

Abstract
Novel combination therapies are desperately needed for combating lung infections caused by bacterial ‘superbugs’. This study aimed to investigate synergistic antibacterial activity of polymyxin B in combination with the cystic fibrosis (CF) drugs KALYDECO™ (ivacaftor) and ORKAMBI™ (ivacaftor+lumacaftor) against Gram-negative pathogens that commonly colonize the CF lung, in particular the problematic Pseudomonas aeruginosa. The in vitro synergistic activity of polymyxin B combined with ivacaftor or lumacaftor was assessed using checkerboard and static time-kill assays against a panel of polymyxin-susceptible and polymyxin-resistant P. aeruginosa isolates from the lungs of CF patients. Polymyxin B, ivacaftor or lumacaftor were ineffective when used individually against polymyxin-resistant (MIC, ≥ 4 mg/L) isolates. However, when used together the combination of clinically-relevant concentrations of polymyxin B (2 mg/L) combined with ivacaftor (8 mg/L) or ivacaftor (8 mg/L)+lumacaftor (8 mg/L) and displayed synergistic killing activity against polymyxin-resistant P. aeruginosa isolates as demonstrated by 100-fold decrease in the bacterial count (CFU/mL) after 24 h. The combinations also displayed excellent antibacterial activity against P. aeruginosa under CF relevant conditions in a sputum medium assay. The combination of lumacaftor (alone) with polymyxin B showed additivity against P. aeruginosa.
The potential antimicrobial mode of action of the combinations against P. aeruginosa was investigated using different methods. Treatment with the combinations induced cytosolic GFP release from P. aeruginosa cells and showed permeabilizing activity in the nitrocefin assay, indicating damage to both the outer and inner Gram-negative cell membranes. Moreover, scanning and transmission electron micrographs revealed that the combinations produce outer membrane damage to P. aeruginosa cells that is distinct from the effect of each compound per se. Ivacaftor was also shown to be a weak inhibitor of the bacterial DNA gyrase and topoisomerase IV with no effect on either human type I or type IIα topoisomerases. Lumacaftor displayed the ability to increase the cellular production of damaging reactive oxygen species.
In summary, the combination of polymyxin B with KALYDECO™ or ORKAMBI™ exhibited synergistic activity against highly polymyxin-resistant P. aeruginosa CF isolates and can be potentially useful for otherwise untreatable CF lung infections.
Keywords: Ivacaftor, lumacaftor, polymyxin, Pseudomonas aeruginosa, cystic fibrosis
Introduction
Antibiotic resistance has evolved into a substantial global health concern.1 Unfortunately, the ‘magic bullet’ antimicrobials we have gratuitously used over the past decades are rapidly losing their caliber.1 Modern medicine is clearly entering a critical period, if bacteria continue developing resistance to multiple antibiotics at the present rate and at the same time the antibiotic pipeline continues to dry up; there could be catastrophic costs to healthcare and society globally.2 Patients with chronic infections such as CF rely on antibiotics on a daily basis and are particularly at risk. One in 22 people of European descent carry a mutant allele for CF making it the most common recessive genetic disease.3 CF is caused by mutations in a gene that encodes the CF trans-membrane conductance regulator (CFTR) protein.3 The defective CFTR gating function leads to an abnormal transmembrane chloride transport, followed by sodium hyper-absorption with water influx into the cell leading to fluid depletion on the epithelial surface of affected organs and a deficiency in bicarbonate secretion. As a consequence, thick mucus accumulation occurs and promotes bacterial lung infections, most commonly due to P. aeruginosa (∼80% of patients).4 Other bacterial pathogens that less commonly colonize and infect the lungs of CF patients include Haemophilus influenzae, Burkholderia cepacia, Burkholderia pseudomallei, Stenotrophomonas maltophilia, Klebsiella pneumoniae and Acinetobacter baumannii.4 P. aeruginosa is often the most problematic to treat, as once chronic infection is established the infecting strain becomes increasingly resistant to antibiotics over the lifetime of CF patients, and 80 to 95% of them die of cardiopulmonary failure due to the infection.4
Ivacaftor (syn. VX-770; KALYDECO™) is the first FDA-approved CFTR modulator, with demonstrated clinical efficacy producing a significant improvement in the lung function of CF patients over placebo in CF patients with certain types of CFTR mutations (which account for 4–5% of cystic fibrosis cases).5 Recently, the FDA approved ivacaftor in combination with lumacaftor (ORKAMBI™). Lumacaftor promotes the transport of the defective CFTR protein to the cell surface, where ivacaftor acts to improve its gating function.6 Specifically, lumacaftor expands the therapeutic window of ivacaftor to include around 28% of CF patients.
Polymyxin B and colistin (polymyxin E) are lipopeptide antibiotics used to treat infections caused by multidrug-resistant (MDR) Gram-negative pathogens.7 Colistin has been commercially available in the clinic as its inactive prodrug colistimethate sodium (CMS) for decades.8 CMS nebulizer solution (Coly-Mycin®) and dry powder pod-inhaler (Colobreathe®) has been approved for the treatment of respiratory infections caused by P. aeruginosa in CF patients;9 however reports of polymyxin-resistant P. aeruginosa isolates from CF patient lung samples are becoming more common.10 Therefore, novel strategies to prolong the efficacy of these important lipopeptide antibiotics are needed. The rise of Gram-negative ‘superbugs’ which are resistant to all available antibiotics implies that even the robust activity of the polymyxins is being lost. The potential synergistic use of antibiotic-nonantibiotic combinations is emerging as a valuable and cost-effective means for improving the clinical efficacy of currently available antibiotics against problematic MDR bacterial pathogens. The focus of the present study was to evaluate the antibacterial synergy of CFTR potentiator drugs KALYDECO and ORKAMBI with polymyxin B against P. aeruginosa that commonly infects the lungs of CF patients.
