (2005), measures a plant extract’s binding activity at α4β2- and α7-nicAchRs, yielding a differential displacement ratio (DDR) indicative of nicAchR selectivity [15]. The DDR was previously utilized to identify plant extracts containing metabolites selective for α7-nicAchRs, although it can be readily applied to identify extracts containing metabolites with equipotent binding activity at α4β2- and α7-nicAchRs (see Methods below) [15]. The latter may fully exploit neuroprotection afforded by nicAchR agonists via activation of both receptor subpopulations associated with NIC’s neuroprotective effects [7, 11, 13, 14].
Several previously uninvestigated plant species were identified in the HTPS as having activity meriting further investigation. Lobelia cardinalis was one of these displaying activity indicative of the presence of metabolites with equipotent binding activity at α4β2- and α7-nicAchRs. Furthermore, the extract from L. cardinalis induced 45Ca2+ uptake via nicAchR activation in SH-SY5Y cells, indicating the metabolite/s present functioned as an agonist/s [22]. Lobelia alkaloids have previously been described as inhibitors of the dopamine transporter (DAT), thus the extract from L. cardinalis was screened for this activity [23–25]. Indeed, the extract significantly inhibited DAT-mediated [3H]-dopamine (DA) uptake in rat striatal synaptosomes. This combination of pharmacological activities is potentially of considerable value for the development of neuroprotective agents targeted on the dopaminergic (DAergic) neurodegeneration that occurs in PD, and psychostimulant-induced DAergic neurotoxicity [6–13, 26–33].
Native Americans knew the potential medicinal value of L. cardinalis, although the uses show no clear relation to the pharmacology described herein. Formulations of the plant were consumed by tribes for a wide variety of purposes, ranging from its use as an emetic, a remedy for the treatment of typhoid, and even as a “love potion” [34]. In addition, a close relative of L. cardinalis, L. inflata, is the source of lobeline [25]. Lobeline has been generated interest as a treatment for neurodegenerative disorders, such as PD and Alzheimer’s disease, as well as neuropsychiatric disease including psychostimulant dependence and attention deficit hyperactivity disorder [25]. Bioactive metabolites originating from species of the Genus Lobelia may hold therapeutic potential for a variety of neurological disorders.
Here, the identification of the major bioactive metabolite present in L. cardinalis, lobinaline, is described, as well as the in vitro characterization of its effects on nicAchRs and the DAT. The alkaloid’s effects on DA uptake in vivo were examined by measuring the clearance of exogenous DA locally applied in the striatum of isoflurane-anesthetized rats using Nafion-coated carbon fiber microelectrodes in combination with high-speed chronoamperometry (HSC) [35–41]. Since excessive free radical production contributes to DAergic neurotoxicity seen in PD and psychostimulant abuse, lobinaline’s capacity to scavenge free radicals was evaluated in the 1,1-diphenyl-2-picrylhydracyl (DPPH) free radical scavenging assay [28, 30, 33, 42–44]. All of the data presented suggest lobinaline is a potential lead compound for the development of multifunctional neuroprotective agents to prevent DAergic neurotoxicity.
2. Materials and Methods
2.1. Chemicals and Supplies
Methanol, hexane, chloroform, ethyl acetate, butanol, acetonitrile, (−)-nicotine (NIC), methyllycaconitine (MLA) citrate salt hydrate, mecamylamine (MEC) hydrochloride, nomifensine, (−)-lobeline hydrochloride, and 2,2-diphenyl-1-picrylhydrazyl (DPPH) were purchased from Sigma Aldrich (St. Louis, MO, USA). Streptomycin (10,000 μg/ml), penicillin (10,000 units/ml), fetal bovine serum (FBS), and Dulbecco’s Modified Eagle Medium (DMEM) were purchased from Life Technologies Corporation (Grand Island, NY, USA). Quercetin was purchased from Chromadex (Irvine, CA, USA). [3H]-epibatidine (S.A. = 30 Ci/mmol), [3H]-cytisine (S.A. = 16 Ci/mmol), [3H]-MLA (S.A. = 60 Ci/mmol), [3H]-GBR12935 (S.A. = 40 Ci/mmol), 45CaCl2 (S.A. = 12.05 mCi/mg), and [3H]-DA (S.A. = 60 Ci/mmol) were purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA). All other chemicals and materials were purchased from Fisher Scientific (Pittsburgh, PA, USA), unless otherwise stated.
2.2. Animals
Adult, male Sprague-Dawley rats (200 – 250 g) purchased from Harlan Laboratories (Indianapolis, IN, USA) were housed in cages in groups of 3 – 4 at the Division of Laboratory Animal Resources at the University of Kentucky. Animals had access to food and water ad libitum. All protocols for the handling, care, and use of animals were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Ken-tucky and were performed in accordance with the National Institute of Health’s Guide for Care and Use of Laboratory Animals.
2.3. Collection of Plant Material
Plant samples were collected as previously described by Littleton et al. (2005) [15]. Briefly, plant samples were collected from field sites by a highly qualified botanist. GPS coordinates were recorded for each accession. Samples were snap-frozen with liquid nitrogen immediately after field collection, and stored at −80°C prior to solvent extraction. Reference samples of each species were deposited at the University of Kentucky Herbarium. Lobelia cardinalis was grown at the University of Kentucky College of Agriculture Spindletop Farm to obtain plant material in bulk.
2.4. Aqueous plant extract library
A Plant extract library was prepared as described previously [15]. Briefly, snap-frozen plant samples collected from the field were removed from storage at −80°C, immediately freeze-dried using a lyophilizer, and finely powdered. Powdered plant material was extracted (100 mg/ml) by suspending samples in aqueous solution (100% Milli-Q H2O) and placing them on a shaker overnight. The following day, aqueous extracts were collected via vacuum filtration, and freeze-dried using a lyophilizer. The resulting residue was re-suspended in an appropriate volume of assay buffer to achieve a final concentration of 100 mg/ml. When necessary, dimethyl-sulfoxide (DMSO) was added (<0.05%) to promote solubility of samples in aqueous solution. The resulting aqueous extracts were stored at −80°C prior to pharmacological screening.
2.5. HTPS: Differential smart screen
HTPS was performed on the plant extract library using a Packard Multiprobe liquid handling system in a 96-well plate format (350 μl well volume) as previously described, with minor modifications [15, 18, 45, 46]. DSS was employed to identify extracts that contained nicAchR ligands with a pharmacological profile unlike that of NIC [15]. Briefly, pure compounds or plant extracts were prepared, and dissolved in a volume of modified Krebs-Ringers buffer (50 mM Tris-HCl, 144 mM NaCl, 1.5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 20 mM HEPES, pH 7.4) necessary to prepare a 100 mg/ml stock solution (see section 2.4). Dilutions of the stock solution were prepared for screening (1:10, 1:20, 1:50, 1:100, 1:200, 1:500, 1:1,000, 1:2,000). Samples were evaluated over this concentration range to determine the dilution that effectively displaced 50% of [3H]-epibatidine (168 pM, 3 hour incubation period) from rat cortical membranes (ID50). [3H]-epibatidine is a potent, high affinity nicAchR ligand with similar binding affinity at most nicAchRs present in the mammalian CNS [47, 48]. Determination of a plant species’ ID50 enabled the identification of extracts containing nicAchR ligands, and provided a “reference point” for subsequent binding studies [15]. Plant extracts that inhibited [3H]-epibatidine binding were tested for their ability to displace [3H]-cytisine (1 nM, 1 hour incubation) and [3H]-MLA (2 nM, 2 hour incubation) at a concentration equal to their ID50 for [3H]-epibatidine displacement. [3H]-cytisine and [3H]-MLA displacement studies were performed in rat cortical and hippocampal membranes, respectively. [3H]-cytisine is a β2-subtype selective ligand, and thus will reflect mainly α4β2-nicAchR binding in the mammalian CNS, whereas [3H]-MLA is α7-nicAchR selective ligand [1, 16–18]. The final protein concentration of membrane preparations in HTPS assays was 150 μg/ml. After reaching equilibrium, membranes were harvested onto 96-well GF/B filtration plates (PerkinElmer Inc., Waltham, MA, USA) and rapidly washed three times with ice-cold 50 mM Tris-HCl buffer (pH 7.4). Filtration plates were allowed to dry overnight before adding 35 μl of scintillation fluid (Microscint 20, Packard Inc.). Plates were then kept in the dark for 2 hours, after which radioactivity was measured using a Packard TopCount® NXT™ microplate scintillation counter. Non-specific binding was measured in the presence of excess NIC (300 μM) and total binding was measured in the presence of radioligand alone. Total specific binding and specific binding in the presence of competitors was calculated by subtracting non-specific binding. Specific binding in the presence of a competitor was expressed as a percentage to total specific binding. Dividing the displacement percentage of [3H]-cytisine by that of [3H]-MLA at a concentration equal to the ID50 for [3H]-epibatidine displacement yields a DDR indicative of nicAchR subtype selectivity. DDR values > 5 indicate the plant extract contains metabolites selective for α4β2-nicAchRs, while DDR values < 1 indicate the presence of metabolites selective for α7-nicAchR, as previously described [4, 15]. Likewise, the DDR can also be used to identify plants extracts containing metabolites with equipotent binding activity at α4β2- and α7-nicAchRs. In theory, a DDR value of ~3 should indicate the presence of nicAchR ligands with equipotent binding affinity at α4β2- and α7-nicAchRs. In support of this hypothesis, anabasine, a nicAchR ligand with relatively equipotent binding affinity at α4β2- and α7-nicAchRs (Ki = 65 and 58 nM, respectively), was previously reported to have a DDR value of 4.16 [1, 15]. Similarly, aqueous extracts from Nicotiana species with greater amounts of NIC than anabasine produce DDR values of > 5, whereas those having greater quantities of anabasine produce a DDR value of ~3 (see Table 1 for comparison) [15, 49]. Therefore, plant extracts that produced DDR values of ~3 were predicted to contain metabolites with equipotent binding affinity at α4β2- and α7-nicAchRs, thus warranting further investigation in the present study. Aqueous extracts from species of interest were evaluated in vitro for their ability to activate nicAchRs and inhibit the DAT (see sections 2.12 and 2.13). Plant species whose extracts displayed a combination of the aforementioned pharmacological activities (presence of a ligand with similar binding affinity at α4β2- and α7-nicAchRs, ability to activate nicAchRs, and inhibition of the DAT) were prioritized for further evaluation.
Table 1.
DDR values, anabasine, and nicotine content of select Nicotiana species
| Species | Nicotine : Anabasine, % of total alkaloids1 | DDR Value2 |
|---|---|---|
| N. tabacum | 94.8 : 0.3 | 9.5 |
| N. undulate | 95.3 : 1.3 | 6.7 |
| N. velutina | 2.8 : 8.1 | 2.9 |
Alkaloid content reported by Saitoh et al. (1984)
DDR values reported by Littleton et al. (2005)
2.6. Fractionation of the L. cardinalis crude methanolic extract
Air-dried aerial portions of L. cardinalis were ground to a coarse powder, and extracted with 9 volumes of methanol (3 volumes per extraction, 24 hours per extraction). The methanolic extract was dried under vacuum using a rotary evaporator, resuspended in water, and partitioned with organic solvents of increasing polarity in the order that follows: hexane, chloroform (CHCl3), ethyl acetate, and butanol. Sodium sulfate was added to the organic fractions to remove any residual water, and each organic fraction was dried under vacuum. The remaining aqueous phase was freeze-dried using a lyophilizer. Fractions were re-suspended in modified Krebs-Ringers buffer (final concentration, 100 mg/ml) and assessed in the DSS (see section 2.5). Additionally, fractions were re-suspended in uptake buffer (final concentration, 100 mg/ml) and assessed for their ability to inhibit DAT-mediated [3H]-DA uptake in rat striatal synaptosomes (see section 2.13).
2.7. pHPLC sub-fractionation of the L. cardinalis CHCl3 fraction
The CHCl3 fraction obtained from the L. cardinalis crude methanolic extract was sub-fractionated via pHPLC using a Waters XBridge Prep C18 (5 μm OBD, 19 × 150 mm) column attached to a Waters 600E Multi-solvent Delivery System coupled to a Waters 2998 Photodiode Array Detector and Waters 2767 Sample Manager, Injector, and Collector. The pHPLC instrument was operated using Waters MassLynx Software (Version 4.1) and FractionLnyx Collection Control Software (Version 4.1). The mobile phase consisted of a mixture of Solvent A (100% Milli-Q water, pH 7.0) and Solvent B (100% acetonitrile, HPLC grade). Separation was performed with the following gradient at a flow rate of 7 ml/minute: initial conditions, 1% B in A; 0 – 6 minutes, linear gradient, 1 – 20% B in A; 6 – 12 minutes, linear gradient, 20 – 40% B in A; 12 – 18 minutes, linear gradient, 40 – 50% B in A; 18 – 24 minutes, linear gradient, 50 – 75% B in A; 24 – 30 minutes, linear gradient, 75 – 95% B in A; 30 – 35 minutes, linear gradient, 95 – 100% B in A; 35 – 43 minutes, isocratic gradient, 100% B. Sub-fractions were dried, and then re-suspended in modified Krebs-Ringer’s buffer or uptake buffer (333 μg/ml) for nicAchR binding studies and [3H]-DA uptake studies, respectively (see section 2.13).
2.8. Isolation of lobinaline
The CHCl3 fraction was obtained from L. cardinalis, as described above. Acid-base extraction was performed on the CHCl3 fraction to obtain lobinaline. Briefly, 100 mg of the dried CHCl3 fraction was dissolved in 50 ml of CHCl3. The solution was acidified with 1 M HCl (final pH, 2 – 3), shaken gently, and the organic phase was discarded. The remaining aqueous phase was washed 2 – 3 times with CHCl3. The aqueous phase was subsequently basified with ammonium hydroxide (final pH, 10 – 12), partitioned between CHCl3 and water, and the resulting organic phase was collected. The basified aqueous layer was extracted two additional times with CHCl3, and each of the organic phases obtained from the basified aqueous phase were combined. Sodium sulfate was added to the combined organic phases to remove any residual water, removed by vacuum filtration, and the organic phase was dried under vacuum using a rotary evaporator. Lobinaline obtained using this method was analyzed using gas chromatographic-mass spectrometric (GC-MS) methods (see section 2.9).
