

Abstract
Medullary thyroid cancer (MTC) can be caused by germline mutations of the RET proto-oncogene or occurs as a sporadic form. It is well known that RET mutations affecting the cysteine-rich region of the protein (MEN2A-like mutations) are correlated with different phenotypes than those in the kinase domain (MEN2B-like mutations). Our aim was to analyse the whole-gene expression profile of MTC with regard to the type of RET gene mutation and the cancer genetic background (hereditary vs sporadic). We studied 86 MTC samples. We demonstrated that there were no distinct differences in the gene expression profiles of hereditary and sporadic MTCs. This suggests a homogeneous nature of MTC. We also noticed that the site of the RET gene mutation slightly influenced the gene expression profile of MTC. We found a significant association between the localization of RET mutations and the expression of three genes: NNAT (suggested to be a tumour suppressor gene), CDC14B (involved in cell cycle control) and NTRK3 (tyrosine receptor kinase that undergoes rearrangement in papillary thyroid cancer). This study suggests that these genes are significantly deregulated in tumours with MEN2A-like and MEN2B-like mutations; however, further investigations are necessary to demonstrate any clinical impact of these findings.
Medullary thyroid cancer (MTC) is currently a useful model for cancer therapy because its strong genetic predisposition provides a rationale for clinical management based on patient genotype. Moreover, following the identification of the tyrosine kinase activity of the RET protein, targeted therapy of MTC has been introduced recently into clinical use, providing an option for patients with advanced disease. Both these issues raise questions about the molecular events, which drive the phenotype of MTC. MTC arises from parafollicular C cells of the thyroid and accounts for a relatively low proportion (3–5%) of all thyroid cancers1,2,3. However, the estimated number of MTC cases worldwide is high since the incidence ranges from 0.11 to 0.21 per 100,000 person/year4. MTC occurs as both a hereditary disease (20–30% of all cases) and a sporadic disease5,6. The hereditary type is a consequence of germline mutations in the RET proto-oncogene and is associated with multiple endocrine neoplasia type 2 syndrome (MEN2), inherited in an autosomal dominant pattern. Historically, MEN2 syndrome has been classified into three distinct variants: MEN2A (OMIM 171400), which occurs in 80% of all hereditary cases7,8, familial medullary thyroid cancer (FMTC) (OMIM 155240), arising in up to 57% of all hereditary cases, and MEN2B (OMIM 162300), in approximately 5% of all hereditary MTCs9. Normally, ligand binding triggers the dimerization and activation of the RET receptor, which induces a signal transduction pathway leading to cell proliferation10,11. In the absence of a ligand, the RET protein is a single unphosphorylated tyrosine kinase receptor, but in cancer cells, single point mutations in the RET proto-oncogene lead to the autophosphorylation of tyrosine residues, which consequently induces the constitutive activation of the RET receptor and a permanent gain-of-function12,13. There is a significant relationship between the site of RET mutation and the phenotype, as well as the clinical course of MEN2 syndrome, and it is believed that RET mutations exhibit different transforming potentials depending on their location within the RET molecule7,8,14,15.
The RET protein is composed of three domains. The extracellular domain includes a ligand binding site, a cadherin-like domain, and a cysteine-rich domain. Mutations located in the extracellular domain may activate the ligand-independent dimerization and autophosphorylation of the RET protein. Mutations located in this region are mainly associated with MEN2A and more rarely with FMTC12,16. The most frequent RET mutations affecting codon 634 (exon 11) are detectable in up to 85% of all MEN2A patients, while mutations in codons 609, 611, 618 and 620 (exon 10) account for 10–15%2,17,18. Mutations in codon 10 are also observed in FMTC syndrome, in which MTC is the only symptom of the disease, often with a late onset and a more benign clinical course. However, the distinction between FMTC and MEN2A is rather debatable, and FMTC is often considered as a phenotypic variant of MEN2A19,20. The second RET protein domain is a hydrophobic region located within the cell membrane, and the third one is an intracellular tyrosine kinase domain (TK), which contains two tyrosine residues (TK1 and TK2). Mutations in this domain (mainly in codon 918 of the TK2 domain) lead to the autophosphorylation of the protein and activate the RET receptor without dimerization, but there is a distinct difference in the transforming activity among those mutations. Mutations affecting the first kinase subdomain (TK1), related to exons 13 and 14, are mainly observed in FMTC syndrome, whereas those involving the second kinase subdomain (TK2) are usually observed in codon 918. Germline mutations are associated with the most aggressive presentation of MTC, MEN2B syndrome, which is characterized by an earlier age of onset, developmental abnormalities including marfanoid habitus with skeletal deformations, intestinal and mucosal ganglioneuromatosis, and pheochromocytoma in 50% of cases. It has been suggested that the activation of the RET receptor due to a germline RET 918 mutation is related to an alteration in the substrate recognition pocket of the catalytic core and facilitates the phosphorylation of novel substrates13,17,21, which may lead to the induction of different downstream pathways. Seventy five percent of MTC patients have no familial history. Somatic RET mutations are observed in 40–70% of sporadic tumours22,23,24,25. Additionally, RAS mutations are detected in a high proportion of RET-negative tumours26,27; however, the role of somatic RAS mutations in MTC development is still not clearly demonstrated. As MEN2B-like mutations are considered the major somatic events in sporadic cancer, o a paradoxical discrepancy occurs between the possibly more aggressive clinical course of somatic RET 918-induced cancer and the generally better prognoses in all sporadic cancers.
It would, therefore, be valuable to compare gene expression profiles not only between hereditary and sporadic MTC but also between sporadic MTC caused by MEN2B-like mutations in RET exon 16 and MEN2A-like mutations in codons 10–11. Only a few similar comparisons have been reported, and the results are ambiguous due to the small number of tumours analysed28,29,30,31. In our study, two null hypotheses were tested:
There is no difference in the gene expression profiles of hereditary and sporadic MTC.
