

Abstract
Many single nucleotide polymorphisms (SNPs) have been associated with lung cancer but lack confirmation and functional characterization. We retested the association of 56 candidate SNPs with lung adenocarcinoma risk and overall survival in a cohort of 823 Italian patients and 779 healthy controls, and assessed their function as expression quantitative trait loci (eQTLs). In the replication study, eight SNPs (rs401681, rs3019885, rs732765, rs2568494, rs16969968, rs6495309, rs11634351, and rs4105144) associated with lung adenocarcinoma risk and three (rs9557635, rs4105144, and rs735482) associated with survival. Five of these SNPs acted as cis-eQTLs, being associated with the transcription of IREB2 (rs2568494, rs16969968, rs11634351, rs6495309), PSMA4 (rs6495309) and ERCC1 (rs735482), out of 10,821 genes analyzed in lung. For these three genes, we obtained experimental evidence of differential allelic expression in lung tissue, pointing to the existence of in-cis genomic variants that regulate their transcription. These results suggest that these SNPs exert their effects on cancer risk/outcome through the modulation of mRNA levels of their target genes.
The study of genetic factors modulating an individual’s predisposition to lung cancer is supported by strong epidemiological evidence obtained from various types of studies. Observational studies have consistently reported an increased risk of lung cancer in first-degree relatives of lung cancer patients1,2,3,4,5. Genome-wide association studies (GWAS) on population-based series identified three main susceptibility loci, at 5p156,7,8, 6p216, and 15q25. The locus at 15q25 harbors genes for three nicotinic acetylcholine receptor subunits (CHRNA3, CHRNA5, and CHRNB4) that have previously been associated with lung cancer risk and nicotine dependence6,9,10. Other GWAS found many single nucleotide polymorphisms (SNPs) that associated with lung cancer risk11,12,13,14,15,16. However, the results obtained in these studies have not generally been confirmed, even in a large consortium study17.
Some genetic variants associated with lung cancer risk have also been associated with prognosis. For instance, a polymorphism (rs6495309) in the promoter of CHRNA3 gene has been reported to be associated with the overall survival of patients with early-stage non–small-cell lung cancer (NSCLC)18. Moreover, rs667282 in CHRNA5 has recently been proposed as a modifier of prognosis in advanced NSCLC19. Several other SNPs, found using a GWAS approach, were proposed to be associated with prognosis or survival of lung cancer patients in different populations20,21,22,23. However, these studies did not identify the same candidate polymorphisms, which may be due (at least in part) to the wide genetic heterogeneity of the human population.
The GWAS cited here, which aimed to find SNPs associated with lung cancer risk or prognosis, identified mostly non-overlapping subsets of SNPs, hindering progress in lung cancer research. One limitation of these studies is that they investigated relatively small, often heterogeneous populations. Therefore, replication of these association studies in other case-control cohorts is warranted. Moreover, to go beyond statistical associations, these candidate SNPs must be investigated for their putative functional roles in lung cancer.
The functional characterization of candidate SNPs from GWAS is particularly challenging, since most of them map in non-coding regions24 and therefore do not exert direct effects on proteins, for example by introducing premature stop codons. These “regulatory SNPs” exert their effects by modifying non-transcribed regions of the genome (e.g. gene promoters, enhancers and silencers) where they alter transcription factor binding and chromatin states, as well as untranslated regions of RNA where they affect RNA splicing25,26. Therefore, these SNPs may modulate the expression of both near and distant genes; when these genes are involved in cancer-related pathways, the SNPs may affect the process of tumorigenesis. For instance, our finding that SNPs in the promoter of CHRNA5 altered this gene’s expression levels in normal lung tissue27 suggested that modulation of transcriptional activity was, at least in part, responsible for the association of the 15q25 locus with lung cancer risk. These results shed light on a possible functional role of these polymorphisms in lung tumorigenesis.
In genetics, chromosomal loci that modulate gene expression–a quantitative trait–are called “expression quantitative trait loci” (eQTLs). eQTL analysis is a powerful genetic approach for testing if modulation of transcriptional activity is a mechanism by which SNPs affect a phenotype28,29. By crossing data on SNP genotypes with those on gene expression, this analysis enables the discovery of meaningful genotype-phenotype relationships30. Hence, the study of eQTLs in lung tissue may provide insight into mechanisms involved in lung tumorigenesis.
This study focused on 56 SNPs previously reported to be associated with lung cancer risk, survival, or factors predisposing to lung cancer, i.e., chronic obstructive pulmonary disease (COPD)31 and nicotine dependence32. First, we attempted to replicate their association with lung cancer risk and survival in a relatively large, uniform and well characterized series of lung cancer patients, with the same tumor histotype (adenocarcinoma) and similar durations of follow-up, and in sex-matched healthy controls. Additionally, we used eQTL analysis to look for the possible involvement of these candidate SNPs in the modulation of gene expression in non-involved lung tissue from lung adenocarcinoma patients, with the aim of identifying their mechanisms of action in lung cancer predisposition and progression.
Results
Patients’ clinical characteristics
This study investigated 823 lung adenocarcinoma patients and 779 sex-matched healthy controls, all from Italy (Table 1). The patients’ median age at surgery was 64 years, but there was a wide age range; the controls’ median age at recruitment was 61 years. The age difference between cases and controls was statistically significant (P = 3.9 × 10−8). Male sex predominated (~70%) among both cases and controls. Ever smokers were more frequent than never smokers in both cases (85%) and controls (98%), and the percentage of ever smokers in controls was significantly higher than in cases (P < 2.2 × 10−16). The higher number of ever smokers in the control group is due to the fact that most of them (621 out of 779) were recruited during a lung cancer screening program. About half of cases were at pathological stage I. The median follow-up period for patients who did not die during the study was 60 months, while at the 60-month follow-up 354 patients (44%) had died.
Table 1. Clinical characteristics of Italian lung adenocarcinoma patients (cases) and healthy controls.
| Characteristic | Cases (n = 823) | Controls (n = 779) | Case-control comparison Pa | Association with overall survivalb | |
|---|---|---|---|---|---|
| HR (95% CI) | P | ||||
| Age, years, median (range) | 64 (29–84) | 61 (32–78) | 3.9 × 10−8 | 1.0 (1.0–1.0) | 0.06 |
| Sex, n | 0.33 | ||||
| Female | 254 | 223 | 1.0 | ||
| Male | 569 | 556 | 1.6 (1.2–2.1) | 1.2 × 10−3 | |
| Smoking habit, n | <2.2 × 10−16 | ||||
| Never | 116 | 15 | 1.0 | ||
| Ever | 689 | 758 | 1.1 (0.8–1.6) | 0.58 | |
| Missing data | 18 | 6 | |||
| Pathological stage, nc | NA | ||||
| I | 410 | NA | 1.0 | ||
| II | 137 | NA | 2.9 (2.2–3.9) | 4.3 × 10−12 | |
| III or IV | 247 | NA | 4.5 (3.5–5.8) | <2.2 × 10−16 | |
| Missing data | 29 | 0 | |||
| Cases with follow-up data, n | 800 | NA | NA | ||
| Median (range) follow-up of patients alive, months | 60 (10–60) | NA | NA | ||
| Dead at the 60-month follow-up, n | 354 | NA | NA | ||
aComparison between cases and controls; Kruskal-Wallis test for the quantitative variable age, chi-square test for categorical variables.
bHR, hazard ratio; CI, confidence interval; Cox multivariate test on cases only.
cPathological stage, assessed after surgery.
NA, not applicable.
To identify clinical characteristics associated with overall survival, multivariate Cox analyses were performed (Table 1). Overall survival associated with sex, with men having shorter survival than women (HR = 1.6; 95% CI, 1.2–2.1; P = 1.2 × 10−3). Overall survival also associated with pathological stage, as expected, but it did not associate with age at surgery or smoking habit.