2.9. GC-MS analysis
GC-MS analyses were performed using a Hewlett Packard 6890 Gas Chromatogram interfaced to a Hewlett Packard 5973 Series Mass Selective Detector, an Agilent Technologies 7683 Series Injector, and a Hewlett Packard 7683 Series Autosampler. ChemStation Software (Version 1.02.06) and the Wiley Spectral Database (Version 4.0) were used for instrument control, data analysis, and structural elucidation. Separation was performed on a HP-5MS column ((5%-phenyl)-methylpolysiloxane; 30.0 m × 320 μm × 0.25 μm). Ultra-high purity helium (flow rate of 1.2 ml/minute) served as the carrier gas. Sample volumes of 1 μl were injected in split mode (split ratio, 10.0:1; split flow 12.3 ml/minute) at an inlet pressure of 1.60 psi. The inlet temperature was held at 250°C. The oven was operated using the following parameters: initial temperature, 80°C; 80°C, 2 minute hold; 10°C/minute to 160°C, 1 minute hold; 60°C/minute to 275°C, 12 minute hold; 60°C/min to 60°C, 0 minute hold; total runtime, 28.50 minutes. The transfer line temperature was held at 280°C. The L. cardinalis CHCl3 pHPLC sub-fraction of interest (based on its DDR value and DAT inhibitory activity) was analyzed via GC-MS to identify the most abundant constituent/s present. Lobinaline isolated from crude plant material was injected at 2 mg/ml, two runs per sample. Purity of the alkaloid was determined by integrating the area under the curve (AUC) of lobinaline’s chromatographic peak (GC-MS run in TIC mode). The identity of the alkaloid was confirmed based on previously reported MS fragmentation data for lobinaline [50].
2.10. [3H]-Epibatidine, [3H]-cytisine, and [3H]-MLA binding
Radioligand displacement studies with pure compounds were performed using methods described previously [51]. Briefly, adult male Sprague-Dawley (200 – 250 g) rats were anesthetized with CO2 and decapitated. Hippocampal and cortical tissues were rapidly dissected, placed in 10 volumes of ice-cold sucrose buffer (0.32 M sucrose, 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride, 0.01% w/v sodium azide, pH 7.4), and homogenized in a glass homogenization tube with a Teflon pestle. The homogenate was centrifuged at 1,000 × g for 10 minutes at 4°C, and the supernatant was placed on ice. The pellet was re-suspended in sucrose buffer, and centrifuged again at 1,000 × g for 10 minutes at 4°C. The two supernatants were combined and the pellet was discarded. The combined supernatants were centrifuged at 50,000 × g for 10 minutes at 4°C. The resulting supernatant was discarded, and the pellet was washed twice with assay buffer (50 mM Tris-HCl, 144 mM NaCl, 1.5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 20 mM HEPES, pH 7.4). The total protein content was measured using the bicinchoninic acid kit (Sigma Aldrich). The final protein concentration was adjusted to 3 mg/ml with assay buffer, and membranes were stored at −80°C prior to further experimentation.
On the day which competition binding studies were performed, membrane preparations were removed from storage at −80°C and thawed on ice. Lobinaline (final concentration, 3.4 mM – 3.4 nM) was dissolved in assay buffer. DMSO (< 0.1%) was added to promote solubility. Solutions containing lobinaline were mixed with membrane (final protein concentration, 1 mg/ml) and radioligand (168 pM [3H]-epibatidine, 3 hour incubation; 2 nM [3H]-cytisine, 2 hour incubation; 2 nM [3H]-MLA, 2 hour incubation) in individual wells in a 96-well plate format. Experiments were performed at room temperature. After reaching equilibrium (2 – 3 hours), membranes were harvested onto 96-well GF/B filter plates (PerkinElmer Inc., Waltham, MA, USA) by vacuum filtration, and rapidly washed three times with ice-cold 50 mM Tris-HCl buffer (pH 7.4). Filter plates were allowed to dry overnight. The following day, 35 μl of scintillation fluid was added to each well (Mircoscint 20, Packard Inc.) and plates were placed in the dark for two hours. Afterward, radioactivity was measured by scintillation counting (2 minutes per well) using a Packard TopCount® NXT™ microplate scintillation counter. In each plate, non-specific binding was measured in the presence of excess NIC (final concentration, 300 μM) and total binding was measured with radioligand alone. Total specific binding and specific binding in the presence of lobinaline was calculated by subtracting non-specific binding. Specific binding in the presence of lobinaline was expressed as a percentage of total specific binding. Binding studies with pHPLC sub-fractions were conducted with essentially the same methods with the exception that only [3H]-MLA binding was performed, whereas the displacement of all three nicAchR ligands was characterized for lobinaline. The CHCl3 sub-fraction that was the most effective at displacing [3H]-MLA was analyzed via GC-MS (see section 2.9) to determine the major constituent/s present.
2.11. [3H]-GBR12935 binding
Competition binding studies were conducted using previously described methods with minor modifications [52–54]. Membranes were prepared as described (see section 2.10) with the exception that striatal tissue was collected, homogenized in ice-cold homogenization buffer (120 mM NaCl, 50 mM Tris-HCl, pH = 7.4), centrifuged at 25,000 × g for 20 minutes at 4°C, then washed thrice with assay buffer (120 mM NaCl, 50 mM Tris-HCl, 0.01% FBS, pH 7.4). Each wash was performed by re-suspending the pellet 10 volumes of assay buffer followed by centrifugation at 25,000 × g for 20 minutes at 4°C. Total protein content was measured using the bicinchoninic acid kit (Sigma Aldrich). The final protein concentration was adjusted to 3 mg/ml with assay buffer. Striatal membranes were stored at −80°C prior to further experimentation.
On the day binding experiments were performed, membranes were removed from storage at −80°C and thawed on ice. Lobinaline (final concentration, 1.0 mM – 0.1 nM) was dissolved in assay buffer. DMSO (< 0.1%) was added to promote solubility. Solutions containing lobinaline were mixed with membrane (final protein concentration, 1 mg/ml) and incubated for 15 minutes prior to the addition of radioligand (1 nM [3H]-GBR12935, 1 hour incubation) in a 96-well plate format. Experiments were performed at room temperature. After reaching equilibrium, membranes were harvested onto 96-well GF/B filter plates (PerkinElmer Inc., Waltham, MA, USA) by vacuum filtration, and rapidly washed three times with ice-cold 50 mM Tris-HCl buffer (pH 7.4). Filter plates were pretreated with a solution of 0.1% polyethyleneimine 1 hour prior to harvesting membranes to reduce non-specific binding. Filter plates were allowed to dry overnight. The following day, 35 μl of scintillation fluid (Microscint 20, Packard Inc.) was added to each well and plates were placed in the dark for two hours. Afterward, radioactivity was measured by scintillation counting (2 minutes per well) using a Packard TopCount® NXT™ microplate scintillation counter. In each plate, non-specific binding was measured in the presence of excess GBR12909 (final concentration, 10 μM) and total binding was measured with radioligand alone. Total specific binding and specific binding in the presence of lobinaline was calculated by subtracting non-specific binding. Specific binding in the presence of lobinaline was expressed as a percentage of total specific binding.
2.12. 45Ca2+ entry in SH-SY5Y cells
Cell-based assays evaluating functional activity of pure compounds or extracts at nicAchRs were conducted using previously described methods with modifications [55–57]. SH-SY5Y human neuroblastoma cells were sub-cultured in polylysine coated petri dishes containing culture medium (DMEM supplemented with 10% FBS, 1 mM glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin) and maintained in a incubator at 37°C in a humidified atmosphere (95% O2/5% CO2). The day prior to performing 45Ca2+ entry studies, SH-SY5Y cells were plated onto 24-well polylysine coated plates containing 300 μl of culture medium at a seeding density of 20,000 cells/well and placed back in the incubator. After allowing 24 hours for cells to adhere, culture medium was aspirated and replaced with 300 μl of assay buffer (130 mM NaCl, 5 mM KCl, 6 mM glucose, 20 mM HEPES, and 1 mM CaCl2, pH 7.4). Pure compounds or extracts were dissolved in assay buffer to allow direct addition to wells. DMSO (final concentration, < 0.1%) was added to promote solubility of extracts or compounds. Control and treatment groups were performed in quadruplicate on each plate. SH-SY5Y cells were pretreated with vehicle, pure compounds, and/or extracts for 1 minute prior to the addition of 45Ca2+ (~5 μCi). 45Ca2+ entry was terminated by aspirating assay buffer, and then washing cells thrice with 1 ml of ice-cold assay buffer. Cells were lysed overnight by the addition 0.5 M NaOH (0.5 ml/well). Lysates were pooled for each group, and two 100 μl aliquots were counted per group. Radioactivity was measured by scintillation counting using a Packard Tri-Carb Liquid Scintillation Counter (Gaithersberg, MD, USA). Basal 45Ca2+ entry was designated as that observed in vehicle treated cells. 45Ca2+ entry in treatment groups was normalized to that observed in vehicle treated controls.
2.13. DAT-mediated [3H]-DA uptake in rat striatal synaptosomes
In vitro [3H]-DA uptake was performed as described previously with minor modifications [58]. Briefly, adult male Sprague-Dawley rats (200–250 g) were anesthetized with CO2 and decapitated. Striata were rapidly dissected and immediately placed into 10 volumes of ice-cold uptake buffer (120 mM NaCl, 3.9 mM KCl, 650 μM MgSO4, 510 μM CaCl2, 190 μM NaHPO4, 100 μM pargyline, 2 mg/ml glucose, 0.2 mg/ml ascorbic acid, 20 mM HEPES, pH 7.4, saturated with 95% O2/5% CO2) containing 0.32 M sucrose. Striatal tissue was homogenized in a glass homogenization tube with a Teflon pestle. The homogenate was centrifuged at 1,000 × g for 10 minutes at 4°C. The resulting supernatant was collected and centrifuged at 16,000 × g for 20 minutes at 4°C. The resulting pellet was washed twice with ice-cold uptake buffer and re-suspended in 10 ml of uptake buffer (synaptosome preparation). Synaptosomes (100 μl) were added to individual wells in a 96-well plate and incubated at 37°C for 10 minutes. Lobinaline was dissolved in 100% DMSO, then diluted with uptake buffer (3 mM – 0.3 mM). The final concentration of DMSO in uptake studies never exceeded 1.0%, which had no significant effect on radiotracer uptake using methods outlined in the present study. Lobinaline (100 μl; final concentration, 1 mM – 0.1 nM) was co-incubated with synaptosomes for 10 minutes at 37°C prior to the addition of [3H]-DA (100 μl). After the 10-minute co-incubation, [3H]-DA was added to each well (final concentration, 15 – 30 nM) and uptake was allowed to proceed for 5 minutes at 37°C. Uptake was terminated by placing 96-well plates on ice, and then immediately harvesting synaptosomes onto a 96-well GF/C filter plates (PerkinElmer Inc.) by vacuum filtration, followed by three rapid washes with ice-cold 50 mM Tris-HCl buffer (pH 7.4). After allowing filtration plates to dry overnight, 35 μl of scintillation fluid (Microscint 20, Packard Inc.) was added to each well and the plate was kept in the dark for 2 hours. Subsequently, radioactivity was measured by scintillation counting using a Packard TopCount® NXT™ microplate scintillation counter. Total uptake was measured in the presence of [3H]-DA alone. Non-specific uptake was determined in the presence of 10 μM GBR-12909. Total specific uptake and specific uptake in the presence of inhibitor was calculated by subtracting non-specific uptake from each, respectively. Specific uptake in the presence of inhibitor was expressed as a percentage of total specific uptake. [3H]-DA uptake studies with conducted with extracts, fractions, or CHCl3 sub-fractions were conducted using the same methods. The CHCl3 sub-fraction that most potently inhibited the DAT was analyzed via GC-MS (see section 2.9) to determine the major constituent/s present.
2.14. DPPH free radical scavenging assay
Lobinaline’s capacity to scavenge free radicals was examined using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radical scavenging assay. The DPPH assay was performed as previously described with minor modifications [59]. Briefly, a DPPH stock solution (600 μM) and stock solutions (1 mg/ml) of lobinaline, lobe-line, and the reference compound quercetin were prepared by dissolving compounds in methanol. Stock solutions of DPPH and test compounds were prepared fresh on the day of the experiment. The assay was performed in a 96-well plate format. DPPH solution (final concentration, 300 μM), or DPPH solution and solutions of pure compounds were (final concentration, 500 - 1 μg/ml) added to each plate in quadruplicate. Plates were immediately covered and placed in the dark for 5 minutes. After 5 minutes, the reaction mixture’s absorbance at 517 nm was measured using a Wallac 1420 VICTOR plate reader (PerkinElmer Inc., MA, USA). DPPH free radical scavenging activity was calculated using the following equation: DPPH Free Radical Scavenging Activity (%) = ((Abs1 − Abs2)/Abs1) × 100, where Abs1 is the absorbance of the solution containing DPPH only and Abs2 is the absorbance of the solution containing DPPH and pure compounds.
2.15. Fractional [3H] release from [3H]-DA preloaded striatal slices
The ability of lobinaline to evoke fractional [3H] release from [3H]-DA preloaded striatal slices was examined as previously described, with minor modifications [23, 60]. Briefly, coronal slices of rat striatum (500 μm, 6 – 8 mg) were incubated in Krebs’ buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl2 1.0 mM NaH2PO4, 1.3 mM CaCl2, 11.1 mM glucose, 25 mM NaHCO3, 0.11 mM ascorbic acid, and 0.004 mM EDTA, pH 7.4, saturated with 95%O2/5%CO2) for 30 minutes in a metabolic shaker at 34°C. Afterward, slices were incubated in Krebs’ buffer containing 0.1 μM [3H]-DA (6 – 8 slices/3 ml) for 30 minutes at 34°C. Subsequently, slices were rinsed and transferred to a glass superfusion chamber maintained at 34°C. Slices were superfused with oxygenated Krebs’ buffer containing the monoamine oxidase (MAO) inhibitor pargyline (10 μM) and the DAT inhibitor nomifensine (10 μM) at 1 ml/minute. The inclusion of a MAO inhibitor reduced [3H] signal arising from [3H]-DA metabolites, and the DAT inhibitor prevented the release of [3H]-DA via DAT reversal, promoting the measurement of fractional [3H] release via exocytosis of [3H]-DA from vesicular stores [23, 61]. Slices were super-fused for 60 minutes, and then five 4-minute samples (4 ml) were collected in scintillation vials to determine the level of basal [3H] outflow ensuring baseline was stable.