There is no difference in the gene expression profiles of MTC caused by MEN2A-like or MEN2B-like mutations.
For the purpose of this study, all mutations in RET exons 10 and 11, both hereditary and sporadic, were classified as MEN2A-like, whereas both germline and somatic mutations in RET exon 16 were classified as MEN2B-like.
Results
Differences in the gene expression profiles of hereditary and sporadic MTC
Differences in the gene expression profiles were analysed in a dataset of 60 MTCs including 22 hereditary and 38 sporadic cases (Set A) (Supplementary Information Table S1). Using unsupervised analysis by hierarchical clustering, we did not observe global differences in the gene expression profiles between patients in whom the disease was caused by germline RET mutation and those with sporadic MTC, either with or without somatic RET mutation (Fig. 1). We then carried out a supervised analysis with 15 probe sets, which detected differential expression between the subtypes (FDR <0.05). The results are summarized in Table 1.
Figure 1. Hierarchical clustering of MTC samples based on microarray results.

Table 1. Genes differentiating between hereditary and sporadic MTC with a FDR <0.05 (Set A).
| Symbol | ProbeSet | Name | Fold-change | p-value | FDR | p-value validation set HG-U133A |
|---|---|---|---|---|---|---|
| PTPRK | 8129418 | protein tyrosine phosphatase, receptor type, K | 0.48 | 0,0000255 | 0,0308 | 0,2 |
| KLF12 | 7972003 | Kruppel-like factor 12 | 0.69 | 0,0000585 | 0,0353 | 0,3 |
| CCL18 | 8006594 | chemokine (C-C motif) ligand 18 (pulmonary and activation-regulated) | 2.88 | 0,0000412 | 0,0351 | 0,4 |
| SLC22A3 | 8123246 | solute carrier family 22 (extraneuronal monoamine transporter), member 3 | 2.12 | 0,0000696 | 0,0387 | 0,4 |
| FHOD3 | 8020973 | formin homology 2 domain containing 3 | 1.65 | 0,0000543 | 0,0353 | 0,6 |
| TAC3 | 7964303 | tachykinin 3 | 1.44 | 0,0000154 | 0,0223 | 0,7 |
| ZNF432 | 8038942 | zinc finger protein 432 | 0.74 | 0,0000536 | 0,0353 | 0,8 |
| LECT1 | 7971838 | leukocyte cell derived chemotaxin 1 | 3.3 | 0,0000976 | 0,0471 | 0,9 |
| TCERG1L | 7937059 | transcription elongation regulator 1-like | 1.4 | 0,0000019 | 0,0123 | not available on HG-U133A |
| DPP10 | 8044700 | dipeptidyl-peptidase 10 (non-functional) | 2.85 | 0,0000034 | 0,0123 | not available on HG-U133A |
| NALCN | 7972601 | sodium leak channel, non-selective | 0.4 | 0,0000114 | 0,0223 | not available on HG-U133A |
| APOA1BP | 7906185 | apolipoprotein A-I binding protein | 1.46 | 0,0000143 | 0,0223 | not available on HG-U133A |
| ZNF404 | 8037430 | zinc finger protein 404 | 0.72 | 0,0000419 | 0,0351 | not available on HG-U133A |
| SPTSSB | 8091799 | serine palmitoyltransferase, small subunit B | 0.35 | 0,0000437 | 0,0351 | not available on HG-U133A |
| KIAA1737 | 7975926 | KIAA1737 | 0.76 | 0,0000922 | 0,0471 | not available on HG-U133A |
Genes differing in expression between MTC tumours caused by different types of RET mutations
The gene expression profiles were compared for 21 MEN2A-like samples (5 somatic and 16 hereditary) and 9 MEN2B-like samples (7 somatic and 2 hereditary) (Set B) (Supplementary Information Table S1). While hierarchical clustering did not indicate any global influence of the type of RET mutation on the gene expression profile (Fig. 1), further analysis using a supervised method revealed ten genes for which the expression was significantly changed (FDR <0.05) in tumours with MEN2A-like mutations compared to tumours with MEN2B-like mutations (Table 2).
Table 2. Comparison of gene expression in samples with MEN2A-like and MEN2B-like RET mutations using probe sets with a FDR <0.05.