Results and Discussion
Antimicrobial synergy of polymyxin-CFTR potentiator combinations against Gram-negative CF lung pathogens
MICs for polymyxin B, ivacaftor, lumacaftor and VX-661 (a developmental CFTR potentiator Vertex Pharmaceuticals compound) alone, and in combination with polymyxin B, are presented in Table 1. Ivacaftor per se were inactive (MIC, >32 mg/L) against all of the Gram-negative isolates tested, except for P. aeruginosa FADDI-PA064 non-mucoid (ivacaftor MIC, 4 mg/L). Lumacaftor and VX-661 per se were inactive (MIC, >32 mg/L) against all of the isolates including FADDI-PA064 non-mucoid. The broth microdilution checkerboard method results showed the polymyxin B-ivacaftor combination displayed synergism (eight isolates) and additivity (eight isolates) against the sixteen polymyxin-resistant P. aeruginosa CF isolates tested (Tables 1 and S2). Across the six polymyxin-susceptible P. aeruginosa isolates synergy was only observed for one isolate and additivity for the remaining five. The polymyxin B-ivacaftor combination was also active against most of the A. baumannii, K. pneumoniae and S. maltophilia isolates, displaying either synergy or additivity in most cases. The polymyxin B-lumacaftor and VX-661 combination largely displayed additivity and indifference across the panel of strains, synergy was only observed with one A. baumannii (ATCC19606), one K. pneumoniae (FADDI-KP081) and one S. maltophilia (FADDI-SM006) isolates. Notably, the polymyxin B-ivacaftor combination displayed a greater degree of synergy against the polymyxin-resistant laboratory strain A. baumannii 19606R compared to its polymyxin-susceptible paired wild-type strain ATCC 19606, where additivity was observed. We have previously reported a unique polymyxin resistance mechanism employed by A. baumannii 19606R which involves the complete loss of lipopolysaccharide (LPS) from the outer membrane.11 As ivacaftor is a hydrophobic compound (logP=5.76), it is likely that a complete loss of LPS from the outer membrane facilitates the ability of ivacaftor to cross the outer membrane barrier of A. baumannii 19606R. This is coincident with our previous report that polymyxin-resistant A. baumannii strains are generally more susceptible to a number of antibiotics than their polymyxin-susceptible parent strains.12 The all the polymyxin B combinations were inactive against all of the H. influenzae, B. cepacia and B. pseudomallei isolates tested. The latter is not surprising as B. cepacia and B. pseudomallei species are intrinsically highly resistant to polymyxins since their LPS has the 4-amino-4-deoxy-L-arabinose modification on the phosphate groups in the lipid A and in the inner core regions.13
Table 1.
The antimicrobial activity of polymyxin B (PMB), ivacaftor (IVA), lumacaftor (LUMA), VX-661 alone and in combination.
| Bacterial isolate | PMB MIC (mg/L) |
IVA MIC (mg/L) |
PMB + IVA FIC indexb |
EFFECT | LUMA MIC (mg/L) |
PMB + LUMA FIC indexb |
EFFECT | VX-661 MIC (mg/L) |
PMB + VX-661 FIC Indexb |
EFFECT | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Poymyxin resistant isolates | |||||||||||
| P. aeruginosa | FADDI-PA064 | >128 | 4 | 0.12 | synergy | >32 | 2 | Indifference | >32 | 2 | indifference |
| FADDI-PA069 | 128 | >32 | 0.18 | synergy | a NA | a NA | a NA | a NA | |||
| FADDI-PA065 | 128 | >32 | 0.37 | synergy | >32 | 2 | indifference | >32 | 0.75 | additivity | |
| FADDI-PA068 | >128 | >32 | 0.56 | additivity | >32 | 2 | indifference | >32 | 1 | indifference | |
| FADDI-CW45 | 128 | >32 | 0.75 | additivity | a NA | a NA | a NA | a NA | |||
| FADDI-CW32 | 128 | >32 | 0.75 | additivity | a NA | a NA | a NA | a NA | |||
| FADDI-CW22 | 128 | >32 | 0.75 | additivity | a NA | a NA | a NA | a NA | |||
| FADDI-PA072 | 64 | >32 | 0.12 | synergy | a NA | a NA | a NA | a NA | |||
| FADDI-PA070 | 64 | >32 | 0.26 | synergy | >32 | 2 | indifference | >32 | 1 | indifference | |
| FADDI-PA066 | 64 | >32 | 0.50 | additivity | >32 | 2 | indifference | >32 | 2 | indifference | |
| FADDI-CW05 | 64 | >32 | 0.75 | additivity | a NA | a NA | a NA | a NA | |||
| FADDI-CW28 | 32 | >32 | 0.13 | synergy | a NA | a NA | a NA | a NA | |||
| FADDI-PA067 | 16 | >32 | 0.26 | synergy | a NA | a NA | a NA | a NA | |||
| FADDI-PA006 | 8 | >32 | 0.75 | additivity | a NA | a NA | a NA | a NA | |||
| FADDI-PA090 | 4 | >32 | 0.50 | additivity | >32 | 0.53 | additivity | >32 | 0.75 | additivity | |
| FADDI-PA014 | 4 | >32 | 0.28 | synergy | a NA | a NA | a NA | a NA | |||
| Polymyxin susceptible isolates | |||||||||||
| ATCC 27853 | 1 | >32 | 0.50 | additivity | >32 | 2 | indifference | >32 | 0.75 | additivity | |
| FADDI-CW42 | 0.5 | >32 | 0.28 | synergy | >32 | 1.25 | indifference | >32 | 1 | indifference | |
| FADDI-PA021 | 0.5 | >32 | 0.56 | additivity | >32 | 2 | indifference | >32 | 0.75 | additivity | |
| FADDI-CW13 | 0.5 | >32 | 0.53 | additivity | >32 | 2 | indifference | >32 | 2 | indifference | |
| FADDI-CW35 | 0.25 | >32 | 0.51 | additivity | >32 | 0.51 | additivity | >32 | 2 | indifference | |
| FADDI-CW34 | 0.25 | >32 | 0.53 | additivity | >32 | 2 | indifference | >32 | 2 | indifference | |
| Polymyxin resistant isolates | |||||||||||
| B. pseudomallei | BP-MSHR 1814 | >256 | >32 | 2 | indifference | >32 | 2 | indifference | >32 | 2 | indifference |
| BP-MSHR 912 | >256 | >32 | 2 | indifference | >32 | 2 | indifference | >32 | 2 | indifference | |
| BP-MSHR 3036 | >256 | >32 | 2 | indifference | >32 | 2 | indifference | >32 | 2 | indifference | |
| Polymyxin resistant isolate | |||||||||||
| B. cepacia | FADDI-BC001 | >128 | >32 | 1 | indifference | >32 | 2 | indifference | >32 | 2 | indifference |
| Polymyxin resistant isolates | |||||||||||
| A. baumannii | ATCC 19606 R | >8 | >32 | 0.28 | synergy | >32 | 0.62 | additivity | >32 | 1 | indifference |
| FADDI-AB181 | >8 | >32 | 0.62 | additivity | >32 | 2 | indifference | >32 | 2 | indifference | |
| FADDI-AB179 | 8 | >32 | 0.25 | synergy | >32 | 2 | indifference | >32 | 1 | indifference | |
| FADDI-AB177 | >4 | >32 | 1 | indifference | >32 | 2 | indifference | >32 | 2 | indifference | |
| FADDI-AB156 | 4 | >32 | 0.25 | synergy | >32 | 2 | indifference | >32 | 1 | indifference | |
| Polymyxin susceptible isolates | |||||||||||
| ATCC 19606 | 2 | >32 | 0.50 | additivity | >32 | 0.25 | synergy | >32 | 0.62 | additivity | |
| FADDI-AB180 | 1 | >32 | 0.52 | additivity | >32 | 0.50 | additivity | >32 | 1 | indifference | |
| FADDI-AB178 | 0.5 | >32 | 0.50 | additivity | >32 | 0.51 | additivity | >32 | 0.53 | additivity | |
| FADDI-AB176 | 0.5 | >32 | 0.53 | additivity | >32 | 0.53 | additivity | >32 | 1 | indifference | |
