

Abstract
Immune cell activation and differentiation occurs concurrently with metabolic reprogramming. This insures that activated cells generate the energy and substrates necessary to perform their specified function. Likewise, the metabolic programs amongst different cells of the immune system vary. By targeting different metabolic pathways, these differences allow for selective regulation of immune responses. Further, the relative susceptibility of cells to a metabolic inhibitor is dictated by their metabolic demands; cellular selectivity is based on demand. Therefore where differences exist in metabolic pathways between healthy and pathogenic cells, there is opportunity for selective regulation with agents lacking intrinsic specificity. There are now a host of studies demonstrating how inhibitors of metabolism (for example glycolysis, glutamine metabolism, and fatty acid oxidation) can regulate immune responses and treat immune mediated pathogenesis. In this brief review we detail how inhibitors of metabolism can be employed to regulate immune responses in both autoimmunity and transplantation.
I-Introduction
In the past decade, the field of immunology has been characterized by a marked increase in our understanding of genetic and signaling programs which define immune cells. More recently, it has become clear that a key component of immune cell regulation and function is the concomitant reprogramming of metabolic pathways (1). The metabolism of a naïve lymphocyte is different from that of a memory cell, is different from that of an effector (and indeed, even effector subsets have great differences in their metabolic profiles) (2). These differences offer promising opportunities for selective regulation of immune subsets. Furthermore, recent studies suggest that even metabolic inhibitors that lack intrinsic specificity can be made to affect only a select subset of cells based on the metabolic demands of those cells. This principle of cellular selectivity based upon cellular demand underlies a great deal of metabolic therapy, and is broadly applicable to a number of different cells and disease types.
The particular metabolism of lymphocytes differs at each stage of development, and has been extensively detailed elsewhere (2-5). In brief, naïve lymphocytes resemble many of the somatic cells in the body, and progress through metabolic pathways in a textbook fashion, relying on glycolysis and subsequent TCA cycling to produce a maximum amount of ATP (2). However, upon activation, there is a dramatic shift in the metabolism of lymphocytes. Similar to the Warburg effect, which was first observed by the biochemist Otto Warburg in cancer cells (6), there is a tremendous increase in glycolysis, even in the presence of abundant oxygen (7). This aerobic glycolysis seems counterintuitive, but is actually a metabolic program undertaken by the cell that prioritizes the early generation of biosynthetic intermediates, which are necessary for activation, proliferation, and effector function. This metabolic change is facilitated by the upregulation of several key transporters, enzymes and signaling pathways (8, 9). Effector cells also have differences in their metabolism between different subsets, with Th1, Th2, and Th17 tending to be more glycolytic, while regulatory T cells (Tregs) rely more on lipid metabolism (8). These subset differences provide another opportunity for differential regulation, based on the select needs of the particular subset. Different effector subsets also have different signaling pathways, such as differential mTOR complex I or II requirements for Th1/Th17 or Th2 cells (10, 11), or increased AMPK activation in Tregs (8), further expanding the potential for selective manipulation of cellular processes based upon their differential metabolic demands.
The transition from effector cell to a memory cell involves further metabolic changes. Memory cells are more reliant on fatty acid oxidation and have increased mitochondrial mass (12, 13). This increase in mitochondria is coupled with the increase in spare respiratory capacity (13). From a metabolic perspective, memory cells are “fueled to last.” Furthermore, these metabolic changes allow the memory T cell to rapidly expand and to take on an effector function upon restimulation (14). From this overview of naïve, effector and memory T cell metabolism, a picture emerges whereby selectively regulating metabolic pathways can lead to the fine-tuned regulation of immune function. In this brief review, we will highlight several instructive examples of how the differential metabolism of innate and adaptive immune cells are beginning to be exploited therapeutically.
II- Targeting signaling by targeting metabolites in innate and adaptive immune cells
The shift in the metabolism of innate immune cells upon activation is similar yet distinct from that which occurs in the adaptive immune system (2, 15). This is advantageous, as it provides opportunities for both concurrent and differential regulation of the two arms of the immune response. Upon activation of macrophages and dendritic cells (DCs) in an inflammatory context, there is a shift from the quiescent state to a Warburg phenotype that is similar to that of activated lymphocytes (16, 17). This is accompanied by a decrease in the flux through the TCA cycle (18, 19). However, the goal of this shift in metabolism is not to support proliferation. Rather, such metabolic changes support full activation and cytokine production (20), as well as other important host defense functions, such as phagocytosis (21). DCs require a shift to aerobic glycolysis in order to mature and become capable of presenting antigen and activating lymphocytes (17, 18, 22). When glycolysis is inhibited by treatment with 2-deoxy-glucose (2DG), a small molecule inhibitor of the first step of glycolysis, hexokinase, macrophages stimulated with LPS display mitigated production of IL1β (19, 23) Underlining the specificity of this metabolic approach, targeting hexokinase with 2DG can selectively block IL-1β production while not altering the transcription of TNFα (19).
In addition to the shift to aerobic glycolysis, there are a number of other metabolic changes that innate cells undergo, which are potential targets for metabolic therapies. The manipulation of these metabolites takes on an added importance because many metabolites also function as signaling molecules, the alteration of which can have profound effects on the downstream immune response. In this context, metabolites are not simply the end product of metabolism, but also can alter cell function by differentially signaling separate biochemical and molecular pathways.