In studies assessing the lobinaline’s ability to concentration-dependently evoke fractional [3H] release, a single concentration of lobinaline (10, 100, or 1000 μM) was perfused through an individual superfusion chamber after collection of basal samples. Slices treated with lobinaline were superfused with a single concentration of the alkaloid for the remainder of the experiment. In each experiment, one chamber was superfused with vehicle only throughout the entire course of the experiment. After the addition of lobinaline, superfusate samples were collected at 4-minute intervals. A repeated-measures design was utilized for superfusion studies to establish a concentration-response for lobinaline-evoked fractional [3H] release.
After establishing a concentration-response for lobinaline in fractional [3H] release studies, experiments were performed to determine whether MEC (10 μM) could antagonize lobinaline-evoked fractional [3H] release. A repeated-measures design was used for studies evaluating the effect of MEC on lobinaline-evoked fractional [3H] release. Slices were perfused for 60 minutes prior to the collection of two 4-minute samples to determine basal [3H] overflow ensuring baseline was stable. After collection of basal samples, chambers were superfused with vehicle or Krebs’ buffer containing MEC (10 μM) for 20 minutes. After being superfused with vehicle or MEC for 20 minutes, lobinaline (100 μM) was added to a single chamber for the remainder of the experiment. In each experiment performed, one chamber was superfused with vehicle only throughout the entire course of the experiment. Superfusate samples were collected in scintillation vials at 4-mintue intervals.
Upon completion of each superfusion experiment, slices were carefully removed from superfusion chambers and solubilized with 1 ml of TS-2 in scintillation vials. The volume and pH of solubilized striatal slices were adjusted to match that of superfusate samples. Radioactivity in the samples collected during superfusion studies was measured by liquid scintillation using a Packard Tri-Carb Liquid Scintillation Counter (Gaithersberg, MD, USA). Fractional [3H] release was calculated by expressing [3H] collected in superfusate samples as a percentage of [3H] present in solubilized striatal slices upon completion of each superfusion study. Fractional [3H] release evoked by various treatments was compared to vehicle treated control slices.
2.16. In vivo electrochemical studies
In vivo electrochemical studies were conducted using previously described methods to evaluate lobinaline’s ability to modulate DAT function in the dorsal striatum of anesthetized rats [39, 41]. Briefly, HSC measurements were performed using a FAST-16 system (Quanteon, L.L.C., Nicholasville, KY, USA). Square wave pulses, 0.00 V to +0.55 V vs. Ag/AgCl reference electrodes, were applied for 100 milliseconds at a frequency of 5 Hz. Reduction and oxidation currents were integrated during the last 80 milliseconds of each pulse and averaged over 1 second. Carbon-fiber microelectrodes (Quanteon, L.L.C., Nicholasville, KY, USA), constructed as previously described, consisted of a single carbon-fiber (outer diameter, 30 – 33 μm) passed through and sealed in a glass capillary tube (exposed fiber length, ~150 μm). Nafion (5% solution, Aldrich Chemical Co., Milwaukee, WI) coating of the exposed carbon-fiber tip (2 – 3 coats cured in a 200°C-oven, 5-minture cure interval per coat) provided excellent selectivity for DA over other anionic interferents (DA over ascorbic acid ≥ 100:1; DA over DOPAC ≥ 100:1), as previously reported [62]. Microelectrodes were calibrated in vitro in phosphate buffered saline (0.05 M) to generate calibration curves for DA (slope > 0.2 nA/μM and L.O.D < 50 nM) for each microelectrode prior to in vivo studies. Electrode responses to DA were linear (r2 ≥ 0.997). Lobinaline was evaluated in vitro prior to in vivo studies to determine whether the alkaloid was electro-active. For in vivo studies, micropipette-microelectrode assemblies were constructed using sticky wax (Kerr, Orange, CA, USA) to affix double-barrel micropipettes (A-M System, Sequim, WA) to microelectrodes. The distance between micropipette and electrode tips ranged from 250 – 350 μm. Care was taken to ensure parallel, vertical alignment of micropipettes and microelectrodes. Double-barrel micropipettes contained a solution of DA (200 μM in 0.9% saline, pH 7.4) in one pipette and lobinaline (1 mM in 0.9% saline solution containing 0.1% DMSO, pH 7.4; vehicle containing DMSO did not affect DA dynamics) in the second pipette. Animals were anesthetized using isoflurane (1 – 3%) and placed securely in a stereotaxic frame. Body temperature was maintained at 37°C throughout the experiment using a heating pad coupled to a rectal thermometer. Ag/AgCl reference electrodes were implanted in the brain parenchyma at a region remote to recording sites through a burr hole and secured using dental acrylic. Skin overlying the cranium of rats was reflected, and the skull and dura overlying recording sites were removed bilaterally. Micropipette-microelectrode assemblies were implanted in the striatum under stereotactic control using the following coordinates: anterior-poster, +1.5 mm; medial-lateral, ±2.2 mm; dorsal-ventral, −3.8 to −5.4 mm. The atlas of Paxinos and Watson (2007) was used to determine coordinates for stereotaxic placement [63]. Solutions of DA (ejection volume, ~100 nl) or lobinaline (ejection volume, ~250 nl) were pressure ejected using a Picospritzer III (Parker instrumentation) and the volume ejected was monitored with a dissecting microscope equipped with a 10 mm reticule [64]. After implantation, the micropipette-microelectrode assembly was left undisturbed to achieve baseline (~30 minutes) before starting the experiment. DA was then pressure ejected at 5-minute intervals until three reproducible signals were obtained (±10%) at each recording site. Lobinaline was then applied slowly (10 – 15 seconds) to minimize disturbance of electrochemical signals. DA was ejected 1 minute following the lobinaline application, and then at 5-minute intervals thereafter (4 – 5 times). The micropipette-microelectrode assembly was lowered 0.5 mm to obtain additional recordings (4 – 6 recordings per hemisphere). Responses to DA were averaged from each animal and data were analyzed using FAST analysis software (Version 6.0; Quanteon, L.L.C., Nicholasville, KY, USA). Two DA signal parameters were obtained: 1) the T80 (80% decay time from peak response), and 2) the clearance rate, the first order rate of decay of the DA signal multiplied times the peak amplitude. The T80 and the clearance rate reflect alterations in DAT function, rather than diffusion or metabolism [41, 65]. Comparisons were made between amplitude matched DA signals pre- and post-lobinaline application.
2.17. Assessment of lobinaline’s oral “druggability”
The oral “druggability” of a molecule is commonly evaluated using Lipinski’s “Rule of Five” [66]. These criteria enable prediction of a lead molecule’s potential as a drug candidate based on its physiochemical properties. Assessment of lobinaline’s druggability based on these criteria was accessed using data readily available at the PubChem website (https://pubchem.ncbi.nlm.nih.gov/) [67, 68].
2.18. Statistical analysis
Statistical analyses, curve fitting, and graphical presentation of data were performed using GraphPad Prism software (Version 6.0; GraphPad Software, San Diego, CA, USA). The statistical significance of treatment-induced 45Ca2+ entry in SH-SY5Y cells was determined using a one-tailed student’s t-test. The statistical significance of DAT inhibition caused by pretreatment of rat striatal synaptosomes with the LCaq and the LCCHCl3 was determined with a one-tailed student’s t-test. ID50, IC50, and EC50 values for high-throughput DSS, [3H]-DA uptakes studies, and DPPH free radical scavenging assays, respectively, were calculated using non-linear regression analysis to fit data to a variable slope, sigmoidal dose-response curve. Ki values were calculated using the Cheng-Prusoff equation for radioligand binding studies using nonlinear regression analysis to fit data to a one-site competition binding model, sigmoidal concentration-response curve. Repeated-measures, two-way analysis of variance (ANOVA) was performed (lobinaline × time interaction) to determine whether lobinaline evoked a significant, dose-dependent increase in fractional [3H] release from [3H]-DA preloaded striatal slices. Repeated-measures, two-way ANOVA was performed (treatment × time interaction) to determine whether lobinaline-evoked fractional [3H] release was MEC-sensitive. Bonferroni’s post-hoc analysis was used to determine time points at which fractional [3H] release was statistically different from vehicle treated controls. DA signals from in vivo electrochemical studies were obtained using Fast analysis software (Version 6.0; Quanteon, L.L.C., Nicholasville, KY, USA) and GraphPad Prism software was used for graphical presentation of data. DAT inhibitors consistently increase the T80, whereas the clearance rate may increase or decrease depending on experimental conditions [65]. Thus, a paired one-tailed student’s t-test and a paired two-tailed student’s t-test were performed to determine the significance of lobinaline’s effects on the aforementioned DA signal parameters, respectively. All data are expressed as the mean ± the standard error of the mean (S.E.M.), unless otherwise stated. A p-value < 0.05 was defined as statistically significant.
3. Results
3.1. Identification of L. cardinalis as a “species of interest” using the high-throughput DSS
In the present study, a library of aqueous plant extracts was screened to identify extracts that contained nicAchR ligands with relatively equipotent binding affinity at α4β2- and α7-nicAchRs. Extracts with a DDR value of ~3 in the DSS were prioritized (see section 2.5). An extract’s DDR value was calculated by dividing the percentage displacement of [3H]-cytisine by that of [3H]-MLA at a concentration equal to the ID50 for [3H]-epibatidine displacement [15]. The aqueous extract from L. cardi-nalis (LCaq) displaced [3H]-epibatidine from rat cortical membranes (ID50 = 1:300, 333 μg/ml), and exhibited a DDR value of 2.96. Thus, L. cardinalis was designated as a “species of interest,” since its DDR value indicated the presence of a nicAchR ligand(s) with relatively equipotent binding affinity at α4β2- and α7-nicAchRs.
3.2. The LCaq activates nicAchRs and inhibits the DAT
The LCaq activated nicAchRs and inhibited the DAT in functional studies performed in vitro (Fig. 1). In a variety of neuronal and non-neuronal cell types, acute treatment with nicAchR agonists (e.g. NIC and DMPP) has been reported to increase 45Ca2+ entry [57, 69]. In contrast, basal levels of intracellular Ca2+ are generally unaltered by treatment with nicAchR antagonists, such as MEC or MLA [22, 57, 69, 70]. Here, 45Ca2+ entry studies were conducted with SH-SY5Y cells due to their neuronal properties, catecholaminergic phenotype, and utility as an in vitro model of DAergic neurotoxicity [71, 72]. Furthermore, SH-SY5Y cells express β2-subunit containing and α7-nicAchRs, reported to mediate many of the neuroprotective effects of nicAchR agonists, as well as α3β4-nicAchRs that are believed to indirectly modulate the activity of the mesolimbic DAergic pathway [6, 7, 9–14, 22, 70]. Ca2+ entry elicited by nicAchR ligands and extracts was measured using 45Ca2+, rather than calcium fluorimetry, to eliminate signal arising from other sources of Ca2+, such as that released from intracellular stores via Ca2+-induced Ca2+ release [22, 69]. In the SH-SY5Y cells, NIC (10 μM) significantly increased 45Ca2+ entry (374.4% increase above basal, p = 0.0001). 45Ca2+ entry was unaffected by MEC (1 μM), a non-selective nicAchR antagonist, in agreement with previous studies [22, 23, 57, 69, 70, 73]. The LCaq (1 mg/ml) significantly increased 45Ca2+ entry (136.8% increase above basal, p < 0.0001), and this effect was completely blocked by MEC. 45Ca2+ entry was significantly increased in SH-SY5Y cells concomitantly treated with the LCaq and NIC (1 mg/ml and 10 μM, respectively; 231.9% increase above basal, p < 0.0001), yet the effect was significantly less (p = 0.0143) than that observed in SH-SY cells treated with NIC alone. The NIC-evoked increase in Ca2+ entry in the current study is consistent with previous studies reporting nicAchR agonist-evoked Ca2+ entry in SH-SY5Y cells, measured using fluorimetric techniques [22]. These results suggest the LCaq likely contains a metabolite(s) that functions as a nicAchR partial agonist. Consistent with this view, the LCaq displays intrinsic activity at nicAchRs present on SH-SY5Y cells, but functions as a nicAchR antagonist in the presence of a nicAchR full agonist (i.e. NIC) [70, 74, 75]. Furthermore, in rat striatal synaptosomes the LCaq significantly and dose-dependently inhibited DAT-mediated [3H]-DA uptake. The LCaq significantly inhibited DAT-mediated [3H]-DA uptake at concentrations of 74.1 μg/ml (35.33% uptake, p < 0.0001), 151.3 μg/ml (11.56% uptake, p < 0.0001) and 3.326 mg/ml (1.485% uptake, p < 0.0001). The unique combination of pharmacological effects (i.e. nicAchR partial agonism and DAT inhibition) merited further investigation of L. cardinalis with the aim of identifying a metabolite or metabolites responsible for the aforementioned activities.
Fig. 1.

Modulation of 45Ca2+ entry in SH-SY5Y cells and DAT-mediated [3H]-DA uptake in rat striatal synaptosomes by the LCaq. Data expressed as the mean ± S.E.M. A) NIC (10 μM) and the LCaq (1mg/ml) significantly increase 45Ca2+ entry in SH-SY5Y cells. The LCaq significantly attenuates NIC-induced 45Ca2+ entry, and MEC (1 μM) pretreatment completely abolishes LCaq-induced 45Ca2+ entry. *** p < 0.001, **** p < 0.0001 vs. vehicle, student’s one-tailed t-test; # p < 0.05 compared to cells treated with NIC alone, student’s one-tailed t-test. B) DAT-mediated [3H]-DA uptake is significantly and dose-dependently inhibited by the LCaq. **** p < 0.0001 vs. vehicle, one-tailed student’s t-test. n = 3 – 4.
