

Abstract
The evidence is now overwhelming that partially assembled nucleosome states (PANS) are as important as the canonical nucleosome structure for the understanding of how accessibility to genomic DNA is regulated in cells. We use a combination of molecular dynamics simulation and atomic force microscopy to deliver, in atomic detail, structural models of three key PANS: the hexasome (H2A·H2B)·(H3·H4)2, the tetrasome (H3·H4)2, and the disome (H3·H4). Despite fluctuations of the conformation of the free DNA in these structures, regions of protected DNA in close contact with the histone core remain stable, thus establishing the basis for the understanding of the role of PANS in DNA accessibility regulation. On average, the length of protected DNA in each structure is roughly 18 basepairs per histone protein. Atomistically detailed PANS are used to explain experimental observations; specifically, we discuss interpretation of atomic force microscopy, Förster resonance energy transfer, and small-angle x-ray scattering data obtained under conditions when PANS are expected to exist. Further, we suggest an alternative interpretation of a recent genome-wide study of DNA protection in active chromatin of fruit fly, leading to a conclusion that the three PANS are present in actively transcribing regions in a substantial amount. The presence of PANS may not only be a consequence, but also a prerequisite for fast transcription in vivo.
Introduction
The organization of genomic DNA into nucleosomes has profound implications for many key cellular processes including replication, transcription, repair, and recombination. Besides its role in packing the genome, nucleosome occupancy at gene promoters inhibits transcription initiation (1), and nucleosomes play a critical role in epigenetic regulation (2). The overall organization of the nucleosome (the repeating subunit of chromatin (3, 4, 5)), as a disklike complex of eight histone proteins (two H2A·H2B dimers and an (H3·H4)2 tetramer), surrounded by ∼200 basepairs of DNA, was known for several decades (6). However, it was not until 1997 that the nucleosome structure was resolved at atomic resolution (7), uncovering key, previously unknown details of the histone-histone and histone-DNA interactions. The availability of the atomistic structure (7, 8) of this fundamental unit of DNA compaction made a critical difference for the efforts to develop the mechanistic understanding of the primary function of the nucleosome as DNA accessibility regulator.
Over the past decade, it became clear that the nucleosome is not a single static structure, but rather a highly dynamic family of interconverting structural states (9); considering just the canonical nucleosome structure is not enough to develop the complete mechanistic understanding of key cellular processes that involve the nucleosome (10, 11). A large number of non-canonical structural states of the nucleosome were proposed and characterized experimentally to various degrees of detail (9, 12, 13, 14, 15, 16), but not at atomistic level.
An important class of non-canonical structures is partially assembled nucleosome states (PANS), some of which were proposed to occur on the nucleosome assembly/disassembly pathways (9, 15, 16, 17); compared to the intact nucleosome core particle (octasome), or its transient, partially unwrapped states (13, 14), the PANS structures lack several histones. Importantly, the existence of PANS is a consequence of the modular nature of the nucleosome, facilitating the histone exchange process, which involves the removal of parts of the nucleosome or the entire nucleosome, followed by replacement with newly synthesized histones including variant histones. The histone exchange, which is also known as histone turnover, has many implications for the composition, structure, and function of different genomic regions and it is necessary to tightly regulate gene expression (18).
Experimental studies point to progressive unfolding of the chromatin structure in anomalously proliferating (cancer) cells (19, 20), which could be associated with PANS formation. Recent genome-wide studies revealed an existence of widespread subnucleosomal structures in dynamic chromatin of living cell (21, 22, 23). These PANS affect DNA accessibility and, most likely, the interaction of transcription factors, histone chaperones, and chromatin-modifying enzymes with chromatin, and can play a role in high-order chromatin organization. In particular, disassembly and remodeling of nucleosomes can be closely associated with supercoiling of DNA, alleviating torsional stress within chromatin (24). Thus, PANS are likely to be as important as the fully assembled nucleosome, and so it is critical that they too are characterized at the same atomistic level of detail.
However, no experimental atomic-resolution models of any PANS are available, which reflects the well-known difficulties associated with capturing transient, highly mobile, and relatively large intermediate structures using traditional atomic resolution techniques such as x-ray crystallography or nuclear magnetic resonance. In particular, it is unclear whether the remaining histones in PANS are arranged in the same way as in the intact nucleosome. Likewise, it is not known how the structural rearrangements after the removal of the histones affect accessibility of amino acid residues—potential target sites for posttranslational modifications. More importantly, the arrangement of the DNA in PANS is not clear as well: in the nucleosome, the DNA is tightly wrapped around the oppositely charged core in a very specific way, but what happens when some of the histones are removed? It could be that the remaining electrostatic attraction is still strong enough to wrap all or most of the DNA around the histone core. Or, it may be that most of the DNA becomes free (not protected by the histone core and thus fully accessible for other proteins). The latter is a particularly important question as it directly ties into the pivotal role that the nucleosome, along with its conformational states, play in regulating DNA accessibility in vivo—the process that likely requires at least partial disassembly of the nucleosome (12, 23, 25). It is well known (26) that chromatin cannot be correctly assembled in vitro under physiological conditions without histone chaperones and ATP-dependent chromatin assembly factors. It is unclear whether, during the reverse process of nucleosome disassembly, the intermediate structures do retain key features of protein-DNA interaction present in the intact nucleosome that would facilitate quick reassembly of the particle. Finally, the nucleosome itself is a dynamic structure (10), and so the much more open PANS are expected to be even more dynamic, characterized by the significant structural fluctuations seen in recent experiments. All of the above aspects of PANS, including the dynamics, would be hard or impossible to infer directly from a static x-ray structure of the intact nucleosome.
Note that only a small subset of possible DNA-histone complexes (12) are likely to be biologically relevant and occur in significant quantities on real nucleosome assembly/disassembly pathways. Specifically, it was shown that during the nucleosome assembly two copies of the H3·H4 dimer bind to the DNA first, forming a tetrasome, followed by sequential addition of each H2A·H2B dimers (27). The nucleosome can disassemble through the reverse reaction that starts with an opening of the (H3·H4)2 tetramer-H2A·H2B dimers interface, followed by the release of H2A·H2B dimers from the DNA (14, 28). It was also shown that H2A·H2B complexes with the DNA are effectively suppressed in vivo, even though the affinity of highly positively charged dimer to the DNA is substantial (29). Some PANS are likely to occur only in specific types of chromatin, e.g., centromeric (30). These considerations limit the number of the most relevant PANS to very few.
Further, because one of our goals here is to demonstrate utility of our atomistic models for interpretation of both in vivo and in vitro experiments (28, 31, 32), we focus on just three key PANS: the disome, the tetrasome, and the hexasome (Table 1). These subnucleosomal structures have been deemed important in vivo, and are already well characterized in vitro, both thermodynamically and by low-resolution methods such as Förster resonance energy transfer (FRET) or small-angle x-ray scattering (SAXS).
Table 1.
Common Nomenclature of Three PANS Investigated in this Work
| Name | Stoichiometrya |
|---|---|
| Disomeb | H3·H4 |
| Tetrasome | (H3·H4)2 |
| Hexasomeb | (H2A·H2B)·(H3·H4)2 |
| Octasomec | 2(H2A·H2B)·(H3·H4)2 |
Including 147-bp DNA fragment.
There are two possible disome and hexasome structures (see the Supporting Material).
Octasome is not considered as PANS and is shown here for reference only.
We use, to our knowledge, a novel approach—a combination of modern molecular modeling techniques (33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46) and atomic force microscopy (AFM) measurements—to add experimentally consistent models of these three new members to the family of atomistic structures of the nucleosome