| FADDI-AB030 | 0.125 | >32 | 0.50 | additivity | >32 | 0.53 | additivity | >32 | 0.62 | additivity | |
| Polymyxin resistant isolates | |||||||||||
| K. pneumoniae | FADDI-KP074 | >128 | >32 | 0.62 | additivity | >32 | 2 | indifference | >32 | 2 | indifference |
| FADDI-KP082 | 128 | >32 | 0.18 | synergy | >32 | 0.5 | additivity | >32 | 0.75 | additivity | |
| FADDI-KP063 | >8 | >32 | 0.25 | synergy | >32 | 2 | indifference | >32 | 2 | indifference | |
| FADDI-KP027 | >8 | >32 | 1 | indifference | >32 | 2 | indifference | >32 | 2 | indifference | |
| FADDI-KP055 | >8 | >32 | 1 | indifference | >32 | 2 | indifference | >32 | 2 | indifference | |
| FADDI-KP064 | >8 | >32 | 1.3 | indifference | >32 | 2 | indifference | >32 | 2 | indifference | |
| Polymyxin susceptible isolates | |||||||||||
| FADDI-KP073 | 2 | >32 | 0.28 | synergy | >32 | 0.51 | additivity | >32 | 0.62 | additivity | |
| FADDI-KP086 | 2 | >32 | 0.50 | synergy | >32 | 0.51 | additivity | >32 | 0.50 | additivity | |
| FADDI-KP033 | 1 | >32 | 0.25 | synergy | >32 | 2 | indifference | >32 | 0.75 | additivity | |
| FADDI-KP033 | 1 | >32 | 0.25 | synergy | >32 | 2 | indifference | >32 | 0.75 | additivity | |
| FADDI-KP015 | 0.5 | >32 | 0.31 | synergy | >32 | 2 | indifference | >32 | 0.50 | additivity | |
| ATCC 13883 | 0.25 | >32 | 0.52 | additivity | >32 | 0.50 | additivity | >32 | 0.75 | additivity | |
| FADDI-KP081 | 0.25 | >32 | 0.56 | additivity | >32 | 0.25 | synergy | >32 | 0.62 | additivity | |
| FADDI-KP077 | 0.25 | >32 | 0.62 | additivity | >32 | 0.50 | additivity | >32 | 0.75 | additivity | |
| Polymyxin resistant isolates | |||||||||||
| S. maltophilia | FADDI-SM001 | >8 | >32 | 0.50 | additivity | >32 | 1 | indifference | >32 | 0.52 | additivity |
| FADDI-SM008 | >8 | >32 | 0.50 | additivity | >32 | 0.5 | additivity | >32 | 2 | indifference | |
| FADDI-SM006 | 8 | >32 | 0.18 | synergy | >32 | 0.5 | additivity | >32 | 0.38 | synergy | |
| FADDI-SM007 | 8 | >32 | 0.50 | additivity | >32 | 0.75 | additivity | >32 | 1 | indifference | |
| FADDI-SM002 | 4 | >32 | 0.62 | additivity | >32 | 0.62 | additivity | >32 | 1 | indifference | |
| Polymyxin susceptible isolates | |||||||||||
| FADDI-SM004 | 2 | >32 | 0.25 | synergy | >32 | 1 | indifference | >32 | 0.75 | additivity | |
| FADDI-SM003 | 1 | >32 | 0.37 | synergy | >32 | 0.62 | additivity | >32 | 1 | indifference | |
| FADDI-SM005 | 0.25 | >32 | 0.37 | synergy | >32 | 0.75 | additivity | >32 | 1 | indifference | |
| Polymyxin resistant isolates | |||||||||||
| H. influenzae | FADDI-HI001 | >16 | >128 | 2 | indifference | >128 | 1 | indifference | >128 | 0.75 | additivity |
| FADDI-HI003 | >16 | >128 | 2 | indifference | >128 | 1 | indifference | >128 | 0.75 | additivity | |
NA, not accessed
FICINDEX = (MIC polymyxin B in combination with ivacaftor/MIC polymyxin B alone) + (MIC ivacaftor in combination with polymyxin B/MIC ivacaftor alone); Synergy FIC<0.5; Additivity FIC=0.5–1.0; Indifference FIC=1–4; Antagonism FIC≥4.
The synergistic killing activity of the polymyxin B-ivacaftor combination in static time-kill studies was assessed against four P. aeruginosa isolates, the polymyxin-susceptible control strain P. aeruginosa ATCC 27853 (polymyxin B MIC, 1 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32 mg/L) and the highly polymyxin-resistant MDR strains FADDI-PA064 (polymyxin B MIC, >128 mg/L; ivacaftor MIC, 4 mg/L; lumacaftor MIC, >32 mg/L), FADDI-PA065 (polymyxin B MIC, >128 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32 mg/L) and FADDI-PA070 (polymyxin B MIC, 64 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32 mg/L) (Figure 1). Clinically relevant concentrations of the drugs were used in the time-kill experiments.7, 14 Polymyxin B, ivacaftor and lumacaftor per se did not exhibit antibacterial activity against P. aeruginosa ATCC 27853, FADDI-PA065 and FADDI-PA070, the bacterial killing curves were essentially indistinguishable from those of the controls. Ivacaftor alone displayed moderate killing kinetics against FADDI-PA064, which is consistent with the superior activity of ivacaftor against this particular polymyxin-resistant strain (FADDI-PA064 ivacaftor MIC, 4 mg/L vs >32 mg/L for all the other P. aeruginosa isolates). Against the polymyxin-susceptible strain ATCC 27853, polymyxin B per se displayed antibacterial activity up to 1 h (before regrowth occurred) at 2 mg/L. The polymyxin B-ivacaftor combination (polymyxin B 2 mg/L-ivacaftor 8 mg/L) demonstrated synergism in the static time-kill studies against all three polymyxin-resistant CF strains with bacterial killing reaching below the limit of detection at 2–6 h and a ≥100-fold reduction in CFU/mL at 24 h compared to polymyxin B alone. Regrowth with polymyxin B-ivacaftor over 24 h is seen in ATCC 27853 and FADDI-PA070, whereas, FADDI-PA064 and FADDI-PA065 show complete killing at 24 h (Figure 1). Notably, the general trend we observe with the FICI data (Table 1) is that the polymyxin B-ivacaftor combination is less effective against the polymyxin-susceptible P. aeruginosa CF isolates. It is therefore tenable to speculate that the regrowth seen with FADDI-PA070 (polymyxin B MIC=64 mg/L) is potentially due to its 2-fold lower MIC (i.e greater polymxyin B susceptibility) compared to FADDI-PA064 (polymyxin B MIC=>128 mg/L) and FADDI-PA065 (polymyxin B MIC=128 mg/L), if we recall that the latter two isolates showed complete killing at 24 h. In the case of the polymyxin B+(ivacaftor/lumacaftor [ORKAMBI]) combination, regrowth over 24 h is seen in ATCC 27853 (the polymyxin-susceptible control isolate), FADDI-PA064 and FADDI-PA070; FADDI-065 is the only isolate that shows complete killing without detectable regrowth at 24 h (Figure 1). Notably, in the case of ATCC 27853 and FADDI-PA064 strains, it appears that lumacaftor is driving the regrowth. We would only go as far to speculate that these complex killing effects may be associated with the different levels of aminoarabinose modified lipid A (a primary mechanism whereby membrane remodeling occurs to modulate the level of polymyxin B resistance in P. aeruginosa CF isolates)15 in the outer membrane of each isolate. To this end, further studies are currently underway to characterize the lipid A dynamics in relation to these complex kinetic killing effects. Overall, the data from the time-kill studies corroborate the findings from the checkerboard experiments, suggesting that polymyxin B in combination with ivacaftor and lumacaftor displays synergistic killing against highly polymyxin-resistant P. aeruginosa CF isolates.