3.3. Putative identification of a multifunctional alkaloid present in L. cardinalis
A crude methanolic extract prepared from air-dried aerial portions of L. cardinalis was subject to bioassay-guided fractionation. Fractions obtained were evaluated in the DSS and for their ability to inhibit the DAT in vitro. The CHCl3 fraction thus obtained displaced [3H]-epibatidine from rat cortical membranes (ID50 = 1:60,000, 1.67 μg/ml), and exhibited a DDR value of 1.59 in the DSS, indicative of the presence of a ligand(s) with comparable affinity at α4β2- and α7-nicAchRs, as compared to nicotine or lobeline (DDR values of 13.00 and 6.27, respectively) [15]. The CHCl3 fraction also caused a significant, dose-dependent inhibition of DAT-mediated [3H]-DA uptake in rat striatal synaptosomes (Fig. 2) at 33.3 μg/ml (37.56% uptake, p < 0.0001) and 3.33 μg/ml (89.67% uptake, p = 0.0013). Thus, pHPLC sub-fractionation of the CHCl3 fraction was performed in an effort to identify a multifunctional plant metabolite responsible for the aforementioned activities. A single CHCl3 sub-fraction was the most effective at displacing [3H]-MLA from rat hippocampal membranes, and was also the most potent inhibitor of the DAT in rat striatal synaptosomes. These results indicated the presence of a putatively novel multifunctional plant metabolite. GC-MS analysis of the CHCl3 sub-fraction revealed lobinaline, the major alkaloid present in L. cardinalis, as the most abundant constituent (AUC = 70.15%) of the sub-fraction [50, 76–79]. The identity of lobinaline, a complex binitrogenous alkaloid, was confirmed based on previously reported structural and MS data [50, 76, 78].
Fig. 2.
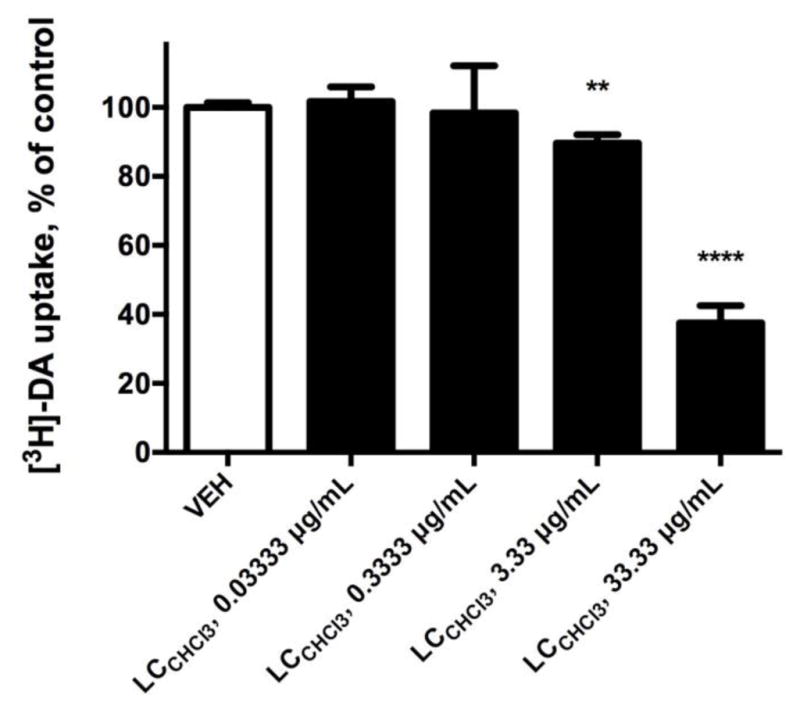
Modulation of DAT-mediated [3H]-DA uptake in rat striatal synaptosomes. Data expressed as the mean ± S.E.M. The L. cardinalis chloroform fraction (LCCHCl3) significantly and dose-dependently inhibited the DAT. ** p < 0.01, **** p < 0.0001 versus vehicle, one-tailed student’s t-test. n = 4.
3.4. Isolation of lobinaline
The crude methanolic extract prepared from air-dried aerial portions of L. cardinalis was fractionated to obtain the CHCl3 fraction. Acid-base extraction was performed on the CHCl3 fraction, yielding lobinaline (purity ≥ 95%). The identity of the alkaloid was confirmed based on previously reported MS data, and its purity was determined by integration of the AUC of the chromatographic peak corresponding to lobinaline on the GC trace [50]. Lobinaline (Fig. 3) was stored in the dark at −20°C prior to further use.
Fig. 3.
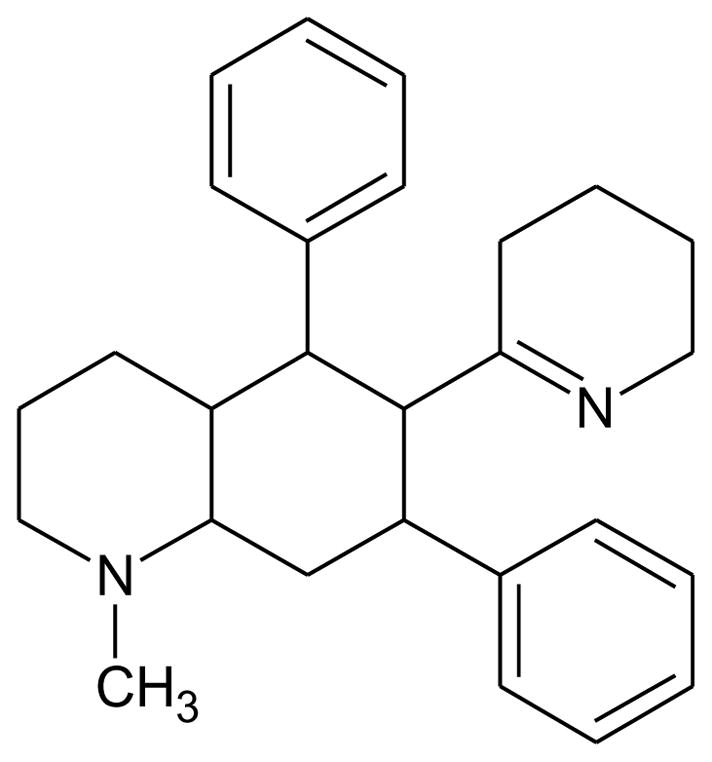
Structure of lobinaline.
3.5. Lobinaline displaces radioligands selective for nicAchRs and the DAT
Lobinaline concentration-dependently inhibited [3H]-epibatidine, [3H]-cytisine, and [3H]-MLA binding at nicAchRs (Ki = 16.22, 1.066, and 67.53 μM, respectively; Fig. 4A – C). Radioligand binding studies with the aforementioned nicAchR ligands were performed as described previously [15, 51]. Competition binding studies with [3H]-epibatidine and [3H]-cytisine were conducted in rat cortical membranes. The former nicAchR ligand is relatively non-selective at neuronal nicAchR subtypes, while the latter is selective for α4β2-nicAchRs [1, 18, 47, 48]. Competition binding studies with the α7-nicAchR selective ligand [3H]-MLA were conducted in rat hippocampal membranes [16, 18]. In contrast to nicotine and lobe-line, lobinaline has similar affinity at α4β2- and α7-nicAchRs (see Table 2 for comparison) [80, 81]. Lobinaline produced a DDR value of 1.32, virtually identical to that produced by the CHCl3 fraction, indicating lobinaline was the main metabolite responsible for the fraction’s nicAchR binding activity. Lobinaline also inhibited binding of [3H]-GBR12935, a highly selective DAT ligand, in rat striatal membranes (Ki = 2.543 μM) [53, 54].
Fig. 4.

Lobinaline displaces nicAchR-selective radioligands. Data are expressed as the mean ± S.E.M. Concentration-dependent inhibition of A) [3H]-epibatidine (Ki = 16.22 μM) and B) [3H]-cytisine (Ki = 1.066 μM) binding at nicAchRs by lobinaline in rat cortical membranes. C) Concentration-dependent inhibition of [3H]-MLA binding (Ki = 67.53 μM) at nicAchRs in rat hippocampal membranes. n = 3 – 4.
Table 2.
NicAchR selectivity of lobinaline, lobeline, and nicotine at α4β2- and α7-nicAchRs
| Ki μM | NicAchR Selectivity | |||
|---|---|---|---|---|
|
| ||||
| Compound | α4β2-nicAchR | α7-nicAchR | (-fold difference)5 | DDR value |
| Lobinaline | 1.066 | 67.54 | 63 | 1.32 |
| Lobeline3 | 0.004 | 6.26 | 1565 | 6.276 |
| Nicotine4 | 0.00096 | 1.448 | 1508 | 13.006 |
Ki’s previously reported by Hojahmat et al. (2010)
Ki’s previously reported by Rueter et al. (2006)
Selectivity based on comparison of Ki’s
DDR values previously reported by Littleton et al. (2005)
3.6. Lobinaline activates nicAchRs and inhibits the DAT in vitro
Lobinaline’s functional effect at nicAchRs was evaluated by assessing its ability to modulate 45Ca2+ entry in SH-SY5Y cells (Fig. 6A). In these experiments, NIC (10 μM) significantly increased 45Ca2+ entry (196.6% increase above basal, p = 0.0008). Treatment of cells with MEC alone (1 μM) did not affect 45Ca2+ entry. At the concentration tested in the current study, lobinaline (1 mM) significantly increased 45Ca2+ entry (41.6% increase above basal, p = 0.0011). Lobinaline-induced 45Ca2+ entry in SH-SY5Y cells was completely abolished by MEC. Concomitant treatment of SH-SY5Y cells with lobinaline and NIC (1mM and 10 μM, respectively) significantly increased 45Ca2+ entry (115.7% increase above basal, p < 0.0001). Similar to the LCaq, concomitant treatment of SH-SY5Y cells with lobinaline and NIC significantly reduced 45Ca2+ entry (p = 0.0347), as compared to cells treated with NIC alone. The observation of nicAchR activation by lobinaline, in combination with its ability to attenuate NIC-induced 45Ca2+ entry, is consistent with the view that lobinaline functions as a partial agonist at nicAchRs expressed by SH-SY5Y cells [70, 74, 75]. In order to assess lobinaline’s efficacy as an inhibitory modulator of the DAT, [3H]-DA uptake was performed in rat striatal synaptosomes. DAT-mediated [3H]-DA uptake was inhibited by lobinaline (IC50 = 11.95 μM; Fig. 6B). Based on these results, and data obtained from binding studies, lobinaline functions as a multifunctional alkaloid with unique pharmacological profile, acting as a nicAchR partial agonist and an inhibitor of the DAT.
Fig. 6.

Lobinaline (LBNA) activates nicAchRs and inhibits the DAT. Data expressed as the mean ± S.E.M. A) NIC (10 μM) and LBNA (1 mM) significantly increase 45Ca2+ entry in SH-SY5Y cells. LBNA significantly attenuates NIC-induced 45Ca2+ entry, and MEC (1 μM) pretreatment completely abolishes lobinaline-induced 45Ca2+ entry. ** p < 0.01, *** p < 0.001, **** p < 0.0001 vs. vehicle, student’s one-tailed t-test. # p < 0.05 vs. cells treated with NIC alone, student’s one-tailed t-test. n = 3 – 4. B.) Lobinaline dose-dependently inhibits the DAT (IC50 = 11.95 μM). n = 3 – 4.
3.7. Lobinaline is a potent DPPH free radical scavenger
Multiple lines of evidence indicate excessive free radical production contributes to DAergic neurotoxicity seen in PD and psychostimulant abuse [28, 30, 33, 42–44]. The DPPH free radical scavenging assay is commonly utilized to assess a molecule’s ability to quench free radicals [59]. Lobinaline acted as a potent free radical scavenger (EC50 = 17.98 μM), as did quercetin (EC50 = 11.19 μM), a plant metabolite previously reported to potently scavenge DPPH free radicals (Fig. 7) [59]. In contrast, lobeline was a weak DPPH free radical scavenger (EC50 = 228.8 μM). Although quercetin’s EC50 is greater than previously reported (6.22 μM), this was expected given the concentration of DPPH used in the present study (300 μM) was 6-fold greater than the former (50 μM) [59].
Fig. 7.
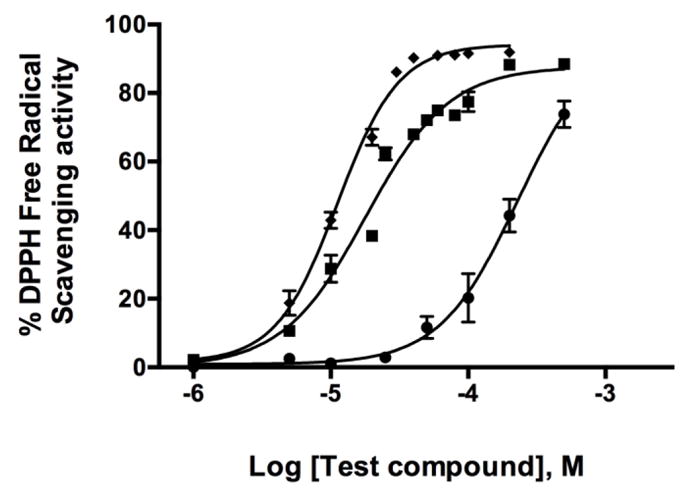
DPPH assay for free radical scavenging activity. Data expressed as the mean ± S.E.M. Quercetin (◆) and lobinaline (■) are potent DPPH free radical scavengers (EC50 = 11.19 and 17.98 μM, respectively). Lobeline (●) is a relatively poor scavenger of DPPH free radicals (EC50 = 228.8 μM). n = 3 – 5.