Association between SNP genotypes and lung cancer risk or prognosis
A total of 64 SNPs, previously reported to associate with lung cancer risk or prognosis, were genotyped in cases and controls (Supplementary Table S1). Quality control filtering of raw data revealed that a 90% genotype call rate was not reached for 6 SNPs (rs1261411, rs2736100, rs6488007, rs6537296, rs503464, and rs55781567). Moreover, for two SNPs (rs3117582 and rs639739), the genotype data did not segregate into distinct clusters. Therefore, 8 SNPs were excluded while 56 SNPs were considered in the subsequent analyses.
To identify SNPs associated with lung cancer risk in our Italian series, we used logistic regression adjusting for sex, age, and smoking habit (Table 2). This replication analysis revealed statistically significant associations between lung adenocarcinoma risk and eight SNPs located on five chromosomes. In particular, we confirmed the association of rs401681 in an intronic region of CLPTM1L gene on chromosome 5, within the 5p15 susceptibility locus; the minor allele of rs401681 associated with a lower lung cancer risk (odds ratio, OR < 1). On chromosome 8, rs3019885 was confirmed to associate with lung cancer risk; with an OR > 1, the minor allele of this SNP is the risk allele. On chromosome 14, we found a weak association between a higher lung adenocarcinoma risk and the minor allele of rs732765, which had previously been associated with poorer survival in NSCLC (see Supplementary Table S1). We replicated the association with lung cancer risk for four SNPs in the nicotinic acetylcholine receptor locus on chromosome 15q25: rs2568494 (previously associated with COPD), rs16969968 (lung cancer risk and survival, and COPD), rs6495309 (lung cancer risk and survival), and rs11634351 (nicotine dependence). Three of these SNPs had an OR > 1, meaning that the minor alleles were risk alleles, while the fourth, rs6495309, had an OR < 1. Finally, we confirmed the association with lung cancer risk for rs4105144 upstream of CYP2A6 gene on chromosome 19, whose minor allele showed a protective role (OR < 1). The strongest statistical association was found at rs2568494 (P = 6.00 × 10−7), located near the IREB2 gene in the nicotinic acetylcholine receptor locus.
Table 2. SNPs associated with lung adenocarcinoma risk in logistic analysis (P < 0.05) in the Italian series of lung adenocarcinoma patients (n = 823) and healthy controls (n = 779)a.
| SNP | Chr. | Position (bp)b | Closest gene | Minor allele | OR (95% CI) | P |
|---|---|---|---|---|---|---|
| rs401681 | 5 | 1,321,972 | CLPTM1L | T | 0.80 (0.69–0.93) | 2.7 × 10−3 |
| rs3019885 | 8 | 117,013,406 | SLC30A8 | G | 1.21 (1.04–1.41) | 0.012 |
| rs732765 | 14 | 74,899,026 | DLST | G | 1.22 (1.02–1.47) | 0.032 |
| rs2568494 | 15 | 78,448,622 | IREB2 | A | 1.44 (1.25–1.67) | 6.00 × 10−7 |
| rs16969968 | 15 | 78,590,583 | CHRNA5 | A | 1.34 (1.16–1.55) | 6.55 × 10−5 |
| rs6495309 | 15 | 78,622,903 | CHRNB4-CHRNA3 | T | 0.83 (0.69–1.00) | 0.049 |
| rs11634351 | 15 | 78,652,376 | CHRNB4 | A | 1.27 (1.10–1.47) | 1.4 × 10−3 |
| rs4105144 | 19 | 40,852,719 | CYP2A6 | T | 0.74 (0.63–0.87) | 2.6 × 10−4 |
aLogistic analysis carried out in PLINK, adjusted for sex, age, and smoking habit, and based on additive effects of SNPs, i.e., an OR > 1 means that the risk of lung adenocarcinoma increases with the number of minor alleles.
bBased on Assembly GRCh38.p5.
Next, to identify SNPs associated with overall survival in lung adenocarcinoma patients, we used a multivariable Cox proportional hazard model, adjusted for sex, age, smoking habit, and pathological stage (stage I versus stage > I). This analysis identified three SNPs on two chromosomes (Table 3). In particular, we confirmed the association with survival for rs9557635, an intronic polymorphism of NALCN gene. Moreover, we found that rs4105144, near CYP2A6 gene on chromosome 19, was associated with survival as well as with risk (see Table 2). Finally, we replicated the association of rs735482 (in the 3’-UTR of ERCC1 gene) with survival. The minor alleles of rs9557635 and rs4105144 were associated with poorer survival (HR > 1), whereas for rs735482 we observed the opposite effect (HR < 1).
Table 3. SNPs associated with overall survival of lung adenocarcinoma patients (n = 823) in a multivariable Cox proportional hazard model at P < 0.05.
| SNP | Chr. | Position (bp)a | Closest genes | Minor allele | HR (95% CI)b | P |
|---|---|---|---|---|---|---|
| rs9557635 | 13 | 101,398,739 | NALCN | A | 1.17 (1.01–1.36) | 0.035 |
| rs4105144 | 19 | 40,852,719 | CYP2A6 | T | 1.21 (1.02–1.44) | 0.025 |
| rs735482 | 19 | 45,408,744 | CD3EAP; ERCC1 | C | 0.78 (0.62–0.98) | 0.035 |
aMap position based on genome assembly GRCh38.p5.
bHazard ratio for additive effects (allele dosage) of the minor allele (genotypes were coded with a numerical value, i.e., 0, 1, or 2, according to the number of minor alleles present) on the risk of dying; Cox’s analysis was adjusted by sex, age, pathological stage (I versus > I), and smoking habit; follow-up until 60 months.
The relationship between these three SNPs and survival was then visualized using Kaplan-Meier curves (Supplementary Fig. S1). Carriers of the minor allele of rs9557635 had worse probability of survival than homozygotes at the common allele (genotype GG, Supplementary Fig. S1A), in agreement with the HR of 1.17 (P = 0.035, see Table 3). For rs4105144, survival curves for heterozygotes and homozygotes at the common allele were almost undistinguishable (Supplementary Fig. S1B), whereas homozygous carriers of the minor allele (genotype TT) had worse survival than the others. For rs735482, heterozygotes had better survival than homozygotes at the major allele (Supplementary Fig. S1C), in agreement with the HR of 0.78 (P = 0.035; the small number (n = 16) of patients homozygous for the minor allele did not allow the effect of this genotype on survival to be assessed).
On the basis of this analysis, we repeated Cox’s analysis after having grouped genotypes according to a dominant or recessive model (Fig. 1). For rs9557635, we compared individuals carrying at least one copy of the minor allele with homozygotes for the common allele, as in a dominant model, and found that minor allele carriers had a higher risk of death (HR = 1.31, P = 0.018) than homozygotes for the common allele (Fig. 1A). For rs4105144 (Fig. 1B), we compared homozygotes at the minor allele to carriers of the common allele, and observed a much stronger statistical association with survival than when we assessed the dosage effect of the minor allele (HR = 1.90, P = 4.6 × 10−5 vs. P = 0.025 in Table 3). This result suggests that, for rs4105144 or a functional variation in tight linkage disequilibrium with it, the minor allele has a recessive effect on overall survival of lung adenocarcinoma patients. Finally, for rs735482 (Fig. 1C), we compared individuals carrying at least one copy of the minor allele with homozygotes for the common allele, and found a significantly better survival (HR = 0.73, P = 0.014) for carriers of the minor allele; this result suggests a possible dominant protective effect of the minor allele on survival of lung adenocarcinoma patients.
Figure 1. Kaplan-Meier survival curves for lung adenocarcinoma patients, according to the genotypes of three SNPs associated with overall survival.