| Symbol | ProbeSet | Mean expression in mutations |
Fold-change | Uncorrected p-value | FDR | |
|---|---|---|---|---|---|---|
| MEN 2A-like | MEN 2B-like | |||||
| NTRK3* | 7991186 | 27.86 | 134.47 | 0.21 | 2.70E-06 | 0.0195 |
| NALCN | 7972601 | 33.13 | 105.66 | 0.31 | 7.10E-06 | 0.0257 |
| ZC3H12C | 7943715 | 88.65 | 52.91 | 1.68 | 1.22E-05 | 0.0294 |
| HMGA2 | 7964736 | 23.13 | 41.51 | 0.56 | 1.69E-05 | 0.0306 |
| HLA-DRB5 | 8125436 | 52.45 | 147.6 | 0.36 | 2.38E-05 | 0.0344 |
| PDGFRL | 8144802 | 215.63 | 135.82 | 1.59 | 4.21E-05 | 0.0393 |
| CDC14B | 8162610 | 57.87 | 35.18 | 1.65 | 4.29E-05 | 0.0393 |
| NNAT* | 8062395 | 291.36 | 44.53 | 6.54 | 4.59E-05 | 0.0393 |
| ZNF658 | 8161346 | 41.25 | 28.16 | 1.46 | 4.91E-05 | 0.0393 |
| HSD17B14 | 8038213 | 129.36 | 78.49 | 1.65 | 5.43E-05 | 0.0393 |
NTRK3 expression and RET mutation type
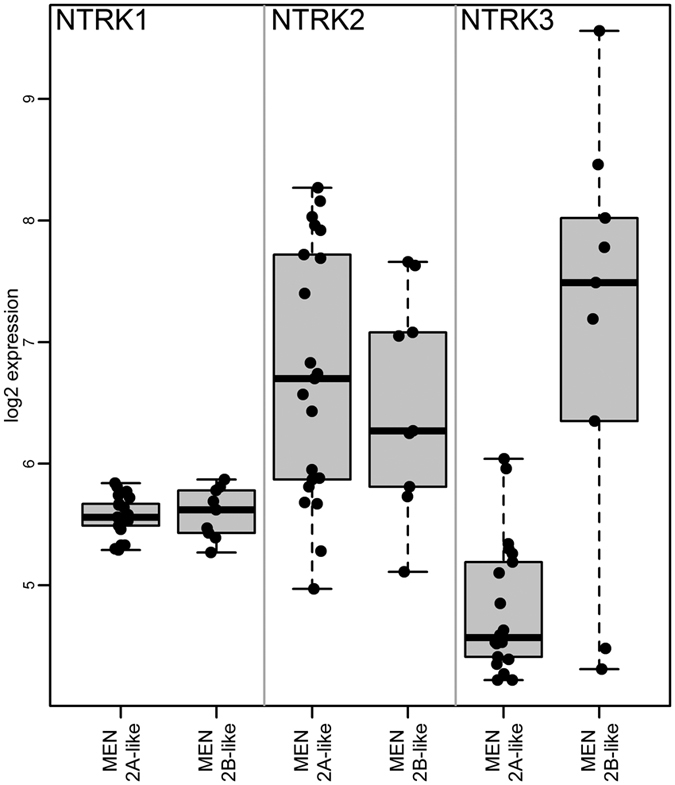
In the microarray supervised data analysis, NTRK3 was the most significantly deregulated gene, almost five times higher in MEN2B-like samples than in MEN2A-like samples. For comparison, the two other variants of the NTRK receptor (NTRK1, NTRK2) were also examined in our transcriptome profiling, but no association with RET mutation type was observed for either of them (NTRK1 was filtered out during the pre-processing step, and NTRK2 was insignificant (FDR = 0.86) (Fig. 2). Twenty-four samples were checked according to the presence of the ETS variant 6 gene (ETV6)-neurotropin receptor 3 (NTRK3) rearrangement (ETV6-NTRK3). The ETV6-NTRK3 fusion gene was not detected in any of the analysed MTC samples (Supplementary Information Fig. S2).
Figure 2. Expression of 3 types of NTRK receptors in MEN2A-like and MEN 2B-like samples in the microarray analysis.
Tumour classification by RET mutation type
To verify how the observed differences delineated the MTC samples with different RET mutation subtypes, we trained a 4-gene classifier (Table 3). Within this classifier, 2 genes (NNAT and PTPRT) were up-regulated for MEN2A-like mutations, and 2 genes (NTRK3 and GABRR1) were up-regulated for MEN2B-like mutations. LOOCV resulted in the correct classification of 20/21 RET MEN2A-like mutation samples and 7/9 RET MEN2B-like mutations, thus showing an accuracy of 90%, a sensitivity of 91%, and a specificity of 87.5% (Table 4).
Table 3. Genes used in classification by type of RET mutation.
| p value | FDR | mean MEN2A-like mutations | mean MEN2B-like mutations | Fold-change | ProbeSet | Symbol |
|---|---|---|---|---|---|---|
| 2.70E-06 | 0.0195 | 27.86 | 134.47 | 0.21 | 7991186 | NTRK3 |
| 4.59E-05 | 0.0393 | 291.36 | 44.53 | 6.54 | 8062395 | NNAT |
| 0.000261 | 0.0957 | 108.38 | 597.72 | 0.18 | 8128087 | GABRR1* |
| 0.000951 | 0.127 | 263.98 | 65.09 | 4.06 | 8066347 | PTPRT* |
Table 4. Performance of the 4-gene classifier for prediction of MEN2A-like or MEN2B-like mutations (see Table 3).
| Accuracy | Sensitivity | Specificity | PPV | NPV |
|---|---|---|---|---|
| 90% | 91% | 87.50% | 95% | 78% |
Validation of the microarray data using independent sets of samples
Hereditary MTC vs sporadic MTC
We validated genes found as significantly deregulated between hereditary and sporadic MTCs, based on the microarray set (Affymetrix Gene 1.0 ST), on independent MTC samples analysed in our previous study32. Among 15 genes, 8 were found to be insignificant. The other 7 genes were not represented on the type of microarrays used in previous experiment (Affymetrix HG-U 133 A). We assessed those genes by RT-qPCR validation and did not see significant changes in expression between sporadic and hereditary forms of MTC.
MEN2A-like vs MEN2B-like mutations
For validation related to differences in the gene expression profiles dependent on RET mutation status, we applied the RT-qPCR method. For this purpose, we used 25 additional MTC samples, 16 with MEN2A-like mutations and 9 with MEN2B-like mutations. From 10 genes that were significantly differentially expressed (FDR <0.05), we were able to analyse 9 because, due to the difficulties with the insufficient quality of amplification, we had to exclude 1 gene (Supplementary Information Table S2). Using the validation set, we confirmed the overexpression of NNAT (Bonferroni adjusted p-value = 0.01, fold change = 3.3) and CDC14B (Bonferroni adjusted p-value = 0.003, fold change = 2.8) genes in samples with MEN2A-like mutations (p < 0,001) (Fig. 3). The expression of the other genes analysed did not show significant differences related to the RET mutation type.