The production of nitric oxide (NO) is sustained by the increased glycolysis following activation (24) and is required for activation-induced inhibition of oxidative phosphorylation (25). In addition, reactive oxygen species (ROS) are associated with priming the NLRP3 inflammasome (26). Treatment with metformin, a diabetes drug that activates AMPK (27), can decrease the production of ROS as a mechanism for modulating immune responses (28). ROS are also key for T cell activation, as lymphocytes that are genetically incapable of producing mitoROS cannot activate NFAT translocation to the nucleus or produce IL2 (29). Another metabolite in flux is citrate, which is pumped out of the mitochondria, and can serve as a substrate for histone acetylation, and thereby facilitate the epigenetic activation of genes crucial to metabolic pathways (30, 31). Histone acetylation has recently been shown to be important in the IL-4 induced polarization of M2 macrophages and is tightly controlled by ATP citrate lyase (Acly) (32). Cytoplasmic citrate can also be used as a substrate for lipid biogenesis, and the production of ROS or NO (33).
Another metabolite that increases upon stimulation is succinate. The increase in glutamine metabolism that accompanies aerobic glycolysis paradoxically increases the succinate present in macrophages after stimulation, even though flux through the TCA cycle is decreased (18). Succinate stabilizes HIF, which leads to increased production if IL1β. Inhibition of glycolysis with 2DG prevents this increase in succinate upon stimulation, and leads to a decrease in IL1β production, as detailed below (19). Recently, an important role for itaconate in regulating macrophage inflammation has been demonstrated (34). Itaconate has a regulatory role in macrophage metabolism, and acts by competitive inhibition of succinate dehydrogenase (Sdh) (35). In this model, itaconate endogenously produced by Irg1 has been shown to inhibit Sdh, which leads to a block in the TCA cycle, and a subsequent increase in succinate accumulation in LPS-stimulated macrophages. Alternatively, in IRG1-/- cells there is an increase in HIF-1α concurrent with a relative decrease in succinate (34). Such findings demonstrate an additional link of the HIF-1α-IL-1β axis to the efficiency and directionality of the electron transport chain.
Acetate also plays a role in guiding cellular differentiation and function. Acetate is increased systemically upon bacterial infection (36). When memory CD8 T cells that were initially primed in increased acetate are restimulated, they are able to mount a more rapid response, and have a more dramatic increase in glycolysis upon restimulation. In addition, adoptive transfer of CD8 memory cells that were initially simulated in acetate is more protective against listeria challenge (36).
Facilitating the metabolic shift that occurs upon activation, cells of the innate immune system undergo transcriptional and signaling pathway changes that dramatically alter their behavior. One such transcriptional change (that was mentioned above) is an increase in HIF upon the induction of aerobic glycolysis. This increase in HIF occurs under normal oxygen tension, is promoted by mTOR signaling, and is necessary for cytokine production upon activation (19, 37). Corresponding to the increased mTOR signaling following activation, there is a decrease in AMPK signaling in innate immune cells upon LPS stimulation. However, metformin treatment activates the AMPK signaling pathway, and this activation is sufficient to decrease IL1β production, as well as increase the production of IL10 (28). In addition to activating AMPK, metformin also inhibits Electron Transport Chain (ETC) complex I (38). Interestingly, the ability of metformin to promote a shift to a more immunosuppressive cytokine profile is also found in rotenone, which also inhibits ETC complex I (28).
As is the case for the various subsets of effector T cells, M1 and M2 macrophages also have different metabolic demands. Transcriptional differences between the classically defined M1 and M2 cells also have important metabolic consequences. The alternatively activated M2 macrophages have a metabolism somewhat similar to Tregs with oxidative phosphorylation providing their energy, as opposed to aerobic glycolysis (39). In addition, M2 macrophages have less iNOS expression (leading to decreased NO production), increased AMPK activity, and less HIF (15). Further, it has been proposed that an IL-4 mediated increase in PGC-1β is the driver of the oxidative phosphorylation that characterizes the metabolism of M2 macrophages (39). This metabolic and signaling phenotype is in contrast to the metabolic changes that occur in more pro-inflammatory innate immune cells, and provides potential differences with which the balance between macrophage subtypes can be manipulated.