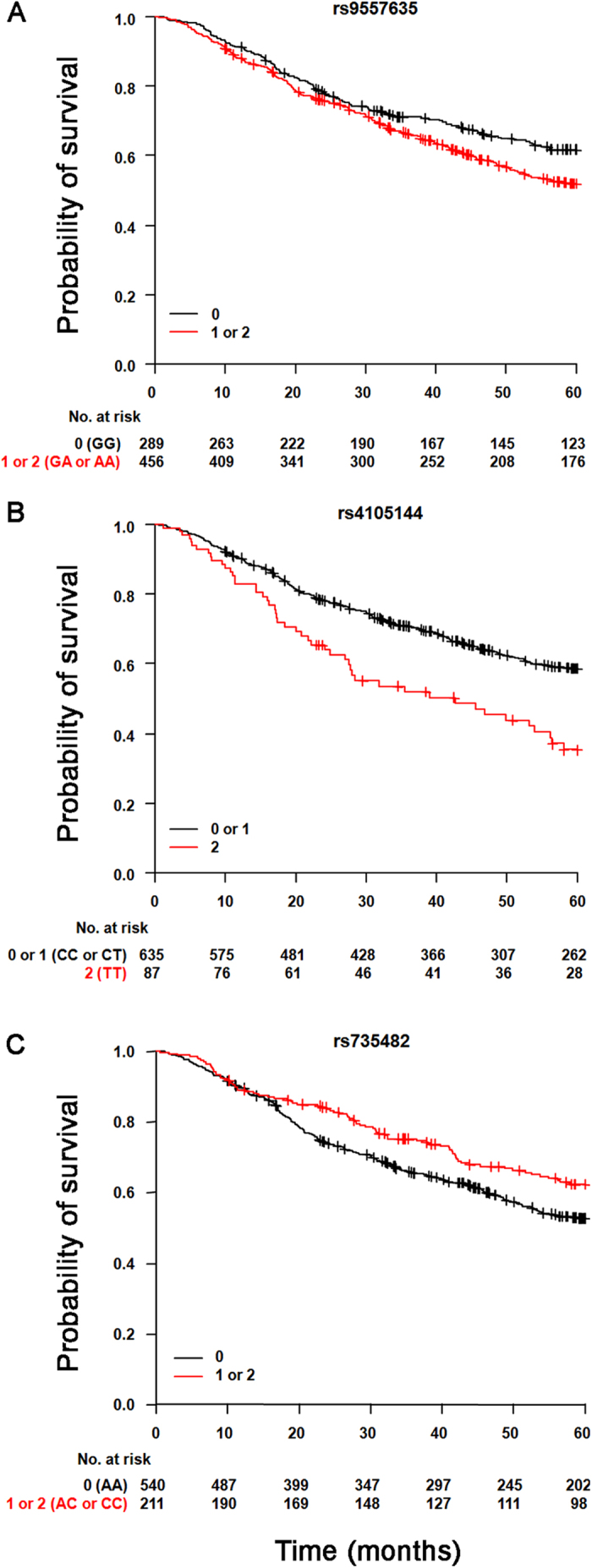
(A) For rs9557635, we tested a dominant model for the minor allele, and therefore compared carriers of the minor allele (red) with homozygotes for the common allele (black). (B) For rs4105144, we tested a recessive model for the minor allele, and so compared homozygotes for the minor allele (red) with carriers for the common allele (black). (C) For rs735482, we tested a dominant model for the minor allele, and therefore compared carriers of the minor allele (red) with homozygotes for the common allele (black). Crosses denote censored samples. Below the figures are reported the number of patients at risk at the specified times of follow-up.
Collectively, this analysis of 56 candidate SNPs previously reported to be associated with lung cancer confirmed, in a relatively large population homogeneous for both histotype and ethnicity, the association for ten SNPs: seven with lung adenocarcinoma risk, two with survival, and one with both risk and survival. Because the original studies that discovered these associations were carried out in populations different from the one studied here, this replication study suggests that these ten SNPs have broad importance in lung adenocarcinoma, irrespective of ethnic group.
Some lung cancer-related SNPs are putative eQTLs
Because most candidate SNPs identified with GWAS map to non-coding regions of the genome, it is plausible that they exert their cellular effects via the modulation of gene expression. To test this possibility for the ten confirmed lung cancer-related SNPs, we carried out an expression quantitative trait loci (eQTL) analysis in the non-involved lung parenchyma of lung adenocarcinoma patients. In particular, we looked for correlations between the genotypes of these SNPs and the expression levels of 10,821 genes in 232 cases. This analysis identified six cis-eQTLs, involving five SNPs associated with the expression of three genes (Table 4). There are more cis-eQTLs than SNPs because one SNP (rs6495309) is associated with expression levels of two transcripts (IREB2 and PSMA4). In detail, IREB2 on chromosome 15 (at ∼78 Mbp, on Assembly GRCh38) was found to be regulated by four SNPs (rs2568494, rs16969968, rs6495309 and rs11634351); in the same locus, rs6495309 is associated also with PSMA4 mRNA levels. Additionally, ERCC1 on chromosome 19 (at ∼45 Mbp) associated with one SNP (rs735482). Two eQTL SNPs are located in the gene they modulate: rs2568494 maps in an intronic region of IREB2 gene and rs735482 maps in the 3′-UTR of ERCC1 gene. This latter SNP also maps in the coding sequence of CD3EAP, a gene that overlaps with ERCC1 but is transcribed in the opposite direction. No trans-eQTLs were found. Altogether, this eQTL analysis provides statistical evidence for the in-cis regulation of three genes by six genetic elements associated with lung cancer risk or survival.
Table 4. cis-eQTLs and their same-chromosome target genes identified in non-involved lung tissue from 232 lung adenocarcinoma patients (analysis limited to 10 confirmed SNPs. eQTLs clustered into two main loci.
| eQTL | SNP | Chr. | Position (bp)a | Target gene | Nominal P | FDR | Betab |
|---|---|---|---|---|---|---|---|
| 1 | rs2568494 | 15 | 78,448,622 | IREB2 | 1.35 × 10−6 | 0.00018 | −0.21 |
| 2 | rs16969968 | 15 | 78,590,583 | IREB2 | 5.70 × 10−6 | 0.00035 | −0.20 |
| 3 | rs11634351 | 15 | 78,652,376 | IREB2 | 0.00158 | 0.035 | −0.15 |
| 4 | rs6495309 | 15 | 78,622,903 | IREB2 | 0.000218 | 0.0098 | 0.21 |
| 5 | rs6495309 | 15 | 78,622,903 | PSMA4 | 0.00100 | 0.027 | 0.092 |
| 6 | rs735482 | 19 | 45,408,744 | ERCC1 | 0.000711 | 0.029 | −0.12 |
aMap position based on genome assembly GRCh38.p5.
bBeta values are from the additive linear regression model (in MatrixEQTL R package), where genotype (expressed as the minor allele count) is assumed to have an additive effect on gene expression. Patients’ sex and age at surgery were used as covariates. A positive value indicates that the expression of the target gene increases with an increase in minor allele count.
We repeated the eQTL analysis considering all 56 SNPs included in the present study (Supplementary Table S2). This analysis found 10 cis-eQTLs, including four of those described above: the associations of rs11634351 and rs6495309 with IREB2 were lost, probably as a consequence of a power reduction in this wider analysis. An additional locus on chromosome 15 (at ~43 Mbp, on Assembly GRCh38) was identified, where rs504417 associated with the expression of ADAL gene. Also rs578776, in the nicotinic acetylcholine receptor locus, was strongly associated with IREB2 expression. Of note, this same SNP, together with rs6495309, associated with mRNA levels of PSMA4, another gene in the chromosome 15q25 locus. On chromosome 19, this wider eQTL analysis identified two additional SNPs (rs1005165 and rs967591) associated with ERCC1 levels; one of them (rs967591) also associated with VASP gene expression. No trans-eQTLs were found.