Figure 1.

Time-kill curves for polymyxin B, ivacaftor, lumacaftor alone and in combinations against three polymyxin-resistant P. aeruginosa CF strains FADDI-PA064 (polymyxin B MIC, >128 mg/L; ivacaftor MIC, 4 mg/L; lumacaftor MIC, >32 mg/L), FADDI-PA065 (polymyxin B MIC, 128 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32 mg/L), FADDI-PA070 (polymyxin B MIC, 64 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32 mg/L) and against a polymyxin-susceptible P. aeruginosa ATCC 27853 strain (polymyxin B MIC, 1 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32 mg/L). Data points are plotted as the mean ± SD of three independent measurements.
We also tested the activity of the combinations against the highly polymyxin-resistant CF isolate P. aeruginosa FADDI-065 under planktonic and biofilm growth conditions in a sputum medium assay, the latter of which is relevant to CF lung conditions (Figure 2). The polymyxin B ivacaftor/lumacaftor combinations performed exceptionally well, reducing cell viability to <2% under the planktonic and <20% under the biofilm growth conditions in the sputum medium, respectively. In comparison, polymyxin B alone was only partially active under the planktonic growth condition and completely inactive under the biofilm growth condition. Overall, these findings would suggest that combination therapy of polymyxins with either KALYDECO™ (ivacaftor) or ORKAMBI™ (ivacaftor+lumacaftor) in CF patients may potentially be effective at clearing problematic polymyxin-resistant MDR P. aeruginosa lung infections. Notably, a recent clinical study showed that P. aeruginosa culture positivity in CF patients was reduced following ivacaftor treatment; whether this was a direct result of an improvement in lung function due to the CFTR regulator activity of ivacaftor or the result of the antibacterial synergy between polymyxin and ivacaftor (many of these patients were likely co-medicated with nebulized or dry powder inhaled colistin) remains unknown.16
Figure 2.
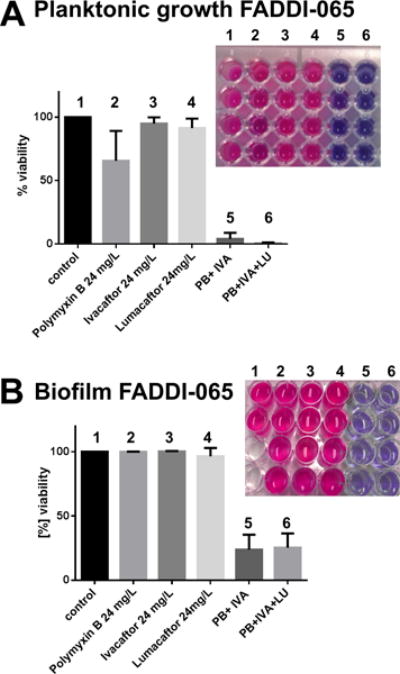
Antimicrobial activity of polymyxin B (24 mg/L=0.18×MIC), ivacaftor (24 mg/L=0.75×MIC), lumacaftor (24 mg/L=0.75×MIC) alone and in combinations against the polymyxin-resistant P. aeruginosa CF strain FADDI-PA065 (polymyxin B MIC=128 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32mg/L) in artificial sputum media under (A) planktonic and (B) biofilm growth conditions. Data points are plotted as the mean ± SD of four independent measurements. The top right-hand corner inset graphics in each bar graph show the raw data from the resazurin fluorescence plate assay; the numbering 1–6 corresponds to the numbering for each condition in the adjacent bar graph. The replicates for each condition are organized vertically in the 96-well plate. The redox sensitive dye resazurin (blue, indicates dead cells) undergoes reduction to resorufin (pink) by NADH from metabolically active (live) cells.
Antibacterial mode of action studies
The Gram-negative outer membrane is a formidable barrier, especially against hydrophobic drugs such as ivacaftor and lumacaftor. Polymyxins exert their antimicrobial action via direct interaction with the lipid A component of the LPS, which leads to disruption of this critical barrier function of the Gram-negative outer membrane7. Polymyxins zone into their primary cellular target, LPS through an initial electrostatic interaction between their cationic L-α,γ-diaminobutyric acid (Dab) side chains (cf. Figure 6B) with the negatively charged phosphate groups of the lipid A component of LPS, displacing divalent cations (Ca2+ and Mg2+) that bridge adjacent LPS molecules in the process and resulting in the disruption of the outer membrane.7 Therefore, the underlying molecular basis for the synergistic activity of the combination may involve the ability of polymyxin B to permeabilise the outer membrane, thereby allowing ivacaftor or lumacaftor to enter the cell and exert its antibacterial action. This hypothesis was tested and supported in the following sections.
Scanning and transmission electron microscopy (SEM and TEM) imaging of P. aeruginosa FADDI-PA065 cells revealed that the bacterial outer membrane structure was drastically disrupted due to the treatment with the combination of polymyxin B with ivacaftor or lumacaftor (Figure 3). This is distinctly different from the cells treated with polymyxin B (8 mg/L) alone where the wide-spread blebbing effect that is commonly associated with polymyxin exposure of Gram-negative cells was observed,17 and the cells treated with ivacaftor (8 mg/L) alone where large outer membrane protrusions were observed. Treatment with lumacaftor alone resulted in no visible outer membrane damage.
Figure 3.