Parallel analysis of transcriptional and metabolomics data has provided further insight into the metabolic circuitry of activated macrophages. Using a combined metabolic and transcriptional analysis, termed CoMBI-T, Jha and colleagues were able to simultaneously examine the interplay between metabolites and the enzymes involved in pathways that use or generate them (40). This approach revealed strikingly different metabolic profiles in M1 and M2 macrophages. In the M1 macrophages, the conversion of isocitrate to α-ketogluterate (AKG) was specifically found to be deficient, due to decreased transcription of the enzyme responsible for that reaction, isocitrate dehydrogenase (Idh1). Decreased AKG production in M1 macrophages requires that citric acid serve as a precursor for itaconate and fatty acid synthesis (40). Irg1, as discussed above, is one of the most strongly upregulated transcripts in M1 macrophages as identified by CoMBI-T analysis, thus leading to increased levels of itaconate in M1 activated macrophages. Dimethyl itaconate administration was able to partially reverse tissue injury in a cardiac ischemia-reperfusion model, in which damage is caused in part by hypoxia-induced ROS production (41). Dimethyl itaconate pretreatment was also shown to decrease ROS production in bone marrow derived macrophages, as well diminish inflammatory cytokine production and inflammasome activation (34). In contrast, M2 macrophages require glutamine metabolism. The generation of UDP-GlcNAc was found to be highly dependent on glutamine, as labelled nitrogen from glutamine made up more than half of the nitrogen in UDP-GlcNAc after just four hours. Glutamine deprivation dramatically decreased M2 generation leading to downregulation specifically in M2 signature gene transcription. This includes downregulation of CCL22, the secretion of which was also dramatically reduced in glutamine free media (40). However, glutamine concentration had no impact on M1 polarization as measured by iNOS expression (40). Furthermore, it has been shown that glutamine deficiency actually increases the production of TNFα, while protecting LPS-stimulated macrophages from lipid toxicity (42)
In addition to metabolites altering macrophage polarization, enzymatic differences can shift the balance between M1 and M2 macrophages. Pyruvate kinase M2 (PKM2) is transcriptionally upregulated upon LPS stimulation of BMDM, and can form a dimer that is enzymatically inactive, but can travel to the nucleus and induce transcription of glycolytic genes by interacting with HIF1α (43). Small molecules DASA-58 and TEPP-46 force PKM2 into a tetrameric conformation (44), which is incapable of promoting HIF transcription, leading to a decrease in IL1β production following LPS stimulation (45). Furthermore, treatment with TEPP-46 decreases M1 macrophage polarization following LPS stimulation, and both DASA-58 and TEPP-46 dramatically decrease activation-induced glycolysis. TEPP-46 administration leads to decreased IL1-β production and increased bacterial loads in the spleen and liver of mice infected with Salmonella typhimurium, due to decreased cellular killing of bacteria (45). Although deleterious in this case, the possibility exists that these small molecules that prevent the activity of PKM2 could be beneficial in treating autoimmune inflammatory diseases. Although there has been less focus on the intervention of innate immune metabolism in treating autoimmunity and destructive inflammation, this is a field ripe for further exploration.
III- Targeting metabolism in BMT
In addition to their role in protective immunity, T lymphocytes make an important contribution to disease pathogenesis in a variety of autoimmune conditions. Normally T cell metabolism upon activation involves a rapid increase in aerobic glycolysis (along with a concurrent transcriptional/translational increase in the machinery involved in these processes), and an increase in the uptake of glutamine to replenish the TCA cycle, in order to derive anaplertoic substrates from that pathway (2). Also, activated T cells display increased shunting of carbons into alternative metabolic circuits, such as the pentose phosphate pathway (46). However, in a number of autoimmune diseases, this metabolism is disrupted. In the case of Graft-versus-Host disease (GVHD) following a bone marrow transplantation (BMT), T cell metabolism is remarkably shifted. The immune response in GVHD is characterized by a constant encounter between the activated lymphocytes and their target tissue. This constant, high level of antigen engagement appears to lead to a different metabolism in the pathogenic donor cells. Their high requirement for ATP means they cannot subsist on oxidative glycolysis alone, and therefore such cells are forced to find an alternate source of fuel. This can be provided by the oxidation of fatty acids (47). When examining the metabolic tracing of 13C labelled glutamine, glucose, and palmitate in T cells from a mouse model of GVHD following allotransplant, it was found that glutamine uptake and incorporation into RNA (as ribose) and fatty acids (as palmitate) are markedly increased as compared to T cells from naïve mice. This is in contrast to the results of labelled glucose tracer incorporation, which shows no difference between ribose or palmitate incorporation, and the use of a palmitate tracer, which demonstrates decreased incorporation across the board in disease mice (48). This indicates that fatty acids are preferentially being catabolized for oxidative phosphorylation, while glutamine is being used as an anabolic substrate, and hints at an alternative metabolism that is specific to pathogenic lymphocytes in GVHD, particularly one that is reliant on lipid oxidation for the vast ATP needs of chronically activated cells. These differences in terms of metabolic demands and reprogramming suggest that metabolic therapy might be able to selectively inhibit activation of the GVHD inducing cells.