Genetic elements located in cis of ERCC1, IREB2, and PSMA4 regulate expression in an allelic-specific manner
To experimentally test the results of the eQTL analysis, we examined if the two putatively regulated genes exhibited differential allelic expression (DAE). We selected lung adenocarcinoma patients who were heterozygous at a marker SNP in each gene, and subgrouped them on the basis of their genotypes at the eQTL SNPs (Fig. 2). cDNA from the patients’ non-involved lung tissue was PCR-amplified in the region spanning the marker SNP, and allelic expression was determined by pyrosequencing the PCR products. Since the marker SNPs were in loose linkage disequilibrium with the eQTL SNPs (data not shown), we stratified the patients into groups according to whether they were heterozygous or homozygous for the eQTL SNPs before assessing DAE.
Figure 2. Workflow for DAE assay.
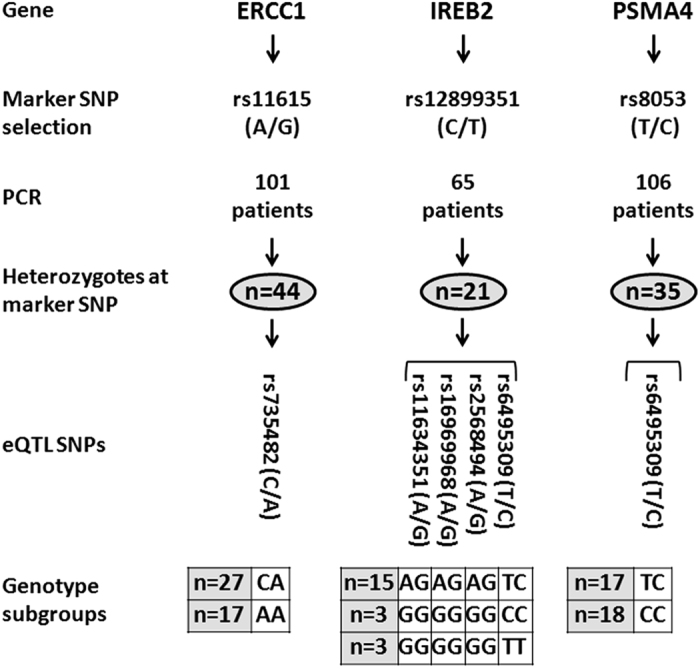
For each putative target gene of eQTL SNPs, we chose a marker SNP in the transcribed region (shown are minor/common alleles). Heterozygous patients were selected by pyrosequencing genomic DNA with SNP-specific PCR primers (Supplementary Table S3). Heterozygotes were then subgrouped according to their genotypes at the eQTL SNPs to create the comparison groups for the DAE assay.
The ratios of the minor to common alleles of marker SNPs in cDNA were compared to those obtained in genomic DNA, in the genotype subgroups for each gene (Fig. 3). For ERCC1 gene (Fig. 3A), the ratios for cDNA were significantly different from those for genomic DNA, indicating DAE, in both subgroups of patients. In particular, in patients heterozygous for rs735482 eQTL SNP there was more expression of the minor than common allele, while in homozygous patients there was less expression of the minor allele (P = 4.0 × 10−4 and P = 8.4 × 10−4, respectively; Wilcoxon’s test on paired log10-transformed allelic ratios). For IREB2 (Fig. 3B), there was evidence of DAE in patients who were heterozygous at all four eQTL SNPs (rs2568494, rs16969968, rs6495309 and rs11634351), with lower expression of the minor than common allele (P = 6.1 × 10−5), but not in the two groups of patients who were homozygous for these SNPs. Similar results were obtained for PSMA4 (P = 0.015, Fig. 3C).
Figure 3. Differential allelic expression in ERCC1, IREB2, and PSMA4 supports the existence of cis-acting genetic regulations.
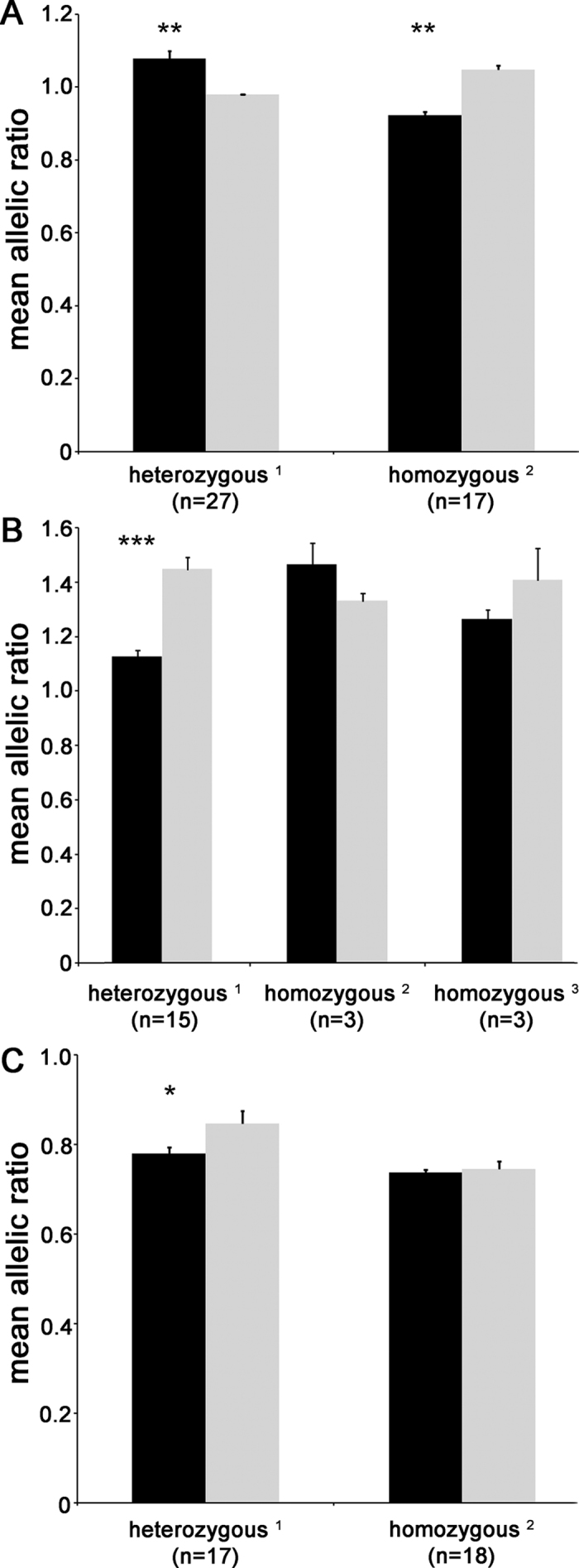
Allelic ratios (minor/common allele) for marker SNPs in cDNA (black bars) and genomic DNA (gray bars) from patients heterozygous for (A) rs11615 in ERCC1 gene, (B) rs12899351 in IREB2 gene, and (C) rs8053 in PSMA4 gene. Marker SNPs map in gene transcribed regions and, thus, can be genotyped in both cDNA and genomic DNA. Data are means and SE. Patients are subgrouped according to their genotypes at the eQTL SNPs for each gene. In particular, the heterozygous1 subgroup consisted of cases that were heterozygous at all eQTL SNPs, while the homozygous2 subgroup had cases that were homozygous for the common allele at all eQTL SNPs, and the third subgroup, homozygous3, formed for IREB2 gene only, consisted of cases that were homozygous for the common allele at three eQTL SNPs (rs2568494, rs16969968, and rs11634351) and homozygous for the minor allele at the fourth eQTL SNP (rs6495309) (see Fig. 2). **P < 0.001; ***P < 0.0001 (Wilcoxon’s signed rank test on paired log10-transformed allelic ratios).