3.8. Lobinaline dose-dependently evokes fractional [3H] release from [3H]-DA preloaded rat striatal slices
The modulation of nicAchRs by lobinaline was also examined by monitoring its ability to evoke fractional [3H] release from rat striatal slices preloaded with [3H]-DA. Superfusion studies were performed in the presence of pargyline (10 μM) and nomifensine (10 μM). The inclusion of a MAO inhibitor reduced the contribution of [3H]-DA metabolites to the [3H] signal recorded, and the use of a DAT inhibitor promoted measurement of [3H]-DA released from vesicular stores, rather than efflux of [3H]-DA via reversal of the DAT [23, 61]. Lobinaline caused a significant, concentration-dependent increase in fractional [3H] release from rat striatal slices preloaded with [3H]-DA (Fig. 8A). Repeated measures, two-way ANOVA revealed a significant main effect of concentration (F(3,26) = 15.50, p < 0.0001), a significant main effect of time (F(11, 286) = 4.865, p < 0.0001), and a significant concentration × time interaction (F(33, 286) = 3.591, p < 0.0001). The lowest concentration of lobinaline that evoked a significant increase in fractional [3H] release compared to vehicle was 100 μM. At concentrations of 100 μM and 1000 μM, lobinaline-evoked fractional [3H] release reached significance (p < 0.05 and p < 0.0001, respectively) 8 minutes after treatment (the second sample collected after lobinaline treatment). Fractional [3H] release evoked by 1000 μM lobinaline reached a maximum (494% greater than vehicle treated, p < 0.0001) 8 minutes post-treatment. Fractional [3H] release evoked by 100 μM lobinaline reached a maximum (186% greater than vehicle treated, p < 0.001) 12 minutes post-treatment, and was no longer significantly different from vehicle treated slices 16 minutes after treatment. In contrast, fractional [3H] release evoked by 1000 μM lobinaline remained significantly elevated above that of vehicle treated slices for the remainder of the experiment (28 minutes).
Fig. 8.

Time course of lobinaline-evoked fractional [3H] release from [3H]-DA preloaded rat striatal slices. Data expressed as the mean ± S.E.M. A) Lobinaline significantly, and dose-dependently increased fractional [3H] release at 100 μM (
 ) and 1,000 μM (■) vs. vehicle treated controls (●). Fractional [3H] release was not significantly increased by 10 μM (
) and 1,000 μM (■) vs. vehicle treated controls (●). Fractional [3H] release was not significantly increased by 10 μM (
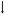 ) lobinaline. Lobinaline was added immediately after the collection of the fifth sample, as indicated by the arrow. * p < 0.05, *** p < 0.001, 100 μM lobinaline vs. vehicle treated slices; ## p < 0.01, ### p < 0.001, #### p < 0.0001 1,000 μM lobinaline vs. vehicle treated slices; Bonferroni’s post-hoc test. n = 4 – 10 rats. B) Effect of MEC on the time course of lobinaline-evoked fractional [3H] release from [3H]-DA preloaded rat striatal slices. Fractional [3H] release was significantly increased by 100 μM lobinaline (
). Fractional [3H] release from striatal slices pretreated with 10 μM MEC prior to the addition of 100 μM lobinaline (
) lobinaline. Lobinaline was added immediately after the collection of the fifth sample, as indicated by the arrow. * p < 0.05, *** p < 0.001, 100 μM lobinaline vs. vehicle treated slices; ## p < 0.01, ### p < 0.001, #### p < 0.0001 1,000 μM lobinaline vs. vehicle treated slices; Bonferroni’s post-hoc test. n = 4 – 10 rats. B) Effect of MEC on the time course of lobinaline-evoked fractional [3H] release from [3H]-DA preloaded rat striatal slices. Fractional [3H] release was significantly increased by 100 μM lobinaline (
). Fractional [3H] release from striatal slices pretreated with 10 μM MEC prior to the addition of 100 μM lobinaline (
 ) was not significantly different from vehicle treated slices (●). MEC was added immediately after the collection of the second sample (small arrow). Lobinaline was added immediately after collection of the seventh sample (large arrow). * p < 0.05, *** p < 0.001, ****p < , 100 μM lobinaline vs. vehicle treated slices; Bonferroni’s post-hoc test. n = 4.
) was not significantly different from vehicle treated slices (●). MEC was added immediately after the collection of the second sample (small arrow). Lobinaline was added immediately after collection of the seventh sample (large arrow). * p < 0.05, *** p < 0.001, ****p < , 100 μM lobinaline vs. vehicle treated slices; Bonferroni’s post-hoc test. n = 4.
After establishing lobinaline’s ability to concentration-dependently evoke fractional [3H] release, studies were performed to evaluate the contribution of nicAchR stimulation to lobinaline-evoked fractional [3H] release. Striatal slices preloaded with [3H]-DA were pretreated with MEC (10 μM) for 20 minutes prior to the addition of lobinaline (100 μM). Fractional [3H] release from vehicle treated striatal slices served as a control. MEC significantly attenuated lobinaline-evoked fractional [3H] release indicating activation of nicAchRs contributes to fractional [3H] release evoked by the alkaloid (Fig. 8B) [23]. Repeated measures, two-way ANOVA revealed a significant main effect of treatment (F(2, 9) = 9.683, p = 0.0057) and a significant main effect of time (F(13, 117) = 2.465, p = 0.0053). The treatment × time interaction was not significant (F(26, 117) = 1.488, p = 0.0793). In this set of experiments, the effect of 100 μM lobinaline alone was generally in agreement with initial studies, albeit the time course of its effect was slightly prolonged. That is, lobinaline-evoked fractional [3H] release reached significance 8 minutes after treatment (160% greater than vehicle, p < 0.05), and was no longer significantly different from vehicle-treated slices after 24 minutes. Additionally, the maximal increase in fractional [3H] release occurred 16 minutes after lobinaline treatment and the maximum (262% greater than vehicle treated controls, p < 0.0001) was greater than that observed in initial studies. When comparing fractional [3H] release from vehicle treated slices and slices pretreated with MEC prior to lobinaline, no significant difference was observed at any time point. These results indicate lobinaline’s effect was MEC-sensitive, providing additional evidence that the alkaloid is a nicAchR agonist. This is in contrast to lobeline, which evokes fractional [3H] release from rat striatal slices that is MEC-insensitive, and was reported to antagonize nicAchRs [23, 61].
3.9. Lobinaline inhibits DA uptake in vivo in the striatum of urethane-anesthetized rats
In vivo electrochemical studies were performed to evaluate modulation of DA uptake in isoflurane-anesthetized rats. In agreement with in vitro [3H]-DA uptake studies, local application of lobinaline in the dorsal striatum significantly prolonged the clearance rate of exogenous DA. A representative trace of exogenous DA clearance recorded pre- and post-lobinaline application is depicted in Fig. 9A. The alkaloid significantly increased the T80 (76.33 ± 39.52 sec., p = 0.0203) and significantly decreased the clearance rate (0.085 ± 0.054 μM/sec., p = 0.0459) 1-minute post-application, as compared to the T80 (33.67 ± 12.45 sec.) and clearance rate (0.127 ± 0.065 μM/sec.) pre-application (Fig. 9B and Fig. 9C). The effects of lobinaline on the aforementioned DA signal parameters are indicative of DAT inhibition. Lobinaline’s effects on DA clearance are short acting and likely competitive, as they are no longer significant 3 – 5 minutes after lobinaline ejection. Lobinaline’s effects on DA clearance are somewhat like those observed after local application of nomifensine in the dorsal striatum of urethane-anesthetized Sprague-Dawley rats, but were not as efficacious, which is consistent with in vitro studies of nomifensine in synaptosomes [54, 65]. In vitro electrochemical studies confirmed lobinaline itself was not electroactive by chronoamperometric recordings used to measure endogenous DA (data not shown). In vivo, lobinaline had no direct effects when locally applied from a micropipette (n = 10; data not shown). Given lobinaline prolonged exogenous DA clearance and failed to cause DA release, the alkaloid appears to act as a DAT inhibitor, rather than a DAT substrate/releasing agent.
Fig. 9.

Effects of lobinaline (LBNA) on exogenous DA clearance in isoflurane-anesthetized rats measured using HSC. Data are expressed as the mean ± S.E.M. A) Representative trace of exogenous DA clearance pre- (solid line) and post-LBNA (dashed line) application. B) LBNA significantly increased (p = 0.0203) the T80 1-minute post application (76.33 ± 39.52 sec.), as compared to the T80 pre-application (33.67 ± 12.45 sec.). * p < 0.05, pre- vs. post-lobinaline application, paired one-tailed student’s t-test. C) LBNA significantly decreased (p = 0.0459) the clearance rate 1-minute post-application (0.085 ± 0.054 μM/sec.), as compared to the clearance rate pre-application (0.127 ± 0.065 μM/sec.). * p < 0.05, pre- vs. post-LBNA application, paired two-tailed student’s t-test. n = 6.
3.10. Lobinaline fits the criteria set forth by Lipinski’s “Rule of Five”
The oral “druggability” of lobinaline, based on its physiochemical properties, was assessed according to Lipinski’s “Rule of Five” [66]. These data are readily available the PubChem website (https://pubchem.ncbi.nlm.nih.gov/) [67, 68]. Lobinaline did not violate any of the criteria set forth by Lipinski’s “Rule of Five”: molecular weight = 386, hydrogen bond donors = 0, hydrogen bond acceptors = 2, cLogP = 4.8, molar fractivity = 82.47. Additionally, based on previously reported in vivo studies, lobinaline displays appropriate pharmacokinetics and low mammalian toxicity in mice relative to lobeline, the most widely studied Lobelia alkaloid [77].
4. Discussion
Plants are a rich source of multifunctional drug leads, as described in recent reviews [3, 82, 83]. In the present study, the multifunctional alkaloid lobinaline was identified as the major bioactive metabolite present in L. cardinalis [50, 76–79]. The alkaloid possesses a unique polypharmacological profile functioning as a nicAchR agonist, DAT inhibitor, and free radical scavenger. These pharmacological effects of lobinaline are previously unreported, and to the best of our knowledge, the alkaloid is the only natural product with a combination of the aforementioned activities. NicAchR agonists, DAT inhibitors, and free radical scavengers attenuate DAergic neurotoxicity in animal models of PD and psychostimulant abuse [6–13, 26–33, 43, 84–89]. To our knowledge, no other drug described thus far possesses all these attributes. Thus, lobinaline may be a particularly valuable lead for the development of multifunctional neuroprotective therapeutics aimed to prevent DAergic neurotoxicity.
Initially, HTPS was conducted on a library of aqueous plant extracts from ~1,000 species in an effort to identify a novel nicAchR agonist with equipotent binding affinity at α4β2- and α7-nicAchRs. L. cardinalis aqueous extract (LCaq) produced a DDR value of 2.96, signifying the extract contained a metabolite/s with similar binding affinity at α4β2- and α7-nicAchRs, as described above (see section 2.5) [4, 15]. In SH-SY5Y cells, the LCaq (1mg/ml) significantly increased 45Ca2+ entry. The LCaq-induced increase in 45Ca2+ entry was abolished by MEC (1 μM), indicating the effect was mediated by nicAchR activation [22, 23, 57, 69, 70, 73]. Furthermore, the LCaq significantly reduced NIC-induced 45Ca2+ entry in SH-SY5Y cells. Collectively, these results indicated the LCaq contained a metabolite/s that functioned as a nicAchR partial agonist with relatively equipotent affinity for α4β2- and α7-nicAchRs [70, 74, 75]. This is in contrast to “typical” nicAchR ligands from plant sources, the majority of which are α4β2-nicAchR selective agonists (e.g. cytisine) or α7-nicAchR antagonists (e.g. MLA) [1, 16–18]. Additionally, the extract inhibited DAT-mediated [3H]-DA uptake in rat striatal synaptosomes. This was an intriguing observation since, to the best of our knowledge, all plant metabolites reported to inhibit the DAT and modulate nicAchRs act as antagonists at the latter [1, 23, 61, 90].
Bioassay-guided fractionation of the L. cardinalis methanolic extract, followed by pHPLC sub-fractionation, indicated both activities resided in a single CHCl3 sub-fraction. GC-MS analysis of the sub-fraction revealed lobinaline was the major constituent present (AUC = 70%), the identity of which was confirmed based on previously reported MS data [50]. Isolation and pharmacological characterization of lobinaline ensued. The alkaloid inhibited binding of the α4β2-nicAchR selective ligand [3H]-cytisine (Ki = 1.066 μM), and the α7-nicAchR selective ligand [3H]-MLA (Ki = 67.53 μM), in rat cortical and hippocampal membranes, respectively, and produced a DDR value of 1.32 [1, 16–18]. Compared to other plant metabolites, such as nicotine and lobeline, lobinaline is relatively non-selective with respect to α4β2- and α7-nicAchRs (see Table 2 for comparison) [80, 81]. The plant metabolite anabasine is also non-selective at α4β2- and α7-nicAchRs (Ki = 65 nM and 58 nM at α4β2- and α7-nicAchRs, respectively) [1]. However, anabasine is a teratogen limiting its clinical utility [91]. In SH-SY5Y cells, lobinaline significantly increased 45Ca2+ entry, an effect that was blocked by MEC. Lobinaline also significantly reduced NIC-induced 45Ca2+ entry in SH-SY5Y cells. The ability of lobinaline to activate nicAchRs and functionally antagonize the effect of a nicAchR full agonist (i.e. NIC) suggests the alkaloid may be a nicAchR partial agonist [70, 74, 75]. Lobeline, on the other hand, antagonizes nicAchRs in a variety of experimental models [23, 61]. Based on these data, lobinaline appears to be distinct from nicotine and lobe-line in terms of its selectivity and functional effects at nicAchRs. Electrophysiological studies (e.g. two-electrode voltage clamping) assessing lobinaline’s efficacy at nicAchRs are necessary to confidently designate lobinaline as a nicAchR partial agonist.