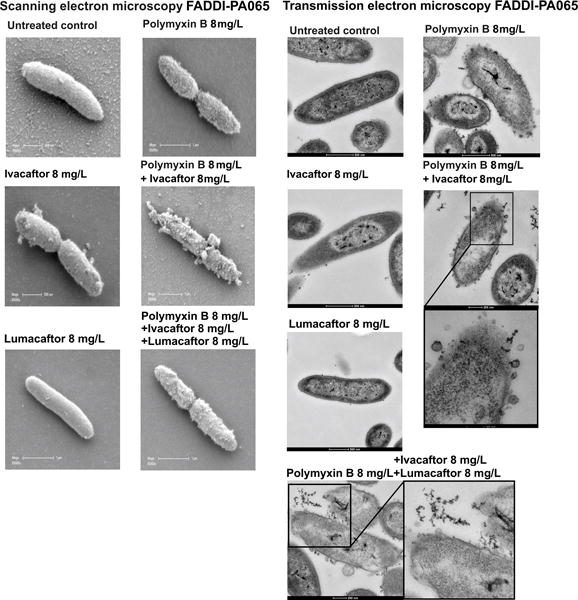
Electron microscopy images of the polymyxin-resistant P. aeruginosa CF isolate FADDI-PA065 (polymyxin B MIC, 128 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC > 32 mg/L) treated with polymyxin B, ivacaftor, lumacaftor alone and in combinations.
Further evidence that the combinations causes damage to the outer membrane comes from the nitrocefin assay data with the β-lactamase positive strain P. aeruginosa FADDI-PA065 (Figure 4A). The assay measures the periplasmic β-lactamase activity using nitrocefin, a chromogenic cephalosporin substrate analogue, which normally penetrates the outer membrane slowly.18 In the presence of outer membrane permeabilizing agents, nitrocefin can more easily permeate the outer membrane barrier. Figure 4 shows that the combinations of polymyxin B with ivacaftor and with ivacaftor-lumacaftor were more effective at permeabilizing the outer membrane than treatment with either drug per se. At this point we would like to emphasize that the nitrocefin assay does not correlate with the killing effect of each drug, and therefore, it is not tenable to draw comparisons with the microbiological antibacterial data. In support of this claim, the time killing kinetics data presented in Figure 1 indicate that the FADDI-065 cells are not killed over 24 h following treatment with each drug alone; at notably 2–4 fold higher concentrations than those used in the nitrocefin assay. Moreover, in light of the SEM/TEM data that clearly shows that ivacaftor causes direct damage to the outer membrane of FADDI-065, it is not surprising that we see a marked response to ivacaftor alone in the nitrocefin assay.
Figure 4.

(A) Outer membrane permeabilizing activity of polymyxin B, ivacaftor, lumacaftor alone and in combinations against P. aeruginosa FADDI-PA065 measured by the nitrocefin assay. Data points are plotted as the mean ± SD of four independent measurements. (B) ROS production in P. aeruginosa FADDI-PA065 cells following treatment measured by the increased fluorescence emission of the oxidative stress sensitive dye CellROX Green. Treatments: 1, untreated cells; 2, lumacaftor 4 mg/L; 3, polymyxin B 2 mg/L; 4, Combination of polymyxin B 2 mg/L+ lumacaftor 4 mg/L.
ROS production is purported to be a common bacterial killing mechanism for antibiotics.19 Figure 4B shows that the combination of polymyxin B with lumacaftor induced a greater increase of ROS production in P. aeruginosa FADDI-PA065 cells compared to treatment with each drug per se, seen as an increase in the fluorescence emission of the oxidative stress sensitive cell dye CellROX Green. Unfortunately, the impact of ivacaftor on ROS production could not be assayed as this compound produces a quencher effect on the detector dye CellROX Green.
Quantitative analysis of confocal images of the P. aeruginosa strain AH298-GFP (expressing green fluorescence protein, GFP) revealed that the combination of polymyxin B with ivacaftor or lumacaftor was more effective at producing outer membrane damage as indicated by the increased cytosolic GFP release compared to treatment with each drug per se, (Figure 5).
Figure 5.

(A) Confocal microscopy images of cytosolic GFP release from P. aeruginosa AH298-GFP treated with polymyxin B, ivacaftor, lumacaftor alone and in combinations. The corresponding densitometric analysis and scanning electron microscopy images of the confocal imaged cells are shown. (B) Bar graph showing the % GFP released following drug treatment relative to the untreated control.
Ivacaftor has been reported to display poor direct antimicrobial activity against coagulase-negative Staphylococcus and methicillin-susceptible S. aureus (MSSA) with an MIC of ∼8 mg/L.20 In line with our results, these authors reported no direct activity for ivacaftor against Gram-negative bacteria such as P. aeruginosa.20 A recent report also noted a decrease in the relative abundance of Streptococcus 1 (S. mitis group) in the airway micro biota of patients receiving ivacaftor therapy.21 Due to the similarity of ivacaftor’s chemical structure to the quinolones (Figure S1B), we tested whether the synergistic activity of ivacaftor was the result of a quinolone-like antimicrobial mode of action (Figure S1A). The antibacterial mode of action of the quinolone antibiotics involves the inhibition of the related DNA gyrase and topoisomerase IV enzymes which are involved in DNA replication.22 The gyrase and supercoiling topoisomerase assay data address two key mechanisms: interfacial poisoning (IFP), i.e., drugs that induce DNA cleavages and catalytic inhibitory compounds (CIC) which simply block catalytic activity of the enzyme. Figure 1A shows that ivacaftor is a relatively weak CIC, but clearly not an IFP. More to it, topoisomerase IV displayed greater sensitivity to ivacaftor, relative to DNA gyrase. The yield of kDNA products (decatenated kDNA) was noticeably reduced by 25 μM ivacaftor (comparing the yields of decatenated kDNA minicircles). In addition, we screened human topoisomerase I and IIα and found ivacaftor had essentially no inhibitory effect on either IFP or CIC (Figure S2). All controls (targeting IFP) worked well, demonstrating the validity of the negative results with ivacaftor. The lack of enzyme inhibition by ivacaftor can be construed in terms of our understanding of quinolone structure-activity relationships (SAR).22 Firstly, ivacaftor has a bulky N-(2,4-di-tert-butyl-5-)hydroxyphenyl substituting R3 of the quinolone nucleus in the place of the carboxylic acid (Figure S1B). Quinolone SAR studies have shown the carboxylic acid moiety is critical for binding to the perturbed DNA, and substitutions of this group produce inactive quinolones22. Secondly, the ivacaftor structure contains an essentially bare quinolone nucleus, which lacks the fundamental substituents at the R1, R6 and R7 positions that are critical for binding to the enzyme-DNA complex.22 Taken together, our findings would suggest that that antibacterial activity of ivacaftor does not involve a quinolone-like mode of action; indeed, quinolones are strong IFP agents while ivacaftor is not (it is a weak CIC). In support of this claim, it is noteworthy to mention that the polymyxin B-ivacaftor combination was active (showing synergy or additivity) against the CW-XX P. aeruginosa strains that are all resistant to the fluoroquinolone antibiotic ciprofloxacin (MICs, 8–16 mg/L) (Table 1). This observation also supports the notion that that the antibacterial activity of ivacaftor involves a novel mode of action, for example, targeting either DNA binding, DNA access or essential ATPase domains. Notably, the action is targeted to the prokaryotic topoisomerases while human enzyme’s appear largely unaffected (Figure S2). Selective targeting of prokaryotic activities, vs. eukaryotic enzymes, further validates at the efficacy of a combinatorial therapeutic inroad.