Discussion
This study focused on 56 SNPs previously reported to be associated with lung cancer in different populations, and comprised both a replication effort and an eQTL analysis. In the replication study, eight SNPs were found to associate with the risk of lung adenocarcinoma in our Italian series. Additionally, three SNPs were found to associate with overall survival in this cohort. Altogether, the replication study confirmed an association with lung cancer for 10 SNPs, as one SNP associated with both risk and survival. In the eQTL analysis, the genotypes of five of these ten SNPs were found to associate with the expression levels of three genes on the same chromosomes, indicating the presence of cis-eQTLs, in 232 samples of non-involved lung tissue from lung adenocarcinoma patients. For these genes (IREB2, PSMA4 and ERCC1), we found evidence of DAE, suggesting that the SNPs exert their effects on lung tissue by modulating gene expression.
In the replication study, eight SNPs associated with lung cancer risk in our case-control cohort. Because the cohort had significantly fewer ever smokers among cases (85%) than controls (98%), these SNPs are likely to be associated directly with lung cancer risk rather than indirectly through an association with smoking behavior.
Among the SNPs that associated with lung cancer risk, four (rs2568494, rs16969968, rs6495309, and rs11634351) were in the nicotinic acetylcholine receptor locus on chromosome 15. rs2568494, previously reported to be associated with COPD33,34, is an A/G variant in IREB2. This SNP is included in the IREB2 AAAT haplotype (rs2568494, rs2656069, rs10851906, rs13180) that has recently been associated with lung cancer risk (OR = 1.5)35. Our data confirm the association of the A allele of rs2568494 with increased lung cancer risk. The second SNP, rs16969968, is a G/A variant in the coding sequence of CHRNA5. This SNP associated with smoking-related traits such as nicotine dependence36, COPD37, smoking cessation treatment benefit38, and smoking quantity10. Our study confirmed the association of its minor allele (A) with increasing lung cancer risk9,39. The third SNP, rs6495309, is an intergenic C/T polymorphism mapping between CHRNA3 and CHRNB4 genes; it previously was associated with lung cancer risk40, survival18 and COPD37. Our study confirms the association of its minor allele (T) with decreasing lung cancer risk. Finally, rs11634351 is a G/A intron variant in CHRNB4 gene that has never been reported to be associated with lung cancer risk, but was found to modulate nicotine dependence and smoking-related phenotypes41. Our finding of a significant association of the A allele of rs11634351 with increased lung cancer risk may be due to its linkage disequilibrium with another variant with a functional role or may indicate the genetic complexity of the locus in modulating lung cancer risk.
Also associating with lung cancer risk was rs401681, a C/T intronic variant in CLPTM1L gene on chromosome 5p15.33; the T allele of rs401681 was previously found to associate with a lower risk of lung cancer in two meta-analyses42,43, and our results confirm these findings. On chromosome 8, we confirmed our previously observed association of the minor allele (G) of rs3019885 with increasing lung cancer risk15. On chromosome 14, a significant association with lung cancer risk was found for the minor allele (G) of rs732765, which, in a Korean study22, was associated with poorer survival of NSCLC patients.
Finally, our study confirmed an association with lung cancer risk for the minor (T) allele of rs410514444. This SNP, which also associates with smoking behavior45, is a T/C variant upstream of CYP2A6, which encodes cytochrome P450 2A646. This cytochrome is the main nicotine-metabolizing enzyme, and it is also involved in the bioactivation of carcinogens present in tobacco smoke47. CYP2A6 has several allelic variants that affect the rate of nicotine metabolism48. Because rs4105144 is in linkage disequilibrium with the slow metabolizing alleles, the association of the minor allele with lower lung cancer risk can be explained by a protective action against carcinogen activation.
In the analysis of overall survival, three SNPs were identified, including rs4105144 which we also confirmed to be associated with lung cancer risk. The association of this SNP with lung cancer survival is a novel finding, and our observation that homozygous carriers of the T allele had poorer survival than carriers of the common allele (P = 4.6 × 10−5) suggests a potential functional involvement of the recessive minor allele. This result contrasts with the protective role of the T allele in lung cancer risk; however, it has already been reported that an increasing number of T alleles associates with poorer survival in gastric cancer patients treated with adjuvant chemotherapy49. The mechanism underlying such association is not known but may involve poor biotransformation of chemotherapeutic agents or endogenous compounds such as retinoic acids and steroids50, which, in turn, might influence survival of lung adenocarcinoma patients.
Our study confirmed the association with lung cancer survival of rs9557635 (in NALCN gene) and found that its minor allele (A) associated with poorer prognosis, as previously reported22. Finally, for rs735482 (mapping in overlapping genes CD3EAP and ERCC1 on chromosome 19), we observed a significant association of the minor allele (C) with better prognosis, differently from studies that associated the same C allele with poorer prognosis22,51. For both these SNPs we observed a dominance effect of the minor allele on survival of lung adenocarcinoma patients.
Overall, our study confirmed few of the previously reported associations of the 56 SNPs with lung cancer risk and survival. This failure is probably due to the fact that most associations were found in heterogeneous populations, characterized by patients with different lung cancer histotypes. Instead, we studied only a single lung cancer histotype (adenocarcinoma), limited to surgically treated patients and to a single country (Italy). An additional explanation is possible confounding effects of somatic mutations on prognosis. Indeed, some somatic mutations, in particular KRAS mutations, associated with poorer survival of NSCLC patients52. However, other mutations (in EGFR, TP53, PIK3CA, and also in KRAS) did not associate with survival in different populations53,54,55,56. Therefore, we hypothesize that germline polymorphisms and somatic mutations interact and cooperate in modulating lung cancer patients’ survival, by acting on or interfering with the same molecular pathways. This possibility should be investigated further.
We previously hypothesized57 that individual predisposition to lung cancer is already detectable in non-involved lung tissue and that polymorphisms which associate with lung cancer risk or prognosis act by modulating gene expression levels in lung tissue. Therefore, we assessed the confirmed ten lung cancer-related SNPs for possible roles as eQTLs, and found a significant (FDR < 0.05) effect of five SNPs in the modulation of mRNA levels of three genes on the same chromosomes, hence functioning as cis-eQTLs. In particular, four risk-associated SNPs in the nicotinic acetylcholine receptor locus on chromosome 15 (rs2568494 in IREB2; rs16969968 in CHRNA5; rs6495309 in CHRNA3/CHRNB4; and rs11634351 in CHRNB4) and one survival-associated SNP (rs735482 in CD3EAP and ERCC1 on chromosome 19), were found to associate with mRNA levels of IREB2 (rs2568494, rs16969968, rs6495309 and rs11634351), of PSMA4 (rs6495309), and of ERCC1 (rs735482) genes. We did not confirm previous reports of nicotinic acetylcholine receptor locus polymorphisms modulating CHRNA5 levels27,58,59. This unexpected result may be due to the existence, in non-involved lung tissue, of CHRNA5 transcript variants that were not detected by the gene expression microarray we used. Indeed, our previous findings of polymorphisms modulating CHRNA5 levels were obtained using qPCR27,58.
To test whether the statistical associations between the five eQTL SNPs and expression levels of the three target genes are attributable to variations in cis-regulatory elements mapping close to these eQTL SNPs, we examined the possibility of differential allelic expression (DAE) of the target genes. Indeed, the allelic-specific transcription of a gene indicates that genetic elements in cis to it (e.g. in its promoter or untranslated regions of its RNA) modulate its expression. Of note, we found the existence of cis regulatory variations for the three genes we tested, i.e. IREB2, PSMA4, and ERCC1. For IREB2 and PSMA4 genes, we detected statistically significant DAE in patients heterozygous at all the eQTL SNPs, whereas no significant association was found in patients homozygous for the same SNPs. Therefore, these results suggest that the eQTL SNPs are in linkage disequilibrium with cis-acting variations, yet unidentified, modulating IREB2 expression level. At the ERCC1 gene, instead, statistically significant DAE was detected in patients both heterozygous and homozygous at the SNP modulating its expression levels. These results might be due to a complex pattern of linkage disequilibrium in the analyzed locus and to the putative existence of more than one functional variation affecting ERCC1 expression. For all target genes herein examined, their functional cis-acting elements have not yet been identified.