Analysis of competing pathways (Figure 1) indicates that metabolic therapy specifically targeting fatty acid metabolism (required to meet the increased demand for ATP) with etomoxir would be ideal, and could selectively inhibit the persistently activated GVHD-inducing lymphocytes. However, great care must be taken to avoid disturbing the proliferation of nonpathogenic donor cells. In a mouse model of bone marrow allotransplant, proliferating bone marrow cells reconstituting the immune system of a lethally irradiated syngeneic host undergo a dramatically different metabolic program than the pathogenic cells, with the healthy cells relying more on glycolysis than oxidative phosphorylation (47). Small molecule inhibitors of the F1F0 ATPase, such as BZ423 and LyC-31138, have demonstrated promise as a metabolic therapy for GVHD (47, 48). These drugs prevent the production of ATP, as well as increase the mitochondrial polarization of the pathogenically activated lymphocytes. In allotransplant BMT models of GVHD, these have been shown to rapidly induce apoptosis in the pathogenic cell population, and significantly decrease cytokine production, leading to decreased GVHD clinical score, and increased survival (47, 48). These ATPase inhibitors act by inducing caspase-regulated apoptosis in the pathogenic cells (47). Importantly, these agents are specific for the pathogenic cells and their rapid requirement for ATP production, and thus do not effect immunological reconstitution of the graft (47). Another therapeutic strategy is targeting fatty acid oxidation with etomoxir, an agent that blocks lipid metabolism at CPT1 (49). Following allotransplant, two weeks of etomoxir treatment was able to decrease clinical GVHD scores from 10 days to a month after the end of treatment. Etomoxir administration was successively able to reduce the proliferation and promote the apoptosis of GVHD inducing CD8 T cells that have divided numerous times, leaving the undivided cells alone (50). This is advantageous, because the highly divided cells were found to be the pathogenic cell population (47). Targeting fatty acid metabolism is a therapeutic strategy that does not impact other cells of the immune system, such as dendritic cells or naïve T cells, and does not inhibit graft reconstitution (50).
Figure 1.
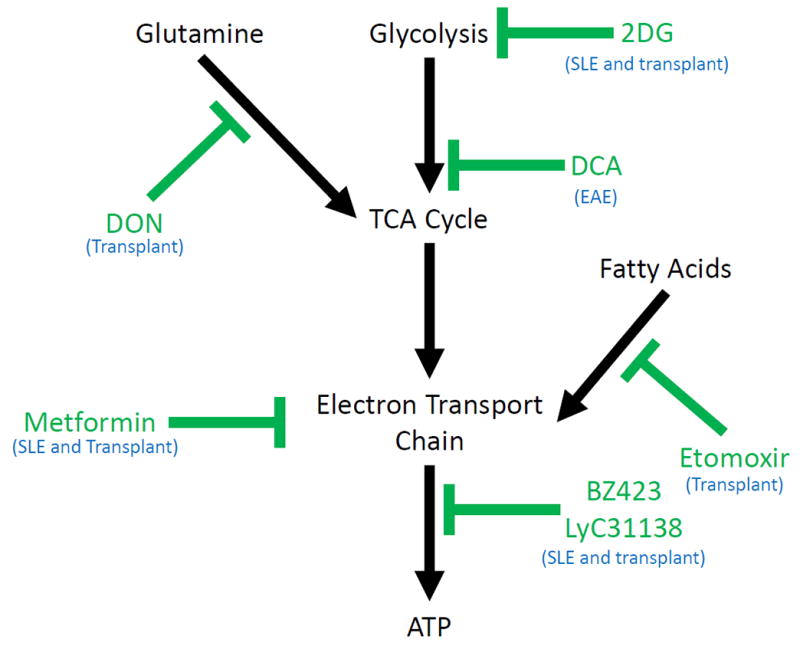
Summary of metabolic pathways vital for T cells responses (black), and drugs that inhibit these pathways (green). The particular condition of autoimmune inflammation the drug has been tested in is in blue.
In spite of these impressive results treating GVHD by blocking lipid metabolism, others have found that the pathogenic T cells in GVHD behave metabolically as normally activated T cells. That is, it has more recently been reported that pathogenic T cells in GVHD increase both their glycolytic and oxidative phosphorylation rates, while decreasing their uptake and oxidation of fatty acids (51). Furthermore, inhibition of glycolysis with 3-PO, a specific inhibitor of PFKB3 (52), improved survival and decreased GVHD clinical scores. This improvement was not seen with 2DG, as it was too toxic for the prolonged treatment necessary (51). It remains to be determined if the differences in observed metabolic phenotypes (reliance on lipid oxidation versus glycolysis) in these studies of GVHD reflect subtle differences between the models with regard to pathogenesis. Most likely both processes will turn out to be playing a role in the GVHD-inducing cells.
IV- Targeting metabolism in solid organ transplantation
In the case of solid organ transplantation, metabolic therapy has also been employed. However, in this scenario the metabolic demands of the effector cells promoting graft rejection are somewhat different from those observed in BMT and thus the anti-metabolic targeting strategy is different (53). For solid organ transplant, it appears that there is far less reliance on lipid metabolism for the generation of ATP, and rejecting cells appear to employ aerobic glycolysis (54). This may be due to the fact that unlike in GVHD, where the donor pathogenic lymphocytes are persistently being activated by host antigen, in the setting of solid organ transplant, the host pathogenic lymphocytes have much less frequent encounter with allo-antigen. As such, CD4 T cell activation in the setting of solid organ transplant more closely resembles the classical metabolic shift that is seen during the normal activation of lymphocytes (2). To this end, it was found that inhibiting glycolysis with 2DG combined with metformin, an inhibitor of complex I of the electron transport chain (38), can substantially decrease the glycolysis of an activated T cell, and by extension its cytokine production and proliferation (55). When these two drugs are combined with 6-diazo-5-oxo-L-norleucine (DON), a glutamine analog that broadly inhibits glutamine uptake and metabolism (56-58), and thus its ability to replenish the TCA cycle, it is possible to more completely abrogate the immune response. In the transplant setting, these three drugs can be combined with great effect. Triple therapy with 2DG, metformin, and DON was capable of suppressing the proliferation of pathogenic CD4 and CD8 effector T cells and was able to markedly inhibit completely MHC mismatched allograft rejection in models of both skin and heart transplantation. Furthermore, due to the fact that Tregs are more reliant on lipid metabolism, the triple therapy inhibited the CD4 effector response, but actually promoted the generation of antigen specific regulatory T cells (55). That is, the metabolic therapy did not lead to the “wholesale” inhibition of immune responses but rather, based on differential metabolism, selectively inhibited effector responses while promoting regulatory T cell responses. Given these properties, current studies are underway to combine metabolic therapy with tolerance inducing therapy (for example costimulatory blockade) in order to try to promote long term tolerance in the absence of long term immunosuppression.