Lobinaline was further evaluated in vitro for its ability to interact with and modulate the DAT. In rat striatal membranes, binding of the highly selective DAT ligand [3H]-GBR12935 was inhibited by lobinaline (Ki = 2.543 μM), and the alkaloid dose-dependently attenuated DAT-mediated [3H]-DA uptake (IC50 = 11.95 μM) in rat striatal synaptosomes [53, 54]. Lobeline also inhibits the DAT, but is less potent (IC50 = 28.2 – 80.0 μM) than lobinaline [23, 80]. In contrast, nicotine is inactive at the DAT in rat striatal synaptosomes [23]. However, in in vivo electrochemical studies performed in rats, systemic administration of nicotine enhanced DAT function [92]. The latter study suggests an intact neurological system is required to examine modulation of DAT activity by nicAchR agonists.
As described above, lobinaline exerts pleiotropic pharmacological effects relevant to the prevention and/or reduction of DAergic neurotoxicity seen in PD and psychostimulant abuse, including nicAchR activation and DAT inhibitory modulation [6–10, 13, 26–31]. Since multiple lines of evidence indicate excessive free radical production contributes to DAergic neurotoxicity seen in the aforementioned pathologies, lobinaline was assessed for its capacity to scavenge free radicals [28, 30, 33, 42–44]. In the DPPH free radical scavenging assay, lobinaline acted as a potent scavenger of free radicals (EC50 = 17.98 μM), as compared to quercetin (EC50 = 11.19 μM), a natural product known for its free radical scavenging and antioxidant activity [59, 83, 88, 89]. Lobeline’s capacity to scavenge DPPH free radicals was relatively poor (EC50 = 228.8 μM). Given quercetin’s neuroprotective effects in cellular and animal models of PD, lobinaline may exert similar effects warranting future investigation of the alkaloid in models of PD [88, 89].
In superfusion studies, lobinaline caused a significant, dose-dependent increase in fractional [3H] release from rat striatal slices preloaded with [3H]-DA. The buffer used in superfusion studies contained a MAO inhibitor and a DAT inhibitor to reduce signal arising from [3H]-DA metabolites and [3H]-DA release via reversal of the DAT, respectively [23, 61]. At the highest concentration examined in the current study (1000 μM), lobinaline-evoked fractional [3H] release remained significantly greater than control for the duration of the experiment. This effect mirror’s that observed for high concentrations of lobeline (10–100 μM) under essentially identical experimental conditions [23]. In contrast, nicotine’s effect on fractional [3H] release is transient, even at high concentrations, likely due to desensitization of nicAchRs [23]. Subsequent experiments revealed lobinaline-evoked fractional [3H] release was MEC-sensitive, providing additional evidence that the alkaloid is an agonist at nicAchRs. Based on these observations, stimulation of nicAchRs underlies lobinaline’s ability to evoke fractional [3H] release from [3H]-DA preloaded striatal slices. Lobeline, on the other hand, is reported to antagonize nicAchRs, and evokes fractional [3H] release that is MEC-insensitive and Ca2+-independent [23].
Lobinaline’s ability to affect vesicular monoamine transporter type-2 (VMAT-2) activity, inhibition of which is believed to underlie lobeline’s effect in similar superfusion studies, was not examined [23]. Furthermore, the [3H] signal measured in studies examining lobeline-evoked fractional [3H] release from [3H]-DA preloaded rat striatal slices was reported to arise predominantly from an increase in [3H]-DOPAC release, rather than [3H]-DA [23]. The authors reached this conclusion upon measuring lobeline-evoked endogenous DA and DOPAC release from rat striatal slices via HPLC coupled with electrochemical detection (HPLC-ECD). Lobinaline’s ability to modulate VMAT-2 and endogenous DA/DOPAC release from rat striatal slices, measured using HPLC-ECD, remain to be explored.
The effect of lobinaline on DAT function was examined in vivo in the striatum of isoflurane-anesthetized rats using HSC coupled to local applications of exogenous DA. This technique allows measurement of DA dynamics in vivo in an intact neurological system with a high degree of spatial and temporal resolution, and has been used extensively to characterize effects of DAT inhibitors [35, 38, 39, 65, 93]. Consistent with in vitro [3H]-DA uptake studies, lobinaline significantly prolonged the clearance rate of exogenous DA 1-minute post-application. Local application of the alkaloid in the dorsal striatum significantly increased the T80 and significantly reduced the clearance rate when compared to amplitude matched DA signals pre-lobinaline application. These observations demonstrate lobinaline’s ability to inhibit DAT function in vivo, although the alkaloid’s effects are transient and are no longer significant 3 – 5 minutes post-application. Alterations in the T80 and the clearance rate reflect modulation of DAT activity, as has been characterized for other DAT inhibitors [39, 65, 94]. Effects somewhat like those of lobinaline were observed following local application of the DAT inhibitor nomifensine in the dorsal striatum of Sprague-Dawley rats, albeit lobinaline was less efficacious, which is consistent with in vitro studies of nomifensine in synaptosomes [54, 65]. In isoflurane-anesthetized rats, lobinaline locally applied in the striatum had no direct effects. DAT substrate-releasing agents, such as amphetamines, induce endogenous DA release [95, 96]. Thus, lobinaline likely functions as a DAT inhibitor, rather than a substrate, although the results are not conclusive. The transient nature of lobinaline’s effects in vivo may result from the alkaloid’s relatively low affinity and/or potency at the DAT. Conformational changes resulting from lobinaline’s interaction with the DAT and mechanism/s underlying lobinaline’s effects on DAergic neurotransmission and DAT function in vivo remain to be thoroughly elucidated.
The multifunctional pharmacological effects of lobinaline are somewhat expected in view of the structural complexity and functional groups present in the decahydroquinoline alkaloid. For example, lobinaline meets all criteria of the “refined” pharmacophore proposed by Inamdar (2011) for DAT inhibitors: 1) ionizable nitrogen, 2) aromatic ring, 3) hydrogen bond acceptor, and 4) two hydrophobic groups [97]. Although these requirements are not absolute, an ionizable nitrogen within ~6 Å of a phenyl moiety is present in the vast majority of DAT inhibitors [97, 98]. Illustrating this point, the lack of a phenyl and/or benzyl substituent in nicotine and anabasine seems to be a reasonable explanation for their inability to inhibit DAT, especially given their structural similarity to the competitive DAT inhibitor/substrate 1-methyl-4-phenlypiperidinium [1, 68, 99]. On the other hand, structural features common to lobinaline and the alkaloids anabasine and anabaseine likely explain lobinaline’s selectivity and activity at select nicAchR subtypes. The 3-(2-piperidyl)pyridine alkaloids anabasine and anabaseine, the latter having a 1,2-unsaturated piperidyl group, are nicAchR agonists with similar affinity for α4β2- and α7-nicAchRs [1, 99]. Although lobinaline lacks a pyridyl functional group, the aromatic character of its phenyl substituents in the vicinity of the 1,2-dehydropiperidine functional group may suffice to create a physiochemical environment adequate to engender similar interactions with nicAchRs. The agonist activity lobinaline displays at nicAchRs is of note, given other decahydroquinolines studied to date antagonize nicAchRs, possibly due to the lack of a tetrahydropiperidyl substituent in those previously investigated [100, 101]. Lobinaline has 5 chiral centers, whose stereogenic configuration in the natural molecule from the plant is unknown. These configurations may be important for one or more of the pharmacological actions that we have reported. Robison et al.[76] achieved a total synthesis of the ring structure but this was in the (easy) all trans configuration. Whether this possesses the same configuration and pharmacology as the molecule from the plant is unknown, but this is a direction for future studies.
A question becomes apparent when considering the pharmacological actions of lobinaline, and the amount of energy that is likely required to synthesize a molecule of its complexity: What benefit does lobinaline afford to the plant? One explanation relates to the effects of nicAchR agonists and DAT inhibitors on insect behavior. Both classes of compounds have been reported to function as naturally occurring insecticides, nicotine and cocaine representing examples of the former and latter [2, 4, 102]. Additionally, modulating two molecular targets may be more effective, due to synergistic effects. Targeting nicAchRs and the DAT also ensures protection against herbivorous insects resistant to one mode-of-action, or the other. Thus, having a “shotgun” approach to fend off herbivorous threats is potentially “safer” for the plant. Furthermore, lobinaline’s ability to function as an antioxidant should reduce oxidative stress arising from excessive exposure to ultraviolet radiation, or free radical species that are generated during normal metabolism [3]. All in all, synthesis of a multifunctional molecule is potentially a more efficient strategy, given a single biosynthetic pathway and resulting metabolite addresses each of the challenges plants encounter, outlined above. Given evolution favors efficiency and effectiveness, natural selection would likely favor the retention and optimization of multifunctional molecules, such as lobinaline, and their respective biosynthetic pathways. Likewise, DAT inhibition and α7 nicAchR agonism, combined with free radical scavenging activity of lobinaline may prove to offer unique therapeutic benefit in future studies when compared to monovalent pharmacotherapuetic strategies.
Only one other molecule which activates nicAchRs and inhibits the DAT has been reported, a synthetic analogue of cocaine, cocaine methiodide (CMI) [1, 90, 103]. However, the clinical utility of CMI is severely limited due its toxic peripheral effects [103]. Previously, a limited number of in vivo studies were performed with lobinaline [77]. Intravenous administration of lobinaline in cats and rabbits was reported to lower blood pressure, whereas respiration was unaffected by the alkaloid [77]. The authors reported minimal lethal doses of 800 and 300 mg/kg for lobinaline and lobeline, respectively, following subcutaneous administration in mice [77]. These initial studies indicate the alkaloid’s pharmacokinetic (PK) and toxicity profile are acceptable for a drug lead, especially considering complications arising from PK and toxicity reportedly underlie failure of ~30% of NCEs in clinical development [104, 105]. Additionally, lobinaline does not violate criteria outlined by Lipinski’s “Rule of Five,” a common assessment used to predict a molecule’s oral “druggability” based on its physiochemical properties [66]. Thus, lobinaline holds considerable value as a potential lead molecule for the development of therapeutics with its combination of effects. One hurdle that may stall optimization of the alkaloid via traditional medicinal chemistry is the lack of a method for the total synthesis of lobinaline, limiting access to a pure starting material [106–108]. Although tracer studies examining lobinaline biosynthesis in planta indicate phenylalanine and lysine are the likely precursors of the alkaloid, a method for the total synthesis of lobinaline remains elusive [79]. Also the presence of five chiral centers in lobinaline would necessitate the separation of enantiomers likely to arise during chemical optimization, creating additional challenges and costs [106]. To address this problem, our group is currently developing a novel plant-based drug discovery platform that favors evolution of plant biosynthetic pathways yielding molecules with desirable interactions at a protein which is a therapeutic molecular target. Proof-of-application studies are being conducted using the DAT as the molecular target, and L. cardinalis as the candidate plant species.
Lobinaline, or congeners with similar pharmacological effects represent promising multifunctional drug leads to prevent DAergic neurotoxicity seen in PD or psychostimulant abuse. For example, DAT inhibitors prevent uptake of endogenous and exogenous neurotoxic substrates of the transporter thought to contribute to DAergic neuron loss in PD, and DAT inhibitors alleviate specific Parkinsonian symptoms in rodent and nonhuman primate models of PD [26, 28, 31–33, 109, 110]. NicAchR agonists selective for α4β2- or α7-nicAchRs are neuroprotective in cellular and animal models of PD, and reduce drug-induced dyskinesia caused by therapeutics used to treat PD [6–13, 111]. In models of psychostimulant abuse, α4β2- and α7-nicAchR selective agonists, as well as DAT inhibitors, attenuate neurotoxicity caused by amphetamines [6, 7, 9, 12, 13, 26, 27, 29, 30, 43]. In animal models of cocaine and amphetamine abuse, “atypical” DAT inhibitors (e.g. JHW-007) reduce psychostimulant self-administration and display low abuse liability [112–117]. DAergic neurotoxicity caused by psychostimulants and neurotoxic DAT substrates utilized to model PD is attenuated by pre-treatment with antioxidants, including ubiquinol (coenzyme Q10) and flavonoids (e.g. baicalein) [84–89]. Thus multifunctional molecules, such as lobinaline, which activate α4β2- and α7-nicAchRs, inhibit the DAT, and function as free radical scavengers may be superior DAergic neuroprotectants that act via a single mechanism of action. This notion is supported by recent reviews indicating multifunctional leads have a higher probability of displaying efficacy with minimal side effects [83, 104, 118]. Furthermore, high affinity binding is not required for multifunctional drugs presumably due to synergistic and/or additive effects arising from their multi-target activities, and this relative lack of potency at a single molecular target may reduce adverse effects [83, 104, 118]. Multi functional drugs may present potential for untoward effects as well. Current studies in our laboratory are examining the rewarding and motor stimulatory effects of lobinaline using in vivo models, and metabolism and bio-availability studies will be examined in future experiments. Collectively, the data presented herein indicate lobinaline’s potential as a lead to develop multifunctional neuroprotective therapeutics for neurological disorders involving DAergic neurodegeneration and/or psychostimulant abuse.
Fig. 5.
Lobinaline displays affinity for the DAT. Data expressed as the mean + S.E.M. A.) Lobinaline concentration-dependently displaces [3H]-GBR12935 (Ki = 2.543 μM) from rat striatal membranes. n = 4
Acknowledgments
This project was supported in part by NIAAA (National Institute on Alcohol Abuse and Alcoholism) grants (5R44AA018226-04) awarded to Dr. John M. Littleton as Principal Investigator. The authors would also like to acknowledge the NIA (National Institute on Aging) and the NIDA (National Institute on Drug Abuse) for grants (5T32AG000242-20 and 2T32DA016176-11, respectively) awarded Dr. Greg A. Gerhardt and Dr. Linda Dwoskin, respectively, as Principal Investigator which supported Dustin P. Brown’s dissertation research.