Our findings of IREB2 DAE in non-involved lung tissue confirm previous results obtained in lung tumor tissue. Fehringer et al.60, who studied two series of lung tumor tissue from NSCLC patients, reported association of rs16969968 with IREB2 levels in one of the two series. The direction of effects (the sign of the beta parameter in Table 4) of the minor alleles on IREB2 mRNA levels in our study are the same as in that study. Together, these results provide convincing evidence that SNPs in the nicotinic acetylcholine receptor locus on chromosome 15 modulate expression levels of IREB2 and, thus, implicate IREB2 expression levels in the individual risk for lung cancer. Because IREB2 encodes an RNA binding protein involved in iron metabolism61, it has been suggested that high levels of iron in the lung are a predisposing factor for high oxidative stress, high inflammation and, therefore, lung cancer60,62. Accordingly, our results suggest that lower expression of IREB2 gene, in the presence of the minor allele of rs2568494 (beta parameter, −0.210; Table 2), confers a higher risk of lung cancer (OR = 1.437).
As far as ERCC1 is concerned, DAE was already reported at both mRNA and protein levels in prostate cancer at rs11615, with the C allele expressed at lower levels than the T allele63. Additionally, a better response to chemotherapy was reported for lung cancer patients carrying the C allele64. ERCC1 is a key player of the nucleotide excision repair (NER) system, which is activated by chemotherapeutic agents that induce DNA damage in cancer cells to kill them65. Therefore, the NER pathway and, in particular, ERCC1 activation are frequently responsible for chemoresistance. Our eQTL and survival results indicate that lower ERCC1 expression in non-involved lung tissue of adenocarcinoma patients, in presence of the C allele, was associated with better outcome. These results can be explained by a lower activity of the NER pathway in patients with lower ERCC1 expression. Although we do not have data about chemotherapy treatment in our patient series, we are confident that patients with advanced stage lung adenocarcinoma underwent chemotherapy since clinical management of these patients usually includes this treatment66,67. Therefore, we can suppose that the better survival observed in patients with allele C of ERCC1 rs11615 polymorphism is, at least in part, due to the reduced functionality of ERCC1 pathway.
In conclusion, our study highlights the relevance of deepening the function of the many loci found associated with human complex traits by GWAS, and points to the role of some of these loci as eQTLs. Our results support the hypothesis that some polymorphisms associated with lung cancer risk or prognosis influence tumor development and progression through the modulation of expression levels of target genes. Functional studies are needed to reach a more complete picture of the molecular mechanisms mediated by such SNP-specific alterations in gene expression, underpinning cancer pathogenesis.
Methods
Population series and biological material
The population series investigated in this study comprised 823 lung adenocarcinoma patients (cases) and 779 healthy controls. Cases were patients who had undergone lobectomy at one of three hospitals in the area around Milan, Italy (Fondazione IRCCS Istituto Nazionale dei Tumori, San Giuseppe Hospital, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico). Samples of peripheral blood and non-involved (apparently normal) lung parenchyma, recovered during lobectomy, had been taken for research purposes. This group included 232 patients for whom lung transcriptome data had already been obtained in a previous study57. Control subjects were recruited among blood donors (n = 158) and participants in a lung cancer screening program (n = 621), and were matched to cases by sex. Control subjects donated a sample of peripheral blood.
After collection, tissue and blood samples were used to extract RNA (from tissue only) and genomic DNA (tissue and blood). These materials were stored in the biobank of Fondazione IRCCS Istituto Nazionale dei Tumori. Methods for the collection of samples and associated clinical data have already been reported15,20,57. The Committees for Ethics of the institutes involved in recruitment (Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy, Ospedale San Giuseppe, Milan, Italy, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Milan, Italy) approved the protocol for collecting samples and clinical data. Cases and controls provided written informed consent for the use of their biological material and data for research purposes. All methods were performed in accordance with the relevant guidelines and regulations.
SNP selection and genotyping
We selected 64 SNPs to genotype based on our previous GWAS results and on GWAS, meta-analyses, and follow-up characterization studies that had been carried out from 2008 to December 31, 2013 (Supplementary Table S1).
For genotyping, we used commercially available or custom TaqMan SNP Genotyping Assays (Supplementary Table S1). SNPs were genotyped using the TaqMan Open Array Genotyping System (Thermo Fisher Scientific, Waltham, USA). DNA samples were loaded at 50 ng/mL and amplified according to the manufacturer’s instructions. For analysis of the genotypes, we used autocalling methods, as implemented in the TaqMan Genotyper software version 1.3. A genotype call rate ≥0.90 was selected as the reliability threshold; this means that SNPs for which it was impossible to determine the genotype for more than 10% of samples were eliminated from consideration.
Detection of eQTLs
To identify which, if any, lung cancer-related SNPs influence gene expression in normal lung tissue (i.e. function as eQTLs), we took advantage of transcriptome data that we had previously generated from the non-involved lung parenchyma of 284 Italian lung adenocarcinoma patients (all smokers)57. In that study, gene expression profiling had been done on HumanHT-12 v4 Expression BeadChip microarrays (Illumina) according to a discovery–validation design. Here, the two datasets of 206 samples (discovery series) and 78 samples (validation series) were combined using the ComBat adjustment method68 implemented in the sva R package69. Probes that were not annotated and those with a detection P value < 0.01 in fewer than 10% of samples were filtered out. When multiple probes mapped to the same transcript, we included only the one with the highest detection rate, defined as the percentage of samples in which the probe had detection P values < 0.01. These expression data (for 10,821 genes) have already been deposited in the Gene Expression Omnibus database (GEO, http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE71181.
For this study, we excluded from analysis 52 of these patients for whom genomic DNA was not available for genotyping. Thus, the eQTL analysis was done with 232 cases. First, the coordinates of the genotyped SNPs and the 10,821 genes with expression data were updated to the human genome assembly GRCh38 (Ensembl release 78) using the biomaRt R package70. Then, using MatrixEQTL R package71, we examined the correlations between the genotype at each SNP and the expression level of each gene, following the standard additive linear regression model which assumes that genotype has an additive effect on gene expression. For this analysis, genotypes were expressed as integers (0, 1, or 2) according to the number of minor alleles at each SNP, and patient’s sex and age at surgery were used as covariates. The statistical significance of the detected eQTLs was first determined by the software using the t statistic, and then adjusted for multiple testing using the Benjamini-Hochberg procedure to obtain the false discovery rate (FDR) of each eQTL. The threshold significance level was set as an FDR < 0.05. SNPs whose genotypes were significantly associated with the expression levels of genes mapping within 1 Mbp of genomic distance were defined as cis-eQTLs, whereas all others were regarded as trans-eQTLs.
Differential allelic expression assay
The genes found to be subject to eQTL modulation were tested for the possibility of differential allelic expression (DAE). For the DAE assay, we selected a single marker SNP located within the transcribed region of each gene of interest, and we compared the allelic frequencies of the marker SNPs between cDNA and genomic DNA from the non-involved lung tissue of lung adenocarcinoma patients who were heterozygous at that SNP (Fig. 2). Marker SNPs had to have an arbitrarily selected minor allele frequency (MAF) >0.25 in the European population and be in a sequence context that permitted the design of a pyrosequencing assay. To identify heterozygous individuals at the marker SNPs, we genotyped subsets of patients chosen on the basis of their eQTL SNPs’ genotypes. SNP genotyping was carried out on a PyroMark Q96 ID system running PyroMark Q96 ID Software (Qiagen). Primer sequences for all PCR steps and pyrosequencing are reported in Supplementary Table S3.