Furthermore, these studies suggest a potentially broader principle in terms of targeting metabolism as a means of regulating immune responses. It is somewhat striking that, in spite of the fact that the metabolic inhibitors employed target metabolic pathways found in all cells, the combination therapy in the setting of transplant rejection had a robust therapeutic index (55). That is, while activated effector cells mediating graft rejection were sensitive to metabolic inhibition, resting immune cells and other healthy tissues were relatively unaffected. Indeed, at higher doses, triple therapy might inhibit proliferating gut epithelial cells or result in bone marrow suppression (59, 60). However, for the purpose of preventing allograft rejection, triple metabolic therapy readily inhibited effector cells without significant side effects. These observations point to cellular selectivity based upon demand. That is, relatively non-specific inhibitors can exert cellular selectivity by differentially affecting cells that necessitate the greatest/extraordinary demand for the particular metabolic pathway (table 1).
Table 1.
Summary table of significant works described herein. For the each drug, its target, the condition of autoimmune inflammation the drug was tested in, its effect, and the literature reference.
| Drug | Pathway Targeted | Disease Model | Effect | Reference |
|---|---|---|---|---|
| 2-Deoxy-D-Glucose (2DG) | Glycolysis (hexokinase) | SLE | Decrease clinical score while leaving normal immune function unaffected (in combination with metformin) |
Yin et al. 2015 Yin et al. 2016 |
| Solid organ transplant | Prolong graft survival (in combination with DON and metformin) | Lee et al. 2015 | ||
| 6-diazo-5-oxo-L-norleucine (DON) | Glutamine uptake and metabolism | Solid organ transplant | Prolong graft survival (in combination with 2DG and metformin) | Lee et al. 2015 |
| BZ423/LyC31138 | ATP synthase | BMT GVHD | Decrease clinical score while preserving graft reconstitution |
Gatza et al. 2011 Glick et al. 2014 |
| Dichloroacetate (DCA) | Pyruvate branch-point (PDHK1) | EAE | Decrease clinical score and pathogenic Th17 formation, while promoting Tregs. | Gerriets et al. 2015 |
| Etomoxir | Lipid metabolism (CPT1α) | BMT GVHD | Decrease clinical score while preserving graft reconstitution | Byersdorfer et al. 2013 |
| Metformin | Electron transport chain (complex I) | SLE | Decrease clinical score while leaving normal immune function unaffected (in combination with 2DG and DON) |
Yin et al. 2015 Yin et al. 2016 |
| Solid organ transplant | Prolong graft survival (in combination with 2DG and DON) | Lee et al. 2015 |
Manipulating the balance between regulatory and effector T cell metabolism is an effective strategy for the treatment of autoimmune inflammation. Pyruvate dehydrogenase (PDH) inhibition by PDH kinase (PDHK) promotes the conversion of pyruvate to lactate over the canonical progression of pyruvate to acetyl-CoA (61). When PDHK1 is inhibited by the addition of dichloroacetate (DCA) (62) in vitro, it has a specific inhibitory effect on the differentiation and function of Th17 cells, while promoting the generation of T regulatory cells (63). This is likely due to differences in Pdhk1 expression in the different cell types, as Th17 have higher Pdhk1 expression levels than Tregs. Therefore, when DCA was used to treat the Th17-driven model of autoimmune inflammation, experimental autoimmune encephalitis (EAE), it was found that the inhibition of PDHK significantly decreased disease scores and progression. Disease alleviation was accompanied by an increase in Treg percentage in the draining lymph node, and a decrease in activated Th17 percentage recovered (63). PDHK1 also plays a crucial role in macrophage polarization, as it is required for M1 macrophage generation and activation in response to the TLR2 agonist PAM, as well as bacterial pathogens that are dependent on TLR2 signaling to induce a response (64). Knockdown of PDHK1 increases the expression of M2 signature genes at early time points after activation, and increases the oxidative respiration that preferentially leads to the M2 polarization. Interestingly, etomoxir pretreatment did not have a deleterious effect on M2 polarization as observed six hours after stimulation, potentially indicating that M2 macrophages do not require fatty acid metabolism until later after activation (64).