Abbreviations
- nicAchR
nicotinic acetylcholine receptor
- DA
dopamine
- DAT
dopamine transporter
- LCaq
Lobelia cardinalis aqueous extract
- DSS
differential smart screen
- DDR
differential displacement ratio
- DAergic
dopaminergic
- HSC
high-speed chronoamperometry
- DPPH
1,1-diphenyl-2-picrylhydrazyl
- NIC
nicotine
- MLA
methyl-lycaconitine
- MEC
mecamylamine
- ID50
50% inhibitory dilution
- IC50
50% inhibitory concentration
- EC50
50% effective concentration
- Abs
absorbance
- MAO
monoamine oxidase
- T80
signal time course
- Tc
clearance rate
Footnotes
Conflict of Interest
Dr. Littleton functions as the Chief Scientific Officer at Naprogenix™ and owns stock in the company. Dr. Rogers is an employee of Naprogenix™. The remaining authors have no conflict of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Daly JW. Nicotinic agonists, antagonists, and modulators from natural sources. Cell Mol Neurobiol. 2005;25(3–4):513–52. doi: 10.1007/s10571-005-3968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steppuhn A, et al. Nicotine’s defensive function in nature. PLoS Biol. 2004;2(8):E217. doi: 10.1371/journal.pbio.0020217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kennedy DO, Wightman EL. Herbal extracts and phytochemicals: plant secondary metabolites and the enhancement of human brain function. Adv Nutr. 2011;2(1):32–50. doi: 10.3945/an.110.000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Littleton J. The future of plant drug discovery. Expert Opinion on Drug Discovery. 2007;2(5):673–683. doi: 10.1517/17460441.2.5.673. [DOI] [PubMed] [Google Scholar]
- 5.Picciotto MR, Kenny PJ. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb Perspect Med. 2013;3(1):a012112. doi: 10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bordia T, et al. The alpha7 nicotinic receptor agonist ABT-107 protects against nigrostriatal damage in rats with unilateral 6-hydroxydopamine lesions. Exp Neurol. 2015;263:277–84. doi: 10.1016/j.expneurol.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrea S, Winterer G. Neuroprotective and neurotoxic effects of nicotine. Pharmacopsychiatry. 2009;42(6):255–65. doi: 10.1055/s-0029-1224138. [DOI] [PubMed] [Google Scholar]
- 8.Piao WH, et al. Nicotine and inflammatory neurological disorders. Acta Pharmacol Sin. 2009;30(6):715–22. doi: 10.1038/aps.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mudo G, Belluardo N, Fuxe K. Nicotinic receptor agonists as neuroprotective/neurotrophic drugs. Progress in molecular mechanisms. J Neural Transm. 2007;114(1):135–47. doi: 10.1007/s00702-006-0561-z. [DOI] [PubMed] [Google Scholar]
- 10.Quik M. Smoking, nicotine and Parkinson’s disease. Trends Neurosci. 2004;27(9):561–8. doi: 10.1016/j.tins.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Janhunen S, Ahtee L. Differential nicotinic regulation of the nigrostriatal and mesolimbic dopaminergic pathways: implications for drug development. Neurosci Biobehav Rev. 2007;31(3):287–314. doi: 10.1016/j.neubiorev.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Quik M, O’Neill M, Perez XA. Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci. 2007;28(5):229–35. doi: 10.1016/j.tips.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Quik M, et al. Multiple roles for nicotine in Parkinson’s disease. Biochem Pharmacol. 2009;78(7):677–85. doi: 10.1016/j.bcp.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buccafusco JJ, et al. Long-lasting cognitive improvement with nicotinic receptor agonists: mechanisms of pharmacokinetic-pharmacodynamic discordance. Trends Pharmacol Sci. 2005;26(7):352–60. doi: 10.1016/j.tips.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Littleton J, Rogers T, Falcone D. Novel approaches to plant drug discovery based on high throughput pharmacological screening and genetic manipulation. Life Sciences. 2005;78(5):467–475. doi: 10.1016/j.lfs.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Lopez MG, et al. Unmasking the functions of the chromaffin cell alpha7 nicotinic receptor by using short pulses of acetylcholine and selective blockers. Proc Natl Acad Sci U S A. 1998;95(24):14184–9. doi: 10.1073/pnas.95.24.14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall M, et al. Characterization of [3H]cytisine binding to human brain membrane preparations. Brain Res. 1993;600(1):127–33. doi: 10.1016/0006-8993(93)90410-o. [DOI] [PubMed] [Google Scholar]
- 18.Davies AR, et al. Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling alpha 7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology. 1999;38(5):679–90. doi: 10.1016/s0028-3908(98)00221-4. [DOI] [PubMed] [Google Scholar]
- 19.Stegelmeier BL, et al. The toxicity and kinetics of larkspur alkaloid, methyllycaconitine, in mice. J Anim Sci. 2003;81(5):1237–41. doi: 10.2527/2003.8151237x. [DOI] [PubMed] [Google Scholar]
- 20.Brust A, et al. Differential evolution and neofunctionalization of snake venom metalloprotease domains. Mol Cell Proteomics. 2013;12(3):651–63. doi: 10.1074/mcp.M112.023135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Celie PH, et al. Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an alpha-conotoxin PnIA variant. Nat Struct Mol Biol. 2005;12(7):582–8. doi: 10.1038/nsmb951. [DOI] [PubMed] [Google Scholar]
- 22.Ridley DL, Pakkanen J, Wonnacott S. Effects of chronic drug treatments on increases in intracellular calcium mediated by nicotinic acetylcholine receptors in SH-SY5Y cells. Br J Pharmacol. 2002;135(4):1051–9. doi: 10.1038/sj.bjp.0704508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teng L, et al. Lobeline and nicotine evoke [3H]overflow from rat striatal slices preloaded with [3H]dopamine: differential inhibition of synaptosomal and vesicular [3H]dopamine uptake. J Pharmacol Exp Ther. 1997;280(3):1432–44. [PubMed] [Google Scholar]
- 24.Nickell JR, et al. Lobelane inhibits methamphetamine-evoked dopamine release via inhibition of the vesicular monoamine transporter-2. J Pharmacol Exp Ther. 2010;332(2):612–21. doi: 10.1124/jpet.109.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Felpin F-X, Lebreton J. History, chemistry and biology of alkaloids from Lobelia inflata. Tetrahedron. 2004;60(45):10127–10153. [Google Scholar]
- 26.Marek GJ, Vosmer G, Seiden LS. Dopamine uptake inhibitors block long-term neurotoxic effects of methamphetamine upon dopaminergic neurons. Brain Res. 1990;513(2):274–9. doi: 10.1016/0006-8993(90)90467-p. [DOI] [PubMed] [Google Scholar]
- 27.Sandoval V, et al. Methylphenidate alters vesicular monoamine transport and prevents methamphetamine-induced dopaminergic deficits. J Pharmacol Exp Ther. 2003;304(3):1181–7. doi: 10.1124/jpet.102.045005. [DOI] [PubMed] [Google Scholar]
- 28.Hornykiewicz O. Biochemical aspects of Parkinson’s disease. Neurology. 1998;51(2 Suppl 2):S2–9. doi: 10.1212/wnl.51.2_suppl_2.s2. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt CJ, Gibb JW. Role of the dopamine uptake carrier in the neurochemical response to methamphetamine: effects of amfonelic acid. Eur J Pharmacol. 1985;109(1):73–80. doi: 10.1016/0014-2999(85)90541-2. [DOI] [PubMed] [Google Scholar]
- 30.Brown JM, Yamamoto BK. Effects of amphetamines on mitochondrial function: role of free radicals and oxidative stress. Pharmacol Ther. 2003;99(1):45–53. doi: 10.1016/s0163-7258(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 31.Storch A, Ludolph AC, Schwarz J. Dopamine transporter: involvement in selective dopaminergic neurotoxicity and degeneration. J Neural Transm. 2004;111(10–11):1267–86. doi: 10.1007/s00702-004-0203-2. [DOI] [PubMed] [Google Scholar]
- 32.Fleming SM, Delville Y, Schallert T. An intermittent, controlled-rate, slow progressive degeneration model of Parkinson’s disease: antiparkinson effects of Sinemet and protective effects of methylphenidate. Behav Brain Res. 2005;156(2):201–13. doi: 10.1016/j.bbr.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Cicchetti F, Drouin-Ouellet J, Gross RE. Environmental toxins and Parkinson’s disease: what have we learned from pesticide-induced animal models? Trends Pharmacol Sci. 2009;30(9):475–83. doi: 10.1016/j.tips.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Sheet PF, Page GC. CARDINAL FLOWER [Google Scholar]
- 35.Gerhardt GA, Burmeister JJ. Encyclopedia of Analytical Chemistry. John Wiley & Sons, Ltd; 2006. Voltammetry In Vivo for Chemical Analysis of the Nervous System. [Google Scholar]
- 36.Gerhardt GA, et al. Nafion-coated electrodes with high selectivity for CNS electrochemistry. Brain Research. 1984;290(2):390–395. doi: 10.1016/0006-8993(84)90963-6. [DOI] [PubMed] [Google Scholar]
- 37.Rose GM, et al. Age-related alterations in monoamine release from rat striatum: an in vivo electrochemical study. Neurobiol Aging. 1986;7(2):77–82. doi: 10.1016/0197-4580(86)90143-0. [DOI] [PubMed] [Google Scholar]
- 38.Cass WA, Gerhardt GA. Direct in vivo evidence that D2 dopamine receptors can modulate dopamine uptake. Neurosci Lett. 1994;176(2):259–63. doi: 10.1016/0304-3940(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 39.Cass WA, et al. Clearance of exogenous dopamine in rat dorsal striatum and nucleus accumbens: role of metabolism and effects of locally applied uptake inhibitors. J Neurochem. 1993;61(6):2269–78. doi: 10.1111/j.1471-4159.1993.tb07469.x. [DOI] [PubMed] [Google Scholar]
- 40.Gerhardt GA, Rose GM, Hoffer BJ. Release of monoamines from striatum of rat and mouse evoked by local application of potassium: evaluation of a new in vivo electrochemical technique. J Neurochem. 1986;46(3):842–50. doi: 10.1111/j.1471-4159.1986.tb13048.x. [DOI] [PubMed] [Google Scholar]
- 41.Cass WA, et al. Clearance of exogenous dopamine in rat dorsal striatum and nucleus accumbens: role of metabolism and effects of locally applied uptake inhibitors. J Neurochem. 1993;61(6):2269–78. doi: 10.1111/j.1471-4159.1993.tb07469.x. [DOI] [PubMed] [Google Scholar]
- 42.Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16(Spec No 2):R183–94. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 43.Cadet JL, Krasnova IN. Molecular bases of methamphetamine-induced neurodegeneration. Int Rev Neurobiol. 2009;88:101–19. doi: 10.1016/S0074-7742(09)88005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riddle EL, Fleckenstein AE, Hanson GR. Mechanisms of methamphetamine-induced dopaminergic neurotoxicity. AAPS J. 2006;8(2):E413–8. doi: 10.1007/BF02854914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koylu E, et al. Sex difference in up-regulation of nicotinic acetylcholine receptors in rat brain. Life Sci. 1997;61(12):PL 185–90. doi: 10.1016/s0024-3205(97)00665-6. [DOI] [PubMed] [Google Scholar]
- 46.Gattu M, Terry AV, Jr, Buccafusco JJ. A rapid microtechnique for the estimation of muscarinic and nicotinic receptor binding parameters using 96-well filtration plates. J Neurosci Methods. 1995;63(1–2):121–25. doi: 10.1016/0165-0270(95)00100-x. [DOI] [PubMed] [Google Scholar]
- 47.Gnadisch D, et al. High affinity binding of [3H]epibatidine to rat brain membranes. Neuroreport. 1999;10(8):1631–6. doi: 10.1097/00001756-199906030-00002. [DOI] [PubMed] [Google Scholar]
- 48.Gerzanich V, et al. Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol. 1995;48(4):774–82. [PubMed] [Google Scholar]
- 49.Saitoh F, Noma M, Kawashima N. The alkaloid contents of sixty Nicotiana species. Phytochemistry. 1985;24(3):477–480. [Google Scholar]
- 50.Clugston DM, MacLean DB, Manske RHF. The examination of lobinaline and some degradation products by mass spectrometry. Canadian Journal of Chemistry. 1967;45(1):39–47. [Google Scholar]
- 51.Lutz JA, et al. A nicotinic receptor-mediated anti-inflammatory effect of the flavonoid rhamnetin in BV2 microglia. Fitoterapia. 2014;98:11–21. doi: 10.1016/j.fitote.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimura M, et al. Syntheses of novel diphenyl piperazine derivatives and their activities as inhibitors of dopamine uptake in the central nervous system. Bioorg Med Chem. 2003;11(8):1621–30. doi: 10.1016/s0968-0896(03)00061-0. [DOI] [PubMed] [Google Scholar]
- 53.Andersen PH. Biochemical and pharmacological characterization of [3H]GBR 12935 binding in vitro to rat striatal membranes: labeling of the dopamine uptake complex. J Neurochem. 1987;48(6):1887–96. doi: 10.1111/j.1471-4159.1987.tb05752.x. [DOI] [PubMed] [Google Scholar]
- 54.Berger P, et al. [3H]GBR-12935: a specific high affinity ligand for labeling the dopamine transport complex. Eur J Pharmacol. 1985;107(2):289–90. doi: 10.1016/0014-2999(85)90075-5. [DOI] [PubMed] [Google Scholar]
- 55.Wada A, et al. Inhibition of Na+-pump enhances carbachol-induced influx of 45Ca2+ and secretion of catecholamines by elevation of cellular accumulation of 22Na+ in cultured bovine adrenal medullary cells. Naunyn Schmiedebergs Arch Pharmacol. 1986;332(4):351–6. doi: 10.1007/BF00500086. [DOI] [PubMed] [Google Scholar]
- 56.Gandia L, et al. Inhibition of nicotinic receptor-mediated responses in bovine chromaffin cells by diltiazem. Br J Pharmacol. 1996;118(5):1301–7. doi: 10.1111/j.1476-5381.1996.tb15537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grando SA, et al. Activation of keratinocyte nicotinic cholinergic receptors stimulates calcium influx and enhances cell differentiation. J Invest Dermatol. 1996;107(3):412–8. doi: 10.1111/1523-1747.ep12363399. [DOI] [PubMed] [Google Scholar]
- 58.Chen R, et al. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci U S A. 2006;103(24):9333–8. doi: 10.1073/pnas.0600905103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saw CL, et al. The berry constituents quercetin, kaempferol, and pterostilbene synergistically attenuate reactive oxygen species: involvement of the Nrf2-ARE signaling pathway. Food Chem Toxicol. 2014;72:303–11. doi: 10.1016/j.fct.2014.07.038. [DOI] [PubMed] [Google Scholar]
- 60.Dwoskin LP, Zahniser NR. Robust modulation of [3H]dopamine release from rat striatal slices by D-2 dopamine receptors. J Pharmacol Exp Ther. 1986;239(2):442–53. [PubMed] [Google Scholar]
- 61.Miller DK, Crooks PA, Dwoskin LP. Lobeline inhibits nicotine-evoked [(3)H]dopamine overflow from rat striatal slices and nicotine-evoked (86)Rb(+) efflux from thalamic synaptosomes. Neuropharmacology. 2000;39(13):2654–62. doi: 10.1016/s0028-3908(00)00140-4. [DOI] [PubMed] [Google Scholar]
- 62.Gerhardt GA, Hoffman AF. Effects of recording media composition on the responses of Nafion-coated carbon fiber microelectrodes measured using high-speed chronoamperometry. J Neurosci Methods. 2001;109(1):13–21. doi: 10.1016/s0165-0270(01)00396-x. [DOI] [PubMed] [Google Scholar]
- 63.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic press; 2007. [DOI] [PubMed] [Google Scholar]
- 64.Gerhardt GA, Palmer MR. Characterization of the techniques of pressure ejection and microiontophoresis using in vivo electrochemistry. J Neurosci Methods. 1987;22(2):147–59. doi: 10.1016/0165-0270(87)90009-4. [DOI] [PubMed] [Google Scholar]
- 65.Zahniser NR, Larson GA, Gerhardt GA. In vivo dopamine clearance rate in rat striatum: regulation by extracellular dopamine concentration and dopamine transporter inhibitors. J Pharmacol Exp Ther. 1999;289(1):266–77. [PubMed] [Google Scholar]
- 66.Lipinski CA, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 67.Li Q, et al. PubChem as a public resource for drug discovery. Drug Discov Today. 2010;15(23–24):1052–7. doi: 10.1016/j.drudis.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.National Center for Biotechnology Information. [accessed May 4, 2015];PubChem Compound Database; CID=419219. http://pubchem.ncbi.nlm.nih.gov/compound/39484.