Figure 3. Expression of CDC14B and NNAT genes in the microarray analysis and the RT-qPCR validation of independent sets of samples.

Despite using two independent amplicon designs and methodologies (Roche Diagnostic GmbH, Mannheim, Germany and TaqMan MGB, Life Technologies, Carlsbad, USA), we were also not able to detect significant differences in the microarray part of the study for NTRK3 expression. We observed very low amplification of the standard curve, and the samples resulted in very low efficiency of amplicons, suggesting technical difficulties with the assay. Therefore, we attempted the validation of the results for NTRK3 using immunohistochemistry.
Immunohistochemistry of the protein product of the NTRK3 gene
We analysed 12 samples: 5 MEN2A-like and 7 MEN2B-like samples. We observed very strong (+++) and strong (++) immunohistochemical staining with anti-TrkC antibody in all MEN2B-like cases. TrkC was localized mainly in the cytoplasm and in the nuclei of tumour cells in 3 cases. Immunostaining revealed that in these slides, TrkC was also present in follicular thyroid cells. On the other hand, all but one MEN 2A-like tumour were negative in TrkC staining for both tumour and follicular cells. Only one sample showed a positive reaction (Supplementary Information, Fig. S1).
Discussion
Differences in the gene expression profiles are usually easily observed when tumour tissue is compared to the corresponding normal tissue or between tumours of different genetic backgrounds (e.g., medullary versus papillary thyroid carcinoma). However, the identification of reliable gene expression biomarkers for tumours with more subtle differences, like distinct founding gene mutations, especially when affecting the same gene, is more difficult and requires much larger datasets to obtain consistent and reproducible results33, and such datasets are often difficult to collect when relatively rare conditions are studied. In MTC, a number of subsets must be considered, dependent on whether the disease is hereditary or sporadic, further complicated by the fact that each of these could be caused by gene mutations in the extracellular or intracellular domain of the RET protein or might not demonstrate RET mutations. Thus far, there have been only a few reports published that are dedicated to MTC, most of which represent limited datasets (23, 13 and 41 MTC cases)28,30,31. Here, we present the results of gene expression profiling in which all MTC subsets are well represented.
Our results clearly demonstrate that there were no distinct differences in the gene expression profiles of hereditary and sporadic MTCs. Therefore, we found a rather homogeneous nature with respect to MTC. Although we found 15 differentially expressed genes characterized by an FDR of <0.05 on microarrays, none of the 14 validated genes were significantly deregulated when tested by an independent group of samples.
Ameur et al.30 suggested that similar pathways might be activated in both sporadic and hereditary MTC and further linked the differences in expression of the genes with the aggressiveness of the tumour. However, they analysed a limited set of samples (4 hereditary MTC and 7 sporadic MTC)30. Conversely, Maliszewska et al.31 argued that familial and sporadic MTC cases are characterized by differential gene expression patterns, but this analysis considered almost all familial cases with a mutation in RET codon 634 without any sporadic cases31.
Similarly, in the case of breast cancer, a landmark genomic study suggested that hereditary and sporadic breast cancers are clearly distinguishable33, but we and others were able to demonstrate subsequently that other sources of heterogeneity might have influenced these conclusions and that hereditary and sporadic breast carcinomas are not in fact dissimilar34,35,36. Differences between hereditary and sporadic cancers may also be attributed to epigenetic events, for example, microRNA silencing37, and their role will probably be further investigated in the future.
Due to the strong genotype-phenotype correlation of the type of RET gene mutation and the clinical outcome, it has been proposed that RET mutations in MEN2A and MEN2B tumours result in changes of distinctive gene expression profiles and signalling pathways28,29,31. We identified 10 genes (FDR <0.05) by microarrays, whose expression distinguished MTC tumours with MEN2A-like mutations from those with MEN2B-like mutations. Among 9 genes validated by RT-qPCR on an independent set of samples, the differences were confirmed for only NNAT and CDC14B, while the other genes were not positively validated by the RT-qPCR.
We observed the highest fold change for the NNAT (neuronatin) gene among all differentially expressed genes, according to the RET mutation type. It is a brain-specific gene with high expression during embryogenesis and is normally silenced in differentiated neurons38. The overexpression of neuronatin is associated with cell proliferation and the shorter survival of glioblastoma patients39 and also promotes medulloblastoma growth40. In our data, the overexpression of the NNAT gene was observed in tumours with MEN2A-like mutations. Most of our MEN2A-like samples harbour a RET634 mutation, a type of mutation which correlates with poor prognoses among all MEN2A-like mutations. Mise et al. verified the high proliferation activity of tumours with MEN2A-like mutations by a transforming assay performed in NIH3T3 fibroblasts and an in vivo test in nude mice14. Mise et al. suggested that MEN2A mutations accelerate cell proliferation, while MEN2B mutations suppressed apoptosis14. They observed that cells with a RETC634R mutation, characteristic for MEN2A, grew rapidly. In vitro tests confirmed the results as the tumour growth was substantially faster in mice injected with NIH3T3 cells transfected with MEN2A-like mutation14. Interestingly, NNAT was also included in the list of genes up-regulated in MEN2A samples by Jain et al.28, but it was insignificant (FDR >0.1).