Conclusions
It is acknowledged that MDR Gram-negative lung infections in CF patients represent a major unmet medical need for effective therapies with minimum adverse effects. The off-label use of nonantibiotic drugs for antibacterial purposes can significantly expedite the discovery of new treatment options for MDR lung infections in this population. The findings from the present study suggest that the ‘unexpected’ combination of polymyxin B with the non-antibiotic CF drugs ivacaftor (KALYDECO™) or ivacaftor-lumacaftor (ORKAMBI™) could potentially prove useful for the treatment of CF lung infections caused by MDR Gram-negative pathogens that are even resistant to the last-line polymxyins. This is especially serendipitous given that both drugs are commonly used in combination in CF patients, i.e., ‘kill two birds with one stone’. Moreover, the present findings suggest ivacaftor and lumacaftor operate via a mode of action (MOA) that is different from that of the polymxyins; which makes the combination even more effective against MDR pathogens, as this scenario presents the pathogen with the difficult task of concomitantly developing resistance against two independent MOAs. Further studies are underway to access the clinical effectiveness of the combination against MDR polymyxin-resistant P. aeruginosa CF lung infections.
Experimental Section
Materials
Polymyxin B (Catalogue number 81334, ≥6,500 IU/mg) was purchased from Sigma-Aldrich (Australia). Ivacaftor, lumacaftor and VX-661 were purchased from (SelleckChem, USA). All other reagents were purchased from Sigma-Aldrich (Australia) and are of the highest commercial grade available.
Bacterial isolates
A total of twenty two clinical isolates (sixteen polymyxin-resistant and five polymyxin-susceptible) of P. aeruginosa, including mucoid and non-mucoid strains, were selected from routine clinical isolates identified from patients with acute exacerbations of CF. Isolates were selected sequentially from different patients as they presented with acute exacerbations; both mucoid and non-mucoid strains came from the same patient. MIC susceptibility screening of fourteen of these CF isolates revealed that five strains were MDR being resistant to at least three of the following agents: aztreonam, ceftazidime, meropenem, piperacillin, ticarcillin, gentamicin, tobramycin, and ciprofloxacin (Table S1).23 A polymyxin-heteroresistant reference strain, P. aeruginosa ATCC 27853 (polymyxin B MIC, 1 mg/L) (American Type Culture Collection, Rockville, USA) was also studied. Resistance to polymyxin B was defined as MICs of ≥4 mg/L. Four polymyxin-resistant (polymyxin B MICs, >128 mg/L) Burkholderia (B. cepacia and B. pseudomallei) strains from CF patients were employed. A total of ten A. baumannii strains were utilised in this study, including polymyxin-susceptible reference strain ATCC 19606 (polymyxin B MIC, 2 mg/L) and its paired polymyxin-resistant isolate ATCC 19606R (polymyxin B MIC, 64 mg/L). Six polymyxin-resistant K. pneumoniae isolates, a polymyxin-susceptible reference strain ATCC 13883 (polymyxin B MIC, 1 mg/L), and eight polymyxin-susceptible strains were employed. Eight clinical S. maltophilia isolates, including three polymyxin-susceptible strains and five polymyxin-resistant strains, were included in this study. Two polymyxin-resistant H. influenza isolates were employed.
In vitro antibacterial synergy
MICs were determined in accordance with the recommendations of the Clinical and Laboratory Standards Institute.24 The time-killing kinetics of polymyxin B per se and in combination with ivacaftor were examined against P. aeruginosa ATCC 27853 (polymyxin B MIC, 1 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32 mg/L), FADDI-PA064 (polymyxin B MIC, >128 mg/L; ivacaftor MIC, 4 mg/L; lumacaftor MIC, >32 mg/L), FADDI-PA065 (polymyxin B MIC, >128 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32 mg/L) and FADDI-PA070 (polymyxin B MIC, 64 mg/L; ivacaftor MIC, >32 mg/L; lumacaftor MIC, >32 mg/L). Aliquots (~20 μL) of early log-phase bacterial suspensions were inoculated into sterile 50 mL glass tubes (Greiner Bio-one, Germany) containing 20 mL of cation-adjusted Mueller-Hinton broth (CaMHB, adjusted with 23 mg Ca2+/L and 12.2 mg Mg2+/L), DMSO (2.5%, v/v) and different concentrations of polymyxin B and ivacaftor. Colonies were counted by a ProtoCOL automated colony counter (Synbiosis, United Kingdom) after the incubation of subcultures for 24 h at 35°C. The detection limit was 20 CFU/mL. The definitions for synergism, additivity, indifference and antagonism are foot-noted in Table 1.
Topoisomerases and gyrase enzyme assays
Purified E. coli DNA gyrase, E. coli topoisomerase IV, human topoisomerase I and human topoisomerase IIα were obtained from TopoGEN, Inc. (Buena Vista, CO, USA). These enzymes were purified to >98% purity as judged by over-loading SDS Polyacrylamide gels. Assays for type II enzymes (DNA gyrase, topoisomerase IV and topoisomerase IIα) werebased on decatenation of kinetoplast DNA (kDNA), which was also purchased from TopoGEN, Inc. The kDNA substrate is composed of a network of interlocked 2.5 kilobase minicircular DNA rings. These minicircular kDNA networks (catenanes) are ideal type II topoisomerase substrates since separation of the large molecular weight kDNA networks from the much smaller 2.5 kilobase minicircles is easily and quickly achieved by agarose gel electrophoresis.25, 24, 25 Decatenation assays contained 200 ng of kDNA and 8 units of enzyme (1 unit decatenates 200 ng kDNA in 30 min at 37°C). DNA gyrase assays were carried out in a 1× gyrase assay buffer (35 mM Tris-HCl, pH 7.5, 24mM KCl, 4 mM MgCl2, 2 mM dithiothreitol, 1.8 mM spermidine, 1 mM ATP, 6.5% (v/v) glycerol and Bovine serum albumin (BSA, 0.1 mg/mL). Topoisomerase IV assays contained 20 mM Hepes-KOH (pH7.6), 50 mM potassium glutamate, 5 mM magnesium acetate, 5mM dithiothreitol, BSA (25 μg/mL). Topoisomerase IIα assays were performed in 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM MgCl2, 0.5 mM dithiothreitol, BSA (30 μg/mL). Reactions were assembled on ice (20ul final volume) and initiated by adding enzyme. After incubating for 30 min at 37°C, reactions were terminated with SDS (1% w/v final), digested with proteinase K (50 μg/mL 15 min at 60°C) and following addition of loading (0.2 vol of 0.125% bromophenol blue, 25% glycerol) samples were loaded on 1% agarose gels containing 0.5 μg/mL ethidium bromide in the gel and running buffer. After electrophoresis (45 min at 3 volts/cm), gels were destained with water (15 min wash) and photographed. Assays for type I enzymes (human topoisomerase I) were based on relaxation of supercoiled DNA (pHOT1, from TopoGEN, Inc.).26 Reactions (20 μL volume) contained 100 ng of pHOT1 plasmid, and 8 units of topoisomerase I (1 unit will relax 100 ng in 30 min at 37°C) in TGS buffer (10 mM Tris-HCl (pH 7.9), 1 mM EDTA, 150 mM NaCl, 0.1% BSA, 0.1 mM Spermidine, 5% glycerol. Reactions were incubated for 30 min at 37°C, terminated with SDS (1% final) and mixed with a leading dye (0.1vol of 0.25% bromophenol blue, 50% glycerol) before loading onto agarose gels. Two gel systems were used for this analysis; half of the reaction volume was loaded onto each gel. One gel contained 0.5 μg/mL ethidium bromide (in gel and running buffer) which is optimal for resolving topo I cleavage products (nicked open circular DNA). The second gel lacked ethidium bromide is allows clear separation of supercoiled DNA and relaxed DNA. The former gel is designed optimally to identify drugs that act as an interfacial poison, inducing DNA cleavages (like camptothecin) while the latter gel system is optimal for detecting relaxation activity and drugs that inhibit enzymatic activity.