V- Targeting metabolism in systemic lupus erythematosus
Another condition of pathogenic T cell activation that has been targeted with metabolic therapy is systemic lupus erythematosus (SLE). Similar to GVHD following BMT, in the case of SLE there is a constant, persistent encounter between the pathogenic lymphocyte (the pathogenesis of SLE is chiefly reliant on CD4 T cells) and its target tissue (65). As a result, pathogenic T cells take on a chronically activated phenotype, and thus have dramatically different metabolic needs than healthy tissue (66). Similar to pathogenic T cells in GVHD, and in contrast to the lymphocytes activated under normal conditions or in the case of solid organ transplant, CD4 T cells in SLE meet their energetic needs mostly through oxidative phosphorylation. Tracer experiments that use 13C uniformly labelled glucose found more oxidation to CO2 in a NZB/W mouse model of lupus, with no differences in glycolysis, or pentose phosphate pathway activity (65). This augmented reliance on oxidative phosphorylation is coupled to increased mitochondrial polarization and mass in the T cells of SLE patients, which is caused in part by decreased mitochondrial autophagy (67). This phenotype can be partially reversed by treatment with 3-PEHPC, a geranylgeranyl transferase inhibitor, which is also sufficient to decrease autoantibody formation and lupus nephritis in both NZB/W and MRL/lpr mouse models of lupus (68).
Further studies have attempted to more fully characterize the metabolism of SLE lymphocytes. Using a triple congenic mouse model of lupus (TC), CD4 T cells were analyzed for metabolic activity ex vivo, and found to have increased basal metabolism prior to the onset of disease, including both glycolysis, as determined by extracellular acidification rate, and oxidative metabolism, as determined by oxygen consumption rate (69). Pre-disease cells had decreased spare respiratory capacity as compared to wild type controls, and although they produced ATP at the same rate, pre-disease cells had a lower ATP charge, indicating increased energy use (69). To gain further insight into the metabolic differences in the lupus mice, extensive gene expression analysis was carried out. As expected, the cells from the lupus mice had higher expression of many genes in the glycolytic pathway, but unexpectedly had an increase in Cpt1a, a key regulator of fatty acid oxidation (69). These metabolic analysis, coupled with gene expression differences, open the opportunity for metabolic therapy in the case of SLE.
To capitalize on the metabolic differences between normal and SLE cells, experiments were undertaken to treat the mice with 2-DG and metformin. When these drugs are used to treat the CD4+ T cells from lupus mice ex vivo, they are capable of preventing the production of interferon gamma. When given in vivo, 2DG and metformin were able to decrease the ECAR and OCR to levels comparable to those of disease free control animals. Furthermore, they do not affect naïve cells or the immune system as a whole, as circulating total antibody levels are unchanged (69). Metabolic therapy is able to prevent disease, as there is a substantially decreased production of autoantibody and formation of pathogenic Tcm cells in the treated mice. These results rely on the synergistic effects of these two drugs, as neither was effective as monotherapy. These results were corroborated in another lupus model (B6.lpr) by the same group, which sought to further refine the possibilities of metabolic intervention. In this model, similar metabolic phenotypes were seen, and the response to metformin and 2-DG treatment was maintained. However, an alternative metabolic therapy, the inhibition of PDHK1 with DCA, was found not to be effective (65).
VI- Conclusions
The purpose of this (brief) review is not to provide a comprehensive overview of the emerging field of immunometabolism. Rather, we have detailed a number of instructive examples in the literature where targeting metabolism is able to inhibit autoimmunity/transplant rejection in lieu of traditional immunosuppressive regimens. The dynamic nature of metabolic programing amongst immune cells is intimately linked to their plasticity and function. We believe that the examples cited mark the beginning of a new approach to treating autoimmune/inflammatory diseases and transplant rejection. That is, targeting metabolism as a means of regulating (not suppressing) immune responses. Indeed, while calcineurin inhibitors (cyclosporine and FK506) suppress both effector and regulatory cells, targeting metabolism can simultaneously inhibit effector responses and promote the generation of regulatory T cells (55). In this regard, metabolic therapy might prove to provide a robust platform for promoting tolerance. Furthermore, a common theme emerging from the studies cited is that of cellular selectivity based upon demand. That is, the concept that even a broad inhibitor like 2-DG which inhibits glycolysis, will selectively inhibit effector cells (that have a markedly enhanced glycolytic demand) yet not adversely affect somatic cells or even T regulatory cells, both of which employ glycolysis but at a much decreased demand when compared to the effector cells.