The second gene, CDC14B (cell division cycle 14B), was also over-expressed in MEN2A-like samples. This gene is a dual-specificity phosphatase that regulates the cell cycle41. The role in the tumour transformation ability of CDC14B gene is controversial. There are studies suggesting that the overexpression of the CDC14B gene induced the oncogenic transformation of transfected NIH3T3 cells and altered their motility42. On the other hand, there are some studies showing the down-regulation or loss of CDC14B expression in many tumours arising from breast, prostate, ovary, liver and brain43 and the inhibition of glioblastoma growth44. The down-regulation of CDC14B also correlated with tumour recurrence and shorter recurrence-free survival in renal cell carcinoma45. In our study, the down-regulated expression of CDC14B was observed in MEN2B-like mutation samples and believed to be clinically more aggressive. Additionally, the somatic M918T mutation correlates with worse MTC outcomes22. These findings might suggest that the expression of CDC14B modulates the prognosis in medullary thyroid cancer.
In our microarray studies, we observed the most significant difference in gene expression between MEN2B-like and MEN2A-like RET mutations, regarding the NTRK3 gene. This tyrosine kinase receptor gene, important for nerve growth factor signal transduction, plays a significant role in neuroendocrine carcinomas46,47. The overexpression of NTRK3 could be associated with the potentially higher clinical aggressiveness of MTC induced by MEN2B-like mutations. This observation is consistent with the results of a McGregor et al. study, where increased strong immunostaining of NTRK3 protein was noted in 87% of 25 analysed MTC samples. The authors correlated NTRK3 expression with aggressive metastatic tumours48, but that study did not consider the RET mutation status. Expression of TrkC promotes breast tumour growth and metastasis49. High expression of NTRK3 was observed in our MEN2B-like mutation cohort in the microarray part of the study, but due to technical reasons, this result could not be confirmed by RT-qPCR. Therefore, we attempted to validate the result with the protein level. Immunohistochemical analysis of NTRK3 expression may be difficult as its expression pattern in thyroid, particularly in MTC, is still not well understood. We consistently observed strong immunostaining in all MEN2B samples, present in both tumour C-cells and the surrounding follicular cells, while TrkC staining was negative in all but one analysed MEN2A-like tumour. However, we need to add that we observed non-specific staining in follicular cells. Positive staining by antibodies of both tumour and normal cells has also been reported by other authors who encountered similar obstacles; in many cases, the non-specific staining in normal cells was not possible to reduce50,51. Therefore, we consider the validation was successful, keeping in mind that most of the tumour samples were previously included in the microarray study. We also checked whether high expression of NTRK3 gene was related to ETV6/NTRK3 rearrangement, as reported in papillary thyroid cancer52,53, but we did not find the ETV6/NTRK3 fusion gene in MTC samples. This is the first study showing the results of ETV6/NTRK3 rearrangement in medullary thyroid cancer.
In our data set, none of the genes with FDR <0.05 proposed by Jain et al.28 were confirmed by comparison between MEN2A-like and MEN2B-like MTC samples; similarly, no differences were observed regarding to the genes reported by Watanabe et al.29 and Maliszewska et al.31.
Detailed analysis of a large MTC dataset supports the conclusion that differences in the gene expression of hereditary and sporadic MTC tumours are minor, if they really exist. This result enforces the hypothesis that hereditary and sporadic MTC activate the same genetic pathway regardless of RET mutation status.
Further, the difference in the tumour transcriptome between RET mutations initiating the MEN2A and MEN2B types of MTC is restricted to a few genes, which are related to cell proliferation, growth and differentiation. However, further investigations are necessary to demonstrate any clinical impact of these intriguing findings.
These results may also suggest that in MTC, not only does genetic predisposition play a significant role but epigenetic regulation could also be an important factor to understand the differences in the phenotypes of MTC subtypes. Importantly, our results indicate a rather homogeneous MTC gene expression profile.
Methods
Patient group
Tumour samples were obtained from 86 MTC patients (52 sporadic and 34 hereditary) (Supplementary Information Table S1) including 60 women and 26 men, with a mean age at diagnosis of 46 years. The proportions of females and males with sporadic MTC (67% and 33%, respectively) were similar to hereditary MTC (73% and 27%, respectively). All samples were obtained intraoperatively in the Maria Sklodowska-Curie Memorial Cancer Center, Gliwice Branch. The study was conducted after the approval of the Bioethics Committee MSC Memorial Cancer Center and Institute of Oncology, Gliwice Branch, was granted. All methods were performed in accordance with the relevant guidelines and regulations approved by the Bioethics Committee. Informed written consent was obtained from all patients or caregivers for the use of their tissues for analysis in this study.
Tissue samples
Tumour tissue removed during surgical resection was snap-frozen on dry ice and stored at −80 °C until use. Total RNA was extracted by the Chomczynski-Sacchi method54 and purified using RNeasy Mini Kits (Qiagen GmbH, Hilden, Germany); its integrity (RIN value) was assessed using a Bioanalyzer 2100 (Agilent Technologies). The average RIN score for RNA samples used in these experiments was 7.13 (5.2–8.6).
RET germline mutations
Genomic DNA was extracted from peripheral blood nucleated cells by the desalting method55. Mutation screening was performed according to a standard algorithm approved by the American and European MTC Management Guidelines19,56, which assumes the analysis of exons 10, 11, 13, 14, 15 and 16. These exons were sequenced directly using Big Dye 1.1 reagent and a 3130xl Genome Analyser (Life Technologies, Carlsbad, USA). We excluded exon 8 from analysis because a mutation at codon 533 of exon 8 is mainly related to a group of patients with Greek and Brazilian ancestry. To assess the possible presence of this type of RET gene mutation in a Polish population, we performed a study in which we screened exon 8 in 100 RET negative patients in codons 10,11 and 13–16, and we did not find any mutation in codon 8.
RET somatic mutations
Genomic DNA was extracted from tissue samples by the DNeasy Blood and Tissue Kit (Qiagen GmbH, Hilden, Germany). All 21 exons of the RET gene were sequenced directly as described above. In patients with detected germline RET mutations, the somatic status was assumed to be identical, and the test was not repeated on tumour DNA.