Next, for each gene, we grouped the heterozygous cases (at the marker SNP) into subgroups depending on their genotypes at the eQTL SNPs. For example, one subgroup consisted of cases that were heterozygous at all eQTL SNPs for the particular gene. The formation of these subgroups was dictated by the genotype combinations that were observed, which depends on the allele frequencies and linkage disequilibrium patterns of the eQTL SNPs.
Then, for all the cases chosen for analysis, we synthesized cDNA from 1 μg of RNA extracted from non-involved lung tissue using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland). To eliminate possible genomic DNA contamination, the cDNA (40 ng) was amplified with cDNA-specific primers (first amplification, Supplementary Table S3). Finally, this PCR product (4% of the reaction volume) and a sample of genomic DNA (40 ng) from the same patient were PCR-amplified using SNP-specific primers, in different tubes at the same time in parallel (second amplification, Supplementary Table S3). These PCR products were pyrosequenced on a PyroMark Q96 ID system running PyroMark Q96 ID Software (Qiagen). The proportions of individual alleles for each SNP were obtained from peak heights using the Pyro Mark MD software package (Qiagen), and allelic ratios (minor/common allele) for cDNA and genomic DNA were calculated for each case.
Statistical analyses
Clinical characteristics were compared between cases and controls using Kruskal-Wallis test for the quantitative variable age and chi-square test for categorical variables. The associations between clinical characteristics and overall survival in cases were assessed using multivariate Cox proportional hazard test, using EZR in R Commander software72.
SNP genotypes were compared between cases and controls to identify genetic variants associated with lung cancer risk. For this purpose, the genotype at each SNP was coded with a numerical value (0, 1, or 2) according to the number of minor alleles present in order to represent the additive effects of the minor allele on the risk of lung cancer. The statistical analysis was done in a logistic model with sex, age, and smoker status as covariates, using PLINK software73. Odds ratios were calculated using the allele counting method, which assumes an additive effect of a variant, i.e., an OR > 1 means that the risk of lung adenocarcinoma increases with each copy of the minor allele. Since this was a validation study, we used a value of P < 0.05 to determine statistical significance.
To identify SNPs associated with overall survival of lung adenocarcinoma patients, we used multivariable Cox proportional hazard models. Again, the genotype at each SNP was coded with a numerical value according to the number of minor alleles present to represent the dosage effect of the minor allele on the risk of dying. In separate analyses, we compared homozygotes for the minor allele (code 2) to carriers of the common allele (code 0 or 1). In all analyses, the data were adjusted for sex, age, pathological stage (I versus > I), and smoker status. A value of P < 0.05 indicated statistical significance. To visualize the relation between carrier status of genotypes and overall survival, we used the Kaplan-Meier method.
For analysis of DAE, allelic ratios were aggregated into means and standard errors (SE), for cDNA and genomic DNA, by genotype subgroup. The data were also log10-transformed for statistical assessment using Wilcoxon’s signed-rank test for paired samples in R package; P < 0.05 indicated statistical significance.
Additional Information
How to cite this article: Pintarelli, G. et al. Genetic susceptibility variants for lung cancer: replication study and assessment as expression quantitative trait loci. Sci. Rep. 7, 42185; doi: 10.1038/srep42185 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
Valerie Matarese, PhD, provided scientific editing. This work was supported in part by a grant from the Italian Association for Cancer Research (AIRC, grant no. IG 14714). The funding organization had no role in design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript.
Footnotes
The authors declare no competing financial interests.
Author Contributions T.A.D. and F.C. conceived the study. M.I., D.T., and L.S. provided biological samples from lung adenocarcinoma patients. S.N. prepared DNA and RNA samples. A.G., S.D.P. and L.C. performed DNA genotyping, under the supervision of P.M. G.P. carried out DAE experiments. C.E.C., M.D. and T.A.D. were involved in data analysis. G.P., F.C. and T.A.D. were involved in experimental design and manuscript preparation. All authors participated in critical revision of the article and gave their final approval of the submitted version.
References
- Tomoshige K. et al. Germline mutations causing familial lung cancer. J. Hum. Genet. 60, 597–603 (2015). [DOI] [PubMed] [Google Scholar]
- Xiong D. et al. A recurrent mutation in PARK2 is associated with familial lung cancer. Am. J. Hum. Genet. 96, 301–308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar A. et al. Hereditary lung cancer syndrome targets never smokers with germline EGFR gene T790M mutations. J. Thorac. Oncol. 9, 456–463 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H. et al. Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J. Natl. Cancer Inst. 106, djt338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissowska J. et al. Family history and lung cancer risk: international multicentre case-control study in Eastern and Central Europe and meta-analyses. Cancer Causes Control 21, 1091–1104 (2010). [DOI] [PubMed] [Google Scholar]
- Wang Y. et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat. Genet. 40, 1407–1409 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi M. T. et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am. J. Hum. Genet. 85, 679–691 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafnar T. et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet. 41, 221–227 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos C. I. et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 40, 616–622 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone N. L. et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: a meta-analysis and comparison with lung cancer and COPD. PLoS Genet. 6, e1001053 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd M. F. et al. Variants in the GH-IGF axis confer susceptibility to lung cancer. Genome Res. 16, 693–701 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinola M. et al. Association of the PDCD5 locus with lung cancer risk and prognosis in smokers. J. Clin. Oncol. 24, 1672–1678 (2006). [DOI] [PubMed] [Google Scholar]
- Spinola M. et al. Genome-wide single nucleotide polymorphism analysis of lung cancer risk detects the KLF6 gene. Cancer Lett. 251, 311–316 (2007). [DOI] [PubMed] [Google Scholar]
- Galvan A. et al. A polygenic model with common variants may predict lung adenocarcinoma risk in humans. Int. J. Cancer 123, 2327–2330 (2008). [DOI] [PubMed] [Google Scholar]
- Galvan A. et al. Genome-wide association study in discordant sibships identifies multiple inherited susceptibility alleles linked to lung cancer. Carcinogenesis 31, 462–465 (2010). [DOI] [PubMed] [Google Scholar]
- Wang Y. et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 46, 736–741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong T. et al. International Lung Cancer Consortium: coordinated association study of 10 potential lung cancer susceptibility variants. Carcinogenesis 31, 625–633 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G. et al. A functional polymorphism on chromosome 15q25 associated with survival of early stage non-small-cell lung cancer. J. Thorac. Oncol. 7, 808–814 (2012). [DOI] [PubMed] [Google Scholar]
- Wang Y., Peng X., Zhu L., Hu L. & Song Y. Genetic variants of CHRNA5-A3 and CHRNB3-A6 predict survival of patients with advanced non-small cell lung cancer. Oncotarget (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan A. et al. Germline polymorphisms and survival of lung adenocarcinoma patients: a genome-wide study in two European patient series. Int. J. Cancer 136, E262–71 (2015). [DOI] [PubMed] [Google Scholar]
- Tang S. et al. Genome-wide Association Study of Survival in Early-stage Non-Small Cell Lung Cancer. Ann. Surg. Oncol. 22, 630–635 (2015). [DOI] [PubMed] [Google Scholar]
- Lee Y. et al. Prognostic implications of genetic variants in advanced non-small cell lung cancer: a genome-wide association study. Carcinogenesis 34, 307–313 (2013). [DOI] [PubMed] [Google Scholar]