Acknowledgments
This work was supported by the NIH (grants AI072677, AI77610 and AI09148)
References
- 1.Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol. 2014;14:435–446. doi: 10.1038/nri3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212:1345–1360. doi: 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacIver NJ, Michalek RD, Rathmell JC. Metabolic Regulation of T Lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 7.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macintyre A, Gerriets V, Nichols A, Michalek R, Rudolph M, Deoliveira D, Anderson S, Abel E, Chen B, Hale L, Rathmell J. The Glucose Transporter Glut1 Is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metabolism. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. 2012;30:39–68. doi: 10.1146/annurev-immunol-020711-075024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L, Jones RG, Choi Y. Enhancing CD8 T Cell Memory by Modulating Fatty Acid Metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Windt, G JW, Everts B, Chang C, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial Respiratory Capacity Is A Critical Regulator Of CD8(+) T Cell Memory Development. Immunity. 2011;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Windt, G JW, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, Chang C, Smith AM, Ai T, Faubert B, Jones RG, Pearce EJ, Pearce EL. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proceedings of the National Academy of Sciences. 2013;110:14336–14341. doi: 10.1073/pnas.1221740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–784. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J. 1986;239:121–125. doi: 10.1042/bj2390121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213:15–23. doi: 10.1084/jem.20151570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LA. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol. 2015;15:18–29. doi: 10.1038/nri3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sbarra AJ, Karnovsky ML. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem. 1959;234:1355–1362. [PubMed] [Google Scholar]
- 22.Everts B, Amiel E, Huang SC, Smith AM, Chang C, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt, G JW, Artyomov MN, Jones RG, Pearce EL, Pearce EJ. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKÎμ supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGettrick AF, O’Neill LA. How metabolism generates signals during innate immunity and inflammation. J Biol Chem. 2013;288:22893–22898. doi: 10.1074/jbc.R113.486464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Everts B, Amiel E, van der Windt GJW, Freitas TC, Chott R, Yarasheski KE, Pearce EL, Pearce EJ. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. 2012;120:1422–1431. doi: 10.1182/blood-2012-03-419747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. Reactive oxygen species inhibitors block priming, but not activation of the NLRP3 inflammasome. Journal of Immunology (Baltimore, Md: 1950) 2011;187:613–617. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, Viollet B, Guigas B. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia. 2011;54:3101–3110. doi: 10.1007/s00125-011-2311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly B, Tannahill GM, Murphy MP, O’Neill LA. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1beta (IL-1beta) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J Biol Chem. 2015;290:20348–20359. doi: 10.1074/jbc.M115.662114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang C, Schumacker PT, Licht JD, Perlman H, Bryce PJ, Chandel NS. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan K. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, Espinosa EC, Lamming D, Manning BD, Zhang Y, Blair IA, Horng T. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife. 2016;5:e11612. doi: 10.7554/eLife.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Infantino V, Convertini P, Cucci L, Panaro M, Di Noia M, Calvello R, Palmieri F, Iacobazzi V. The mitochondrial citrate carrier: a new player in inflammation. Biochem J. 2011;438:433–436. doi: 10.1042/BJ20111275. [DOI] [PubMed] [Google Scholar]
- 34.Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC, Griss T, Weinheimer CJ, Khader S, Randolph GJ, Pearce EJ, Jones RG, Diwan A, Diamond MS, Artyomov MN. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016;24:158–166. doi: 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, Koseki H, Cabrales P, Murphy AN, Hiller K, Metallo CM. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J Biol Chem. 2016;291:14274–14284. doi: 10.1074/jbc.M115.685792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balmer M, Ma E, Bantug G, Grählert J, Pfister S, Glatter T, Jauch A, Dimeloe S, Slack E, Dehio P, Krzyzaniak M, King C, Burgener A, Fischer M, Develioglu L, Belle R, Recher M, Bonilla W, Macpherson A, Hapfelmeier S, Jones R, Hess C. Memory CD8+ T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity. 2016;44:1312. doi: 10.1016/j.immuni.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 37.Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282:20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 38.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 39.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metabolism. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ, Driggers EM, Artyomov MN. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu C, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He L, Weber KJ, Schilling JD. Glutamine Modulates Macrophage Lipotoxicity. Nutrients. 2016;8 doi: 10.3390/nu8040215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole R, Pandey A, Semenza G. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anastasiou D, Yu Y, Israelsen WJ, Jiang J, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, Yang H, Mattaini KR, Metallo CM, Fiske BP, Courtney KD, Malstrom S, Khan TM, Kung C, Skoumbourdis AP, Veith H, Southall N, Walsh MJ, Brimacombe KR, Leister W, Lunt SY, Johnson ZR, Yen KE, Kunii K, Davidson SM, Christofk HR, Austin CP, Inglese J, Harris MH, Asara JM, Stephanopoulos G, Salituro FG, Jin S, Dang L, Auld DS, Park H, Cantley LC, Thomas CJ, Vander Heiden G M. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ, O’Neill LA. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21:65–80. doi: 10.1016/j.cmet.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang R, Dillon C, Shi L, Milasta S, Carter R, Finkelstein D, McCormick L, Fitzgerald P, Chi H, Munger J, Green D. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, Glick GD, Ferrara JL. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med. 2011;3:67ra8. doi: 10.1126/scitranslmed.3001975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glick GD, Rossignol R, Lyssiotis CA, Wahl D, Lesch C, Sanchez B, Liu X, Hao LY, Taylor C, Hurd A, Ferrara JL, Tkachev V, Byersdorfer CA, Boros L, Opipari AW. Anaplerotic metabolism of alloreactive T cells provides a metabolic approach to treat graft-versus-host disease. J Pharmacol Exp Ther. 2014;351:298–307. doi: 10.1124/jpet.114.218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopaschuk GD, Wall SR, Olley PM, Davies NJ. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ Res. 1988;63:1036–1043. doi: 10.1161/01.res.63.6.1036. [DOI] [PubMed] [Google Scholar]
- 50.Byersdorfer CA, Tkachev V, Opipari AW, Goodell S, Swanson J, Sandquist S, Glick GD, Ferrara J. Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood. 2013;122:3230–3237. doi: 10.1182/blood-2013-04-495515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen HD, Chatterjee S, Haarberg KM, Wu Y, Bastian D, Heinrichs J, Fu J, Daenthanasanmak A, Schutt S, Shrestha S, Liu C, Wang H, Chi H, Mehrotra S, Yu XZ. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J Clin Invest. 2016;126:1337–1352. doi: 10.1172/JCI82587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schoors S, De Bock K, Cantelmo A, Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong B, Quaegebeur A, Goveia J, Bifari F, Wang X, Blanco R, Tembuyser B, Cornelissen I, Bouché A, Vinckier S, Diaz-Moralli S, Gerhardt H, Telang S, Cascante M, Chesney J, Dewerchin M, Carmeliet P. Partial and Transient Reduction of Glycolysis by PFKFB3 Blockade Reduces Pathological Angiogenesis. Cell Metabolism. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 53.Lo Y, Lee C, Powell JD. Insight into the role of mTOR and metabolism in T cells reveals new potential approaches to preventing graft rejection. Current Opinion in Organ Transplantation. 2014;19:363–371. doi: 10.1097/MOT.0000000000000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Priyadharshini B, Turka LA. T Cell Energy Metabolism as a Controller of Cell Fate in Transplantation. Current Opinion in Organ Transplantation. 2015;20:21–28. doi: 10.1097/MOT.0000000000000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee CF, Lo YC, Cheng CH, Furtmuller GJ, Oh B, Andrade-Oliveira V, Thomas AG, Bowman CE, Slusher BS, Wolfgang MJ, Brandacher G, Powell JD. Preventing Allograft Rejection by Targeting Immune Metabolism. Cell Rep. 2015;13:760–770. doi: 10.1016/j.celrep.2015.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shapiro RA, Clark VM, Curthoys NP. Inactivation of rat renal phosphate-dependent glutaminase with 6-diazo-5-oxo-L-norleucine. Evidence for interaction at the glutamine binding site. J Biol Chem. 1979;254:2835–2838. [PubMed] [Google Scholar]
- 57.Ortlund E, Lacount MW, Lewinski K, Lebioda L. Reactions of Pseudomonas 7A Glutaminase-Asparaginase with Diazo Analogues of Glutamine and Asparagine Result in Unexpected Covalent Inhibitions and Suggests an Unusual Catalytic Triad Thr-Tyr-Glu. Biochemistry (N Y) 2000;39:1199–1204. doi: 10.1021/bi991797d. [DOI] [PubMed] [Google Scholar]
- 58.Thangavelu K, Chong QY, Low BC, Sivaraman J. Structural Basis for the Active Site Inhibition Mechanism of Human Kidney-Type Glutaminase (KGA) Scientific Reports. 2014;4:3827. doi: 10.1038/srep03827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cooney DA, Jayaram HN, Milman HA, Homan ER, Pittillo R, Geran RI, Ryan J, Rosenbluth RJ. DON, CONV and DONV-III. Pharmacologic and toxicologic studies. Biochem Pharmacol. 1976;25:1859–1870. doi: 10.1016/0006-2952(76)90190-8. [DOI] [PubMed] [Google Scholar]
- 60.Sklaroff RB, Casper ES, Magill GB, Young CW. Phase I study of 6-diazo-5-oxo-L-norleucine (DON) Cancer Treat Rep. 1980;64:1247–1251. [PubMed] [Google Scholar]
- 61.Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van d B, Verdegaal EME, Cascante M, Shlomi T, Gottlieb E, Peeper DS. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109–112. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- 62.Stacpoole PW. The pharmacology of dichloroacetate. Metab Clin Exp. 1989;38:1124–1144. doi: 10.1016/0026-0495(89)90051-6. [DOI] [PubMed] [Google Scholar]
- 63.Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, Rathmell JC. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, Liu G. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J Immunol. 2015;194:6082–6089. doi: 10.4049/jimmunol.1402469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, Morel L. Glucose Oxidation Is Critical for CD4+ T Cell Activation in a Mouse Model of Systemic Lupus Erythematosus. J Immunol. 2016;196:80–90. doi: 10.4049/jimmunol.1501537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wahl DR, Petersen B, Warner R, Richardson BC, Glick GD, Opipari AW. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus. 2010;19:1492–1501. doi: 10.1177/0961203310373109. [DOI] [PubMed] [Google Scholar]
- 67.Caza TN, Talaber G, Perl A. Metabolic regulation of organelle homeostasis in lupus T cells. Clin Immunol. 2012;144:200–213. doi: 10.1016/j.clim.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caza TN, Fernandez DR, Talaber G, Oaks Z, Haas M, Madaio MP, Lai ZW, Miklossy G, Singh RR, Chudakov DM, Malorni W, Middleton F, Banki K, Perl A. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann Rheum Dis. 2014;73:1888–1897. doi: 10.1136/annrheumdis-2013-203794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7:274ra18. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]