Datasets
Analyses were carried out on four data sets: two were analysed by microarrays (Sets A, B), one was analysed by RT-qPCR (Set C), and one was analysed by immunohistochemistry assay (Set D) (Supplementary Information Table S1)
Microarray analysis Sets
Set A (Cancer Genetic Background Analysis Set) included 60 MTC samples to compare gene expression profiles between hereditary and sporadic MTC. Twenty-two were hereditary MTCs (including 2 families with 2 samples each), 19 were sporadic MTCs with somatic RET mutations, 15 were sporadic MTCs without somatic RET mutations, and 4 were sporadic MTC samples with unknown RET somatic mutation status (Supplementary Information Table S1). Array profiling was performed following the manufacturer’s protocol with minor modifications. Briefly, 250 ng of total RNA was processed as described in the GeneChip WT PLUS Reagent Kit Manual “Target Preparation for GeneChip Whole Transcript (WT) Expression Arrays, P/N 703174 Rev. 2” using only half the cDNA as template for the IVT reaction. cRNA was hybridized to the GeneChip Gene 1.0 ST arrays (Affymetrix). Washing, staining and scanning were performed according to the “GeneChip Expression Wash, Stain and Scan User Manual for Cartridge Arrays, P/N 702731 Rev. 3”.
Set B (Mutational Analysis Set) included 30 samples from Set A for comparison of gene expression profiles between tumours with MEN2A-like and MEN2B-like mutations. This set contained 21 MTC samples with MEN2A-like mutations (exons 10 and 11) and 9 MTC samples with MEN2B-like mutations (only exon 16), both germline and somatic.
Quantitative Real-Time PCR Set
Set C (Validation Set, gene level) was used for the RT-qPCR validation of the differences of gene expression profile obtained by microarray analysis. It contained 25 Set A-independent MTC samples, including 2 samples of one family; (16 MEN2A-like and 9 MEN2B-like mutations; 13 sporadic and 12 hereditary MTCs) (Supplementary Information Table S1). Total RNA was extracted as described above. Reverse transcription was performed using Qiagen Omniscript RT Kits and 200 ng of input RNA in a final volume of 20 μl. All genes were amplified by the 7900HT Fast Real-Time PCR system (Life Technologies, Carlsbad, USA). The PCR reaction involved a first step at 50 °C (2 min; activation and incubation with AmpErase UNG) and 95 °C (10 min; activation of AmpliTaq Gold polymerase), followed by 40 cycles of amplification (95 °C, 15 s; 60 °C, 1 min). Each reaction contained 10 μl of TaqMan Universal Mastermix (Life Technologies, Carlsbad, USA), 200 nM of each primer, 2.8 μl of RNase-free water and 5 μl of diluted cDNA template. For amplicon design, we used the Roche Universal Probe Library, as well as the TaqMan MGB set for the NTRK3 gene (Supplementary Information Table S2). The conditions for selecting the amplicons were as follows: 1) a sequence for all transcripts covered by the microarray probe, 2) an amplicon unique to the human transcriptome database (confirmed by BLAST), and 3) no known SNPs in any of the primer or probe sequences. A standard curve was generated for each amplicon using 8 concentrations in duplicates; a linear regression slope indicated amplification efficiency. For the normalization of the RT-qPCR data, we chose the EIF3S10, HADHA and UBE2D2 genes. The normalization factor was obtained using the GeNorm applet for Microsoft Excel.
Immunohistochemistry for TrkC
Set D (Validation Set, protein level) contained 12 MTC samples analysed in the microarray and RT-qPCR study (8 samples from Set A, 3 from Set C and 1 independent sample). In total, sections from 5 samples with MEN2A-like and 7 with MEN2B-like mutations were examined. To uncover antigens, tumour samples embedded by standard methods were deparaffinized and rehydrated with 3 in 1 Dako TRS High pH buffer, blocked in 3% H2O2, and washed with PBS. The samples were incubated with anti-human TrkC mouse monoclonal antibody (clone 75219, R&D Systems; 1:20 dilution). For visualization, we used the EnVision Flex+ system (Dako, K8002).
Detection of ETV6-NTRK3 rearrangements
Tumour DNA was amplified using primers previously described by Leeman-Neill et al.53. In all, 24 DNA samples were available for analysis (Supplementary Information Table S1).
Statistical analysis
Microarray data analysis
Raw microarray data from 60 MTC samples (Set A) were pre-processed by the RMA method using Affymetrix Expression Console software. The subsequent data analysis was performed in an R/Bioconductor environment. Control probe sets and low-variability probe sets (less than 15% of samples with at least 1.5-fold change in either direction from the median) were filtered out. Next, unsupervised analysis by hierarchical clustering was carried out, using the Ward agglomeration method operated on Euclidean distance measures. The selection of probe sets, which significantly distinguished between the analysed groups, was performed using the Welch t-test. The Benjamini-Hochberg false discovery rate (FDR) was used to assess the multiple testing errors. Sample classification was performed using support vector machine (svm) methods with a linear kernel, and the performance of the classifier was estimated by leave-one-out cross-validation (LOOCV). A gene was included in the classifier in each cross-validation step when it satisfied the significance criteria of a Welch t-test p value lower than 0.001 and a 4-fold change (up or down-regulated).
Real Time quantitative PCR data analysis
Normalized RT-qPCR data were assessed by the non-parametric Mann-Whitney U test adjusted by Bonferroni correction.