- 69.Villarroya M, et al. Measurement of Ca2+ Entry Using 45Ca2+ In: Lambert D, editor. Calcium Signaling Protocols. Humana Press; 1999. pp. 137–147. [DOI] [PubMed] [Google Scholar]
- 70.Chatterjee S, et al. Partial agonists of the alpha3beta4* neuronal nicotinic acetylcholine receptor reduce ethanol consumption and seeking in rats. Neuropsychopharmacology. 2011;36(3):603–15. doi: 10.1038/npp.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie HR, Hu LS, Li GY. SH-SY5Y human neuroblastoma cell line: in vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin Med J (Engl) 2010;123(8):1086–92. [PubMed] [Google Scholar]
- 72.Presgraves SP, et al. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox Res. 2004;5(8):579–98. doi: 10.1007/BF03033178. [DOI] [PubMed] [Google Scholar]
- 73.Clarke PB, Reuben M. Release of [3H]-noradrenaline from rat hippocampal synaptosomes by nicotine: mediation by different nicotinic receptor subtypes from striatal [3H]-dopamine release. Br J Pharmacol. 1996;117(4):595–606. doi: 10.1111/j.1476-5381.1996.tb15232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mihalak KB, Carroll FI, Luetje CW. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol. 2006;70(3):801–5. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- 75.Zhu BT. Mechanistic explanation for the unique pharmacologic properties of receptor partial agonists. Biomed Pharmacother. 2005;59(3):76–89. doi: 10.1016/j.biopha.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 76.Robison MM, et al. The skeletal structure of lobinaline. J Org Chem. 1966;31(10):3206–13. doi: 10.1021/jo01348a027. [DOI] [PubMed] [Google Scholar]
- 77.Manske RHF. LOBINALINE, AN ALKALOID FROM LOBELIA CARDINALIS L. Canadian Journal of Research. 1938;16b(12):445–448. [Google Scholar]
- 78.Robison MM, et al. The stereochemistry and synthesis of the lobinaline ring system. J Org Chem. 1966;31(10):3220–3. doi: 10.1021/jo01348a029. [DOI] [PubMed] [Google Scholar]
- 79.Gupta RN, Spenser ID. Biosynthesis of Lobinaline. Canadian Journal of Chemistry. 1971;49(3):384–397. [Google Scholar]
- 80.Hojahmat M, et al. Lobeline esters as novel ligands for neuronal nicotinic acetylcholine receptors and neurotransmitter transporters. Bioorg Med Chem. 2010;18(2):640–9. doi: 10.1016/j.bmc.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rueter LE, et al. A-85380: a pharmacological probe for the preclinical and clinical investigation of the alphabeta neuronal nicotinic acetylcholine receptor. CNS Drug Rev. 2006;12(2):100–12. doi: 10.1111/j.1527-3458.2006.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gomes NG, et al. Plants with neurobiological activity as potential targets for drug discovery. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(8):1372–89. doi: 10.1016/j.pnpbp.2009.07.033. [DOI] [PubMed] [Google Scholar]
- 83.Koeberle A, Werz O. Multi-target approach for natural products in inflammation. Drug Discov Today. 2014;19(12):1871–82. doi: 10.1016/j.drudis.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 84.Virmani A, Gaetani F, Binienda Z. Effects of metabolic modifiers such as carnitines, coenzyme Q10, and PUFAs against different forms of neurotoxic insults: metabolic inhibitors, MPTP, and methamphetamine. Ann N Y Acad Sci. 2005;1053:183–91. doi: 10.1196/annals.1344.016. [DOI] [PubMed] [Google Scholar]
- 85.Virmani A, et al. Metabolic syndrome in drug abuse. Ann N Y Acad Sci. 2007;1122:50–68. doi: 10.1196/annals.1403.004. [DOI] [PubMed] [Google Scholar]
- 86.Kita T, et al. Protective effects of phytochemical antioxidants against neurotoxin-induced degeneration of dopaminergic neurons. J Pharmacol Sci. 2014;124(3):313–9. doi: 10.1254/jphs.13r19cp. [DOI] [PubMed] [Google Scholar]
- 87.Phillipson OT. Management of the aging risk factor for Parkinson’s disease. Neurobiol Aging. 2014;35(4):847–57. doi: 10.1016/j.neurobiolaging.2013.10.073. [DOI] [PubMed] [Google Scholar]
- 88.Bournival J, Quessy P, Martinoli MG. Protective effects of resveratrol and quercetin against MPP+-induced oxidative stress act by modulating markers of apoptotic death in dopaminergic neurons. Cell Mol Neurobiol. 2009;29(8):1169–80. doi: 10.1007/s10571-009-9411-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karuppagounder SS, et al. Quercetin up-regulates mitochondrial complex-I activity to protect against programmed cell death in rotenone model of Parkinson’s disease in rats. Neuroscience. 2013;236:136–48. doi: 10.1016/j.neuroscience.2013.01.032. [DOI] [PubMed] [Google Scholar]
- 90.Francis MM, et al. Specific activation of the alpha 7 nicotinic acetylcholine receptor by a quaternary analog of cocaine. Mol Pharmacol. 2001;60(1):71–9. doi: 10.1124/mol.60.1.71. [DOI] [PubMed] [Google Scholar]
- 91.Green BT, et al. Plant toxins that affect nicotinic acetylcholine receptors: a review. Chemical research in toxicology. 2013;26(8):1129–1138. doi: 10.1021/tx400166f. [DOI] [PubMed] [Google Scholar]
- 92.Middleton LS, Cass WA, Dwoskin LP. Nicotinic receptor modulation of dopamine transporter function in rat striatum and medial prefrontal cortex. J Pharmacol Exp Ther. 2004;308(1):367–77. doi: 10.1124/jpet.103.055335. [DOI] [PubMed] [Google Scholar]
- 93.Hoffman AF, Gerhardt GA. In vivo electrochemical studies of dopamine clearance in the rat substantia nigra: effects of locally applied uptake inhibitors and unilateral 6-hydroxydopamine lesions. J Neurochem. 1998;70(1):179–89. doi: 10.1046/j.1471-4159.1998.70010179.x. [DOI] [PubMed] [Google Scholar]
- 94.Gulley JM, Larson GA, Zahniser NR. Using High-Speed Chronoamperometry with Local Dopamine Application to Assess Dopamine Transporter Function. [PubMed] [Google Scholar]
- 95.Joyce BM, Glaser PE, Gerhardt GA. Adderall produces increased striatal dopamine release and a prolonged time course compared to amphetamine isomers. Psychopharmacology (Berl) 2007;191(3):669–77. doi: 10.1007/s00213-006-0550-9. [DOI] [PubMed] [Google Scholar]
- 96.Glaser PE, et al. Differential effects of amphetamine isomers on dopamine release in the rat striatum and nucleus accumbens core. Psychopharmacology (Berl) 2005;178(2–3):250–8. doi: 10.1007/s00213-004-2012-6. [DOI] [PubMed] [Google Scholar]
- 97.Inamdar AP. Ligand-based pharmacophore studies in the dopaminergic system. 2011 [Google Scholar]
- 98.Enyedy IJ, et al. Pharmacophore-based discovery of substituted pyridines as novel dopamine transporter inhibitors. Bioorg Med Chem Lett. 2003;13(3):513–7. doi: 10.1016/s0960-894x(02)00943-5. [DOI] [PubMed] [Google Scholar]
- 99.Kem WR, et al. Anabaseine is a potent agonist on muscle and neuronal alpha-bungarotoxin-sensitive nicotinic receptors. J Pharmacol Exp Ther. 1997;283(3):979–92. [PubMed] [Google Scholar]
- 100.Tsuneki H, et al. Marine alkaloids (−)-pictamine and (−)-lepadin B block neuronal nicotinic acetylcholine receptors. Biol Pharm Bull. 2005;28(4):611–4. doi: 10.1248/bpb.28.611. [DOI] [PubMed] [Google Scholar]
- 101.Daly JW, et al. Decahydroquinoline alkaloids: noncompetitive blockers for nicotinic acetylcholine receptor-channels in pheochromocytoma cells and Torpedo electroplax. Neurochem Res. 1991;16(11):1207–12. doi: 10.1007/BF00966697. [DOI] [PubMed] [Google Scholar]
- 102.Nathanson JA, et al. Cocaine as a naturally occurring insecticide. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(20):9645–9648. doi: 10.1073/pnas.90.20.9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hill ER, et al. Potencies of cocaine methiodide on major cocaine targets in mice. PLoS One. 2009;4(10):e7578. doi: 10.1371/journal.pone.0007578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4(11):682–90. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 105.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3(8):711–5. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 106.Feher M, Schmidt JM. Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry. J Chem Inf Comput Sci. 2003;43(1):218–27. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- 107.Salim AA, Chin Y-W, Kinghorn AD. Drug Discovery from Plants. In: Ramawat KG, Merillon JM, editors. Bioactive Molecules and Medicinal Plants. Springer; Berlin Heidelberg: 2008. pp. 1–24. [Google Scholar]
- 108.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44(1):235–49. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 109.Dekundy A, et al. Effects of dopamine uptake inhibitor MRZ-9547 in animal models of Parkinson’s disease. J Neural Transm. 2014 doi: 10.1007/s00702-014-1326-8. [DOI] [PubMed] [Google Scholar]
- 110.Huot P, et al. UWA-121, a mixed dopamine and serotonin re-uptake inhibitor, enhances L-DOPA anti-parkinsonian action without worsening dyskinesia or psychosis-like behaviours in the MPTP-lesioned common marmoset. Neuropharmacology. 2014;82:76–87. doi: 10.1016/j.neuropharm.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 111.Di Paolo T, et al. AQW051, a novel and selective nicotinic acetylcholine receptor alpha7 partial agonist, reduces l-Dopa-induced dyskinesias and extends the duration of l-Dopa effects in parkinsonian monkeys. Parkinsonism Relat Disord. 2014;20(11):1119–23. doi: 10.1016/j.parkreldis.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 112.Batman AM, et al. The selective dopamine uptake inhibitor, D-84, suppresses cocaine self-administration, but does not occasion cocaine-like levels of generalization. Eur J Pharmacol. 2010;648(1–3):127–32. doi: 10.1016/j.ejphar.2010.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rothman RB, et al. Dopamine transport inhibitors based on GBR12909 and benztropine as potential medications to treat cocaine addiction. Biochem Pharmacol. 2008;75(1):2–16. doi: 10.1016/j.bcp.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Desai RI, et al. Pharmacological characterization of a dopamine transporter ligand that functions as a cocaine antagonist. J Pharmacol Exp Ther. 2014;348(1):106–15. doi: 10.1124/jpet.113.208538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li L, et al. The stereotypy-inducing effects of N-substituted benztropine analogs alone and in combination with cocaine do not account for their blockade of cocaine self-administration. Psychopharmacology (Berl) 2013;225(3):733–42. doi: 10.1007/s00213-012-2862-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schmitt KC, et al. INTERACTION OF COCAINE-, BENZTROPINE-, AND GBR12909-LIKE COMPOUNDS WITH WILDTYPE AND MUTANT HUMAN DOPAMINE TRANSPORTERS: MOLECULAR FEATURES THAT DIFFERENTIALLY DETERMINE ANTAGONIST BINDING PROPERTIES. Journal of neurochemistry. 2008;107(4):928–940. doi: 10.1111/j.1471-4159.2008.05667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Howell LL, Wilcox KM. The dopamine transporter and cocaine medication development: drug self-administration in nonhuman primates. J Pharmacol Exp Ther. 2001;298(1):1–6. [PubMed] [Google Scholar]
- 118.Leung EL, et al. Network-based drug discovery by integrating systems biology and computational technologies. Brief Bioinform. 2013;14(4):491–505. doi: 10.1093/bib/bbs043. [DOI] [PMC free article] [PubMed] [Google Scholar]