- Wu X. et al. Genome-wide association study of genetic predictors of overall survival for non-small cell lung cancer in never smokers. Cancer Res. 73, 4028–4038 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. I. et al. RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano M. T. et al. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 47, 1393–1401 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falvella F. S. et al. Promoter polymorphisms and transcript levels of nicotinic receptor CHRNA5. J. Natl. Cancer Inst. 102, 1366–1370 (2010). [DOI] [PubMed] [Google Scholar]
- Monteiro A. N. & Freedman M. L. Lessons from postgenome-wide association studies: functional analysis of cancer predisposition loci. J. Intern. Med. 274, 414–424 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson W., Liang L., Abecasis G., Moffatt M. & Lathrop M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 10, 184–194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra H. J. & Franke L. From genome to function by studying eQTLs. Biochim. Biophys. Acta 1842, 1896–1902 (2014). [DOI] [PubMed] [Google Scholar]
- Takiguchi Y., Sekine I., Iwasawa S., Kurimoto R. & Tatsumi K. Chronic obstructive pulmonary disease as a risk factor for lung cancer. World J. Clin. Oncol. 5, 660–666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscat J. E., Ahn K., Richie J. P. Jr & Stellman S. D. Nicotine dependence phenotype and lung cancer risk. Cancer 117, 5370–5376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell S. L. et al. The role of IREB2 and transforming growth factor beta-1 genetic variants in COPD: a replication case-control study. BMC Med. Genet. 12, 24-2350-12-24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arja C. et al. Genetic determinants of chronic obstructive pulmonary disease in South Indian male smokers. PLoS One 9, e89957 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolkowska-Suchanek I. et al. Susceptibility loci in lung cancer and COPD: association of IREB2 and FAM13A with pulmonary diseases. Sci. Rep. 5, 13502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone S. F. et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum. Mol. Genet. 16, 36–49 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui K., Ge X. & Ma H. Four SNPs in the CHRNA3/5 alpha-neuronal nicotinic acetylcholine receptor subunit locus are associated with COPD risk based on meta-analyses. PLoS One 9, e102324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. S. et al. Genetic variation (CHRNA5), medication (combination nicotine replacement therapy vs. varenicline), and smoking cessation. Drug Alcohol Depend. 154, 278–282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R. J. et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452, 633–637 (2008). [DOI] [PubMed] [Google Scholar]
- Timofeeva M. N. et al. Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Hum. Mol. Genet. 21, 4980–4995 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broms U. et al. Analysis of detailed phenotype profiles reveals CHRNA5-CHRNA3-CHRNB4 gene cluster association with several nicotine dependence traits. Nicotine Tob. Res. 14, 720–733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D. P., Yang C. L., Zhou X., Ding J. A. & Jiang G. N. Association between CLPTM1L polymorphisms (rs402710 and rs401681) and lung cancer susceptibility: evidence from 27 case-control studies. Mol. Genet. Genomics 289, 1001–1012 (2014). [DOI] [PubMed] [Google Scholar]
- Zhang X. L. et al. Decreased risk of developing lung cancer in subjects carrying the CLPTM1L rs401681 (G > A) polymorphism: evidence from a meta-analysis. Genet. Mol. Res. 13, 1373–1382 (2014). [DOI] [PubMed] [Google Scholar]
- Timofeeva M. N. et al. Genetic polymorphisms in 15q25 and 19q13 loci, cotinine levels, and risk of lung cancer in EPIC. Cancer Epidemiol. Biomarkers Prev. 20, 2250–2261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorgeirsson T. E. et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat. Genet. 42, 448–453 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukola A. et al. A Genome-Wide Association Study of a Biomarker of Nicotine Metabolism. PLoS Genet. 11, e1005498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossini A., De Almeida Simao T., Albano R. M. & Pinto L. F. CYP2A6 polymorphisms and risk for tobacco-related cancers. Pharmacogenomics 9, 1737–1752 (2008). [DOI] [PubMed] [Google Scholar]
- Bloom J. et al. The contribution of common CYP2A6 alleles to variation in nicotine metabolism among European-Americans. Pharmacogenet Genomics 21, 403–416 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J. H. et al. Associations between CYP2A6 polymorphisms and outcomes of adjuvant S-1 chemotherapy in patients with curatively resected gastric cancer. Gastric Cancer. (2015). [DOI] [PubMed] [Google Scholar]
- Di Y. M., Chow V. D., Yang L. P. & Zhou S. F. Structure, function, regulation and polymorphism of human cytochrome P450 2A6. Curr. Drug Metab. 10, 754–780 (2009). [DOI] [PubMed] [Google Scholar]
- Jeon H. S. et al. A functional variant at 19q13.3, rs967591G > A, is associated with shorter survival of early-stage lung cancer. Clin. Cancer Res. 19, 4185–4195 (2013). [DOI] [PubMed] [Google Scholar]
- Ying M., Zhu X. X., Zhao Y., Li D. H. & Chen L. H. KRAS Mutation as a Biomarker for Survival in Patients with Non-Small Cell Lung Cancer, A Meta-Analysis of 12 Randomized Trials. Asian Pac. J. Cancer. Prev. 16, 4439–4445 (2015). [DOI] [PubMed] [Google Scholar]
- Zhang Z. et al. Prognostic value of epidermal growth factor receptor mutations in resected non-small cell lung cancer: a systematic review with meta-analysis. PLoS One 9, e106053 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T., Yatabe Y., Onozato R., Kuwano H. & Mitsudomi T. Prognostic implication of EGFR, KRAS, and TP53 gene mutations in a large cohort of Japanese patients with surgically treated lung adenocarcinoma. J. Thorac. Oncol. 4, 22–29 (2009). [DOI] [PubMed] [Google Scholar]
- Bauml J. et al. Determinants of survival in advanced non-small-cell lung cancer in the era of targeted therapies. Clin. Lung Cancer. 14, 581–591 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler M. et al. PIK3CA mutations in non-small cell lung cancer (NSCLC): genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget 6, 1315–1326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan A. et al. Gene expression signature of non-involved lung tissue associated with survival in lung adenocarcinoma patients. Carcinogenesis 34, 2767–2773 (2013). [DOI] [PubMed] [Google Scholar]
- Falvella F. S. et al. Transcription deregulation at the 15q25 locus in association with lung adenocarcinoma risk. Clin. Cancer Res. 15, 1837–1842 (2009). [DOI] [PubMed] [Google Scholar]
- Nguyen J. D. et al. Susceptibility loci for lung cancer are associated with mRNA levels of nearby genes in the lung. Carcinogenesis 35, 2653–2659 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehringer G. et al. Association of the 15q25 and 5p15 lung cancer susceptibility regions with gene expression in lung tumor tissue. Cancer Epidemiol. Biomarkers Prev. 21, 1097–1104 (2012). [DOI] [PubMed] [Google Scholar]
- Rouault T. A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2, 406–414 (2006). [DOI] [PubMed] [Google Scholar]
- Ghio A. J. et al. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am. J. Respir. Crit. Care Med. 178, 1130–1138 (2008). [DOI] [PubMed] [Google Scholar]
- Woelfelschneider A. et al. A distinct ERCC1 haplotype is associated with mRNA expression levels in prostate cancer patients. Carcinogenesis 29, 1758–1764 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J. et al. HapMap-based study of a region encompassing ERCC1 and ERCC2 related to lung cancer susceptibility in a Chinese population. Mutat. Res. 713, 1–7 (2011). [DOI] [PubMed] [Google Scholar]
- McNeil E. M. & Melton D. W. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 40, 9990–10004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt W. E. et al. 2nd ESMO Consensus Conference in Lung Cancer: locally advanced stage III non-small-cell lung cancer. Ann. Oncol. 26, 1573–1588 (2015). [DOI] [PubMed] [Google Scholar]
- Reck M. et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 25 Suppl 3, iii27–39 (2014). [DOI] [PubMed] [Google Scholar]
- Johnson W. E., Li C. & Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
- Leek J. T., Johnson W. E., Parker H. S., Jaffe A. E. & Storey J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S., Spellman P. T., Birney E. & Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin A. A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 48, 452–458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.