Imaging methods
Scanning and transmission electron microscopy were performed as we have previously described.27 Confocal imaging of the release of cytosolic green fluorescence protein (GFP) release was performed using the P. aeruginosa strain Pa AH298-GFP which expresses GFP under the control of the growth-rate dependent rrnBp1 promoter, kindly provided by Professor Philip S. Stewart.28 Pa AH298-GFP cells were grown on chamber slides and treated with polymyxin (8 mg/L), ivacaftor (8 mg/L) and lumacaftor (8 mg/L) alone or in combination of polymyxin-ivacaftor and polymyxin-ivacaftor-lumacaftor for 1 h at 37°C. Cell morphology was assessed using an LSM780 confocal microscope (Zeiss) equipped with a 63 × oil objective, excitation/emission filters used were Ex/Em 475/520–560 nm. NIH ImageJ software was used to for quantitative data analysis. Per slide (23.065 × 23.065 micron) 120–130 bacterial cells were counted. The percentage of GFP release was calculated as %GFP release=untreated control cells/treated cells × 100%.
Nitrocefin assays
The outer membrane permeabilizing activity of the test compounds was assessed by the β-lactamase nitrocefin assay. Briefly, the β-lactamase positive P. aeruginosa strain FADDI-PA065 was sub-cultured on a nutrient agar and incubated at 35°C overnight. One colony was randomly selected and grown overnight in 10 mL CAMBH at 37°C. The culture was grown to exponential phase and harvested by centrifugation (10000 × g; 10 min). The pellet was washed twice in PBS, pH 7.2 and resuspended in PBS to an OD=0.50. Nitrocefin was prepared to a concentration of 2 mM in DMSO. The assay was performed in 96-well plates. The 200 μL reaction mixture consisted of 100 μL bacterial suspension, 96 μL drug (2 mg/L polymyxin B alone or in combination with 4 mg/L ivacaftor) and 4 μL of 2 mM nitrocefin solution. The absorbance (OD 495 nm) was kinetically measured at room temperature for every minute. The values reported are the mean of four independent measurements± standard deviation (SD).
Artificial sputum media (ASM) biofilm assay
The ASM assays were performed with P. aeruginosa strain FADDI-PA065 in a 24-well plate format. ASM culture medium ideal mimics the components of CF patient sputum, and includes f egg yolk, amino acids, mucin and DNA combined in ratios as described in detail by Kirchner et al., prepared fresh and filter sterilized.18, 29 P. aeruginosa strain FADDI-065 was sub-cultured on a nutrient agar and incubated at 35°C overnight and one colony was randomly selected and grown overnight in 10 mL CAMBH at 37°C. The overnight culture was diluted in CAMBH to an OD of 0.05±0.01 and then further diluted 1:100 in fresh ASM. A total volume of 1.8 mL was added to each well. Plates were secured with parafilm and incubated for 3 days under aerobic conditions at 37°C in which the P. aeruginosa biofilms were developed. After 3 days polymyxin B, ivacaftor, and lumacaftor, alone or in combination were added and further incubated for 24 hours. For each antibiotic concentration four replicates were conducted. After the 24 h incubation, biofilms were disrupted with 100 μL of 100 mg/mL, cellulase (diluted in 9.6 g/L citrate; pH adjusted to 4.6 with NaOH) and incubated for 1 h at 37°C. Biofilms were further manually disrupted by pipetting. To determine the cell viability 100 μL of 0.02% (v/v) resazurin was added to each well and further incubated for 2 h at 37°C. Fluorescence (F) of each well was measured using an ENVISION plate reader (PerkinElmer, Australia) set at an excitation wavelength of 530 nm and an emission wavelength of 590 nm. Fluorescence was corrected by the subtraction of the background noise. The cell viability was calculated as (mean F of treated biofilms/mean F untreated control) × 100%.
Detection of cellular ROS production
ROS production in exponential phase P. aeruginosa FADDI-PA065 cells (OD 600 nm 0.1) following antibiotic treatment were measured using the oxidative stress sensitive dye CellROX Green (Lifetechnologies, Australia). CellROX Green fluorescence emission was measured at an excitation wavelength of 485 nm on a Cary Eclipse Fluorescence spectrophotometer (Varian, Australia) set. The emission spectrum was collected between 490–600 nm and slit widths were set to 5 nm for both the excitation and emission monochromators.
Supplementary Material
Acknowledgments
We are grateful to Professor John Turnidge from the University of Adelaide for allowing us to reproduce the data in Supplementary Table 1 that were generated in his laboratory and previously reported23. We also are thankful for the CW-P. aeruginosa clinical strains obtained from Prof Anton Y. Peleg and A/Prof Denis Spelman from the Alfred Hospital, Melbourne and the B. pseudomallei isolates provided by Professor Bart Currie (Menzies School of Health Research). This study is supported by a research grant to J.L and T.V. from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (R01AI111965). J. L. is an Australian National Health and Medical Research Council (NHMRC) Senior Research Fellow, and T. V. is an Australian NHMRC Industry Career Development Level 2 Research Fellow. E.K.S acknowledges support from the US Cystic Fibrosis Foundation. Y.D. acknowledges support from the NIAID (R01AI104895).