Additional Information
How to cite this article: Oczko-Wojciechowska, M. et al. Differences in the transcriptome of medullary thyroid cancer regarding the status and type of RET gene mutations. Sci. Rep. 7, 42074; doi: 10.1038/srep42074 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The authors would like to thank Professor Ronald Hancock for his valuable revision of the manuscript and Professor James. A. Fagin for the vector with ETV6-NTRK3 rearrangement (pMSCV-ETV6-NTRK3). The authors would like to also thank Aleksandra Pfeifer for her excellent statistical assistance. This research was supported by Polish National Science Centre grant no. NN401410639 and the National Centre for Research and Development project under the program “Prevention practices and treatment of civilization diseases” NCBiR [MILESTONE]: STRATEGMED2/267398/4/NCBR/2015.
Footnotes
The authors declare no competing financial interests.
Author Contributions M.O.W., M.S. and J.K. were responsible for the conception and design of the study. A.P., D.R. and B.W. were responsible for the RET sequencing, A.C. for the resection of human thyroids, J.K., S.S.U. and T.G. for the clinical evaluations, M.O.W. and M.K. for the microarray experiments, E.C. and D.L. for the histopathological evaluation of specimens, M.K. for the RT-QPCR analysis, B.N. and E.C. for the immunohistochemistry experiment, M.S. and T.S. for the data analysis, M.O.W., J.K., M.W., M.J. and B.J. for the data interpretation; M.O.W., M.S., J.K., B.J. and M.W. wrote the manuscript; B.J. supervised the project. All authors reviewed the manuscript.
References
- Sippel R. S., Kunnimalaiyaan M. & Chen H. Current management of medullary thyroid cancer. Oncologist 13, 539–547 (2008). [DOI] [PubMed] [Google Scholar]
- Frank-Raue K., Rondot S. & Raue F. Molecular genetics and phenomics of RET mutations: Impact on prognosis of MTC. Molecular and Cellular Endocrinology 322, 2–7 (2010). [DOI] [PubMed] [Google Scholar]
- Roy M., Chen H. & Sippel R. S. Current understanding and management of medullary thyroid cancer. Oncologist 18, 1093–100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacini F., Castagna M. G., Brilli L. & Pentheroudakis G. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 23 Suppl 7, vii110-9 (2012). [DOI] [PubMed] [Google Scholar]
- Wells S. a, Pacini F., Robinson B. G. & Santoro M. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J. Clin. Endocrinol. Metab. 98, 3149–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- GH S., Friess H. & Peros G. The genetic basis of hereditary medullary thyroid cancer: clinical implications. Cancer Sci 99, 1940–1949 (2008). [DOI] [PubMed] [Google Scholar]
- Eng C. et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 276, 1575–1579 (1996). [PubMed] [Google Scholar]
- Mulligan L. M. et al. Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the International RET Mutation Consortium. J. Intern. Med. 238, 343–346 (1995). [DOI] [PubMed] [Google Scholar]
- Romei C. et al. Multiple endocrine neoplasia type 2 syndromes (MEN 2): Results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur. J. Endocrinol. 163, 301–308 (2010). [DOI] [PubMed] [Google Scholar]
- Wells S. A. & Santoro M. Targeting the RET pathway in thyroid cancer. Clin. Cancer Res. 15, 7119–7123 (2009). [DOI] [PubMed] [Google Scholar]
- Phay J. E. & Shah M. H. Targeting RET receptor tyrosine kinase activation in cancer. Clin. Cancer Res. 16, 5936–5941 (2010). [DOI] [PubMed] [Google Scholar]
- Mulligan L. M. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer 14, 173–186 (2014). [DOI] [PubMed] [Google Scholar]
- Eng C. RET proto-oncogene in the development of human cancer. J. Clin. Oncol. 17, 380–393 (1999). [DOI] [PubMed] [Google Scholar]
- Mise N., Drosten M., Racek T., Tannapfel A. & Pützer B. M. Evaluation of potential mechanisms underlying genotype-phenotype correlations in multiple endocrine neoplasia type 2. Oncogene 25, 6637–6647 (2006). [DOI] [PubMed] [Google Scholar]
- Mulligan L. M. et al. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat. Genet. 6, 70–74 (1994). [DOI] [PubMed] [Google Scholar]
- Santoro M., Melillo R. M., Carlomagno F., Vecchio G. & Fusco A. Minireview: RET: normal and abnormal functions. Endocrinology 145, 5448–5451 (2004). [DOI] [PubMed] [Google Scholar]
- Machens A. et al. Genotype-phenotype correlations in hereditary medullary thyroid carcinoma: oncological features and biochemical properties. J. Clin. Endocrinol. Metab. 86, 1104–1109 (2001). [DOI] [PubMed] [Google Scholar]
- Raue F. et al. Clinical utility gene card for: multiple endocrine neoplasia type 2. Eur. J. Hum. Genet. 20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloos R. T. et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 19, 565–612 (2009). [DOI] [PubMed] [Google Scholar]
- Moers A. M. et al. Familial medullary thyroid carcinoma: not a distinct entity? Genotype-phenotype correlation in a large family. Am. J. Med. 101, 635–641 (1996). [DOI] [PubMed] [Google Scholar]
- Knowles P. P. et al. Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem. 281, 33577–33587 (2006). [DOI] [PubMed] [Google Scholar]
- Elisei R. et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J. Clin. Endocrinol. Metab. 93, 682–687 (2008). [DOI] [PubMed] [Google Scholar]
- Eng C. et al. Mutation of the RET protooncogene in sporadic medullary thyroid carcinoma. Genes. Chromosomes Cancer 12, 209–212 (1995). [DOI] [PubMed] [Google Scholar]