Abbreviations
- ASM
Artificial Sputum Media
- CAMBH
Cation adjusted Muller Hinton Broth
- MIC
minimum inhibitory concentration
- CFU
colony-forming units
- GFP
green fluorescence protein
- MOA
mode of action
- ROS
reactive oxygen species
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Lodato EM, K W. Background Paper 6.1 Antimicrobial resistance. World Health organization; 2013. [Google Scholar]
- 2.Cars O, Hogberg LD, Murray M, Nordberg O, Sivaraman S, Lundborg CS, So AD, Tomson G. Meeting the challenge of antibiotic resistance. BMJ. 2008;337:a1438. doi: 10.1136/bmj.a1438. [DOI] [PubMed] [Google Scholar]
- 3.Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 4.Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15(2):194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordonez C, Elborn JS. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–72. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–31. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Velkov T, Roberts KD, Nation RL, Thompson PE, Li J. Pharmacology of polymyxins: new insights into an ‘old’ class of antibiotics. Future Microbiol. 2013;8(6):711–24. doi: 10.2217/fmb.13.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yun B, Azad MA, Wang J, Nation RL, Thompson PE, Roberts KD, Velkov T, Li J. Imaging the distribution of polymyxins in the kidney. J Antimicrob Chemother. 2014;70(8):827–829. doi: 10.1093/jac/dku441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Velkov T, Abdul Rahim N, Zhou QT, Chan HK, Li J. Inhaled anti-infective chemotherapy for respiratory tract infections: Successes, challenges and the road ahead. Advanced drug delivery reviews. 2015;85:65–82. doi: 10.1016/j.addr.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moskowitz SM, Brannon MK, Dasgupta N, Pier M, Sgambati N, Miller AK, Selgrade SE, Miller SI, Denton M, Conway SP, Johansen HK, Hoiby N. PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob Agents Chemother. 2012;56(2):1019–30. doi: 10.1128/AAC.05829-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moffatt JH, Harper M, Harrison P, Hale JD, Vinogradov E, Seemann T, Henry R, Crane B, St Michael F, Cox AD, Adler B, Nation RL, Li J, Boyce JD. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother. 2010;54(12):4971–7. doi: 10.1128/AAC.00834-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Nation RL, Owen RJ, Wong S, Spelman D, Franklin C. Antibiograms of multidrug-resistant clinical Acinetobacter baumannii: promising therapeutic options for treatment of infection with colistin-resistant strains. Clin Infect Dis. 2007;45(5):594–8. doi: 10.1086/520658. [DOI] [PubMed] [Google Scholar]
- 13.Isshiki Y, Kawahara K, Zahringer U. Isolation and characterisation of disodium (4-amino-4-deoxy-beta-L-arabinopyranosyl)-(1–>8)-(D-glycero-alpha-D-talo-oct-2-ulopyranosylona te)-(2–>4)-(methyl 3-deoxy-D-manno-oct-2-ulopyranosid)onate from the lipopolysaccharide of Burkholderia cepacia. Carbohydr Res. 1998;313(1):21–7. doi: 10.1016/s0008-6215(98)00179-7. [DOI] [PubMed] [Google Scholar]
- 14.Assessment report KALYDECO ivacaftor EMEA/H/C/002494//0000
- 15.Ernst RK, Moskowitz SM, Emerson JC, Kraig GM, Adams KN, Harvey MD, Ramsey B, Speert DP, Burns JL, Miller SI. Unique lipid a modifications in Pseudomonas aeruginosa isolated from the airways of patients with cystic fibrosis. J Infect Dis. 2007;196(7):1088–92. doi: 10.1086/521367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heltshe SL, Mayer-Hamblett N, Burns JL, Khan U, Baines A, Ramsey BW, Rowe SM. Pseudomonas aeruginosa in Cystic Fibrosis Patients With G551D-CFTR Treated With Ivacaftor. Clin Infect Dis. 2014 doi: 10.1093/cid/ciu944. [Epub ahead of print] 2014/11/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koike M, Iida K, Matsuo T. Electron microscopic studies on mode of action of polymyxin. Journal of bacteriology. 1969;97(1):448–52. doi: 10.1128/jb.97.1.448-452.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Callaghan CH, Morris A, Kirby SM, Shingler AH. Novel method for detection of beta-lactamases by using a chromogenic cephalosporin substrate. Antimicrob Agents Chemother. 1972;1(4):283–8. doi: 10.1128/aac.1.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130(5):797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 20.Reznikov LR, Abou Alaiwa MH, Dohrn CL, Gansemer ND, Diekema DJ, Stoltz DA, Welsh MJ. Antibacterial properties of the CFTR potentiator ivacaftor. J Cyst Fibros. 2014 doi: 10.1016/j.jcf.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernarde C, Keravec M, Mounier J, Gouriou S, Rault G, Ferec C, Barbier G, Hery-Arnaud G. Impact of the CFTR-potentiator ivacaftor on airway microbiota in cystic fibrosis patients carrying a G551D mutation. PLoS One. 2015;10(4):e0124124. doi: 10.1371/journal.pone.0124124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu DT, Fernandes PB. Structure-activity relationships of the fluoroquinolones. Antimicrob Agents Chemother. 1989;33(2):131–5. doi: 10.1128/aac.33.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Turnidge J, Milne R, Nation RL, Coulthard K. In vitro pharmacodynamic properties of colistin and colistin methanesulfonate against Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob Agents Chemother. 2001;45(3):781–5. doi: 10.1128/AAC.45.3.781-785.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.CLSI. Performance Standards for Antimicrobial Susceptibility Testing: Twentieth Informational Supplement (M100-S20) CLSI; Wayne, PA, USA: 2010. [Google Scholar]
- 25.Spitzner JR, Chung IK, Muller MT. Eukaryotic topoisomerase II preferentially cleaves alternating purine-pyrimidine repeats. Nucleic Acids Res. 1990;18(1):1–11. doi: 10.1093/nar/18.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller MT, Pfund WP, Mehta VB, Trask DK. Eukaryotic type I topoisomerase is enriched in the nucleolus and catalytically active on ribosomal DNA. The EMBO Journal. 1985;4(5):1237–1243. doi: 10.1002/j.1460-2075.1985.tb03766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdul Rahim N, Cheah SE, Johnson MD, Yu H, Sidjabat HE, Boyce J, Butler MS, Cooper MA, Fu J, Paterson DL, Nation RL, Bergen PJ, Velkov T, Li J. Synergistic killing of NDM-producing MDR Klebsiella pneumoniae by two ‘old’ antibiotics-polymyxin B and chloramphenicol. The Journal of antimicrobial chemotherapy. 2015 doi: 10.1093/jac/dkv135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Werner E, Roe F, Bugnicourt A, Franklin MJ, Heydorn A, Molin S, Pitts B, Stewart PS. Stratified growth in Pseudomonas aeruginosa biofilms. Applied and environmental microbiology. 2004;70(10):6188–96. doi: 10.1128/AEM.70.10.6188-6196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirchner S, Fothergill JL, Wright EA, James CE, Mowat E, Winstanley C. Use of artificial sputum medium to test antibiotic efficacy against Pseudomonas aeruginosa in conditions more relevant to the cystic fibrosis lung. J Vis Exp. 2012;(64):e3857. doi: 10.3791/3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.