- Moura M. M. et al. Correlation of RET somatic mutations with clinicopathological features in sporadic medullary thyroid carcinomas. Br. J. Cancer 100, 1777–1783 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal N. et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J. Clin. Endocrinol. Metab. 98, E364–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boichard A. et al. Somatic RAS mutations occur in a large proportion of sporadic RET-negative medullary thyroid carcinomas and extend to a previously unidentified exon. J. Clin. Endocrinol. Metab. 97, E2031–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oczko-Wojciechowska M. et al. The prevalence of somatic RAS mutations in medullary thyroid cancer - A Polish population study. Endokrynol. Pol. 66, 121–125 (2015). [DOI] [PubMed] [Google Scholar]
- Jain S. et al. Expression profiles provide insights into early malignant potential and skeletal abnormalities in multiple endocrine neoplasia type 2B syndrome tumors. Cancer Res. 64, 3907–3913 (2004). [DOI] [PubMed] [Google Scholar]
- Watanabe T. et al. Characterization of gene expression induced by RET with MEN2A or MEN2B mutation. Am. J. Pathol. 161, 249–256 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameur N. et al. Aggressive inherited and sporadic medullary thyroid carcinomas display similar oncogenic pathways. Endocr. Relat. Cancer 16, 1261–1272 (2009). [DOI] [PubMed] [Google Scholar]
- Maliszewska A. et al. Differential gene expression of medullary thyroid carcinoma reveals specific markers associated with genetic conditions. Am. J. Pathol. 182, 350–62 (2013). [DOI] [PubMed] [Google Scholar]
- Oczko-Wojciechowska M. et al. [Gene expression profile of medullary thyroid carcinoma–preliminary results]. Endokrynol. Pol. 57, 420–6 (2006). [PubMed] [Google Scholar]
- Hedenfalk I. et al. Gene-expression profiles in hereditary breast cancer. N. Engl. J. Med. 344, 539–548 (2001). [DOI] [PubMed] [Google Scholar]
- Lisowska K. M. et al. BRCA1-related gene signature in breast cancer: the role of ER status and molecular type. Front. Biosci. (Elite Ed). 3, 125–136 (2011). [DOI] [PubMed] [Google Scholar]
- Dudaladava V. et al. Gene Expression Profiling in Hereditary, BRCA1-linked Breast Cancer: Preliminary Report. Hered. Cancer Clin. Pract. 4, 28–38 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneux G. & Smalley M. J. The cell of origin of BRCA1 mutation-associated breast cancer: a cautionary tale of gene expression profiling. J. Mammary Gland Biol. Neoplasia 16, 51–55 (2011). [DOI] [PubMed] [Google Scholar]
- Pavicic W., Perkiö E., Kaur S. & Peltomäki P. Altered methylation at microRNA-associated CpG islands in hereditary and sporadic carcinomas: a methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA)-based approach. Mol. Med. 17, 726–735 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph R., Tsang W., Dou D., Nelson K. & Edvardsen K. Neuronatin mRNA in PC12 cells: downregulation by nerve growth factor. Brain Res. 738, 32–8 (1996). [DOI] [PubMed] [Google Scholar]
- Xu D. S. et al. Neuronatin in a subset of glioblastoma multiforme tumor progenitor cells is associated with increased cell proliferation and shorter patient survival. PLoS One 7, e37811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu I.-M. et al. Coexpression of neuronatin splice forms promotes medulloblastoma growth. Neuro. Oncol. 10, 716–724 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. P. et al. The dual-specificity phosphatase CDC14B bundles and stabilizes microtubules. Mol. Cell. Biol. 25, 4541–51 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa M., Guillamot M., Bueno M. J. & Malumbres M. The Cdc14B phosphatase displays oncogenic activity mediated by the Ras-Mek signaling pathway. Cell Cycle 10, 1607–17 (2011). [DOI] [PubMed] [Google Scholar]
- Bassermann F. et al. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 134, 256–67 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeano F. et al. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene 32, 998–1009 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Choi J.-W., Lee J.-H. & Kim Y.-S. Loss of CDC14B expression in clear cell renal cell carcinoma: meta-analysis of microarray data sets. Am. J. Clin. Pathol. 141, 551–8 (2014). [DOI] [PubMed] [Google Scholar]
- Araki K. et al. Frequent overexpression of the c-kit protein in large cell neuroendocrine carcinoma of the lung. Lung Cancer 40, 173–180 (2003). [DOI] [PubMed] [Google Scholar]
- Marchetti A. et al. Frequent mutations in the neurotrophic tyrosine receptor kinase gene family in large cell neuroendocrine carcinoma of the lung. Hum. Mutat. 29, 609–616 (2008). [DOI] [PubMed] [Google Scholar]
- McGregor L. M. et al. Roles of trk family neurotrophin receptors in medullary thyroid carcinoma development and progression. Proc. Natl. Acad. Sci. USA 96, 4540–4545 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W. et al. TrkC plays an essential role in breast tumor growth and metastasis. Carcinogenesis 31, 1939–47 (2010). [DOI] [PubMed] [Google Scholar]
- Yan P., Benhattar J., Seelentag W., Stehle J. C. & Bosman F. T. Immunohistochemical localization of hTERT protein in human tissues. Histochem Cell Biol 121, 391–397 (2004). [DOI] [PubMed] [Google Scholar]
- Nichols D. W. et al. A testing algorithm for determination of HER2 status in patients with breast cancer. Ann. Clin. Lab. Sci. 32, 3–11 (2002). [PubMed] [Google Scholar]
- Ricarte-Filho J. C. et al. Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers. J. Clin. Invest. 123, 4935–44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeman-Neill R. J. et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer 120, 799–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P. & Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–9 (1987). [DOI] [PubMed] [Google Scholar]
- Maniatis T., Fritsch E. F. & Sambrook J. Molecular cloning: a laboratory manual. (1982). [Google Scholar]
- Fugazzola L., De Leo S. & Perrino M. The optimal range of RET mutations to be tested: European comments to the guidelines of the American Thyroid Association. Thyroid Res. 6 Suppl 1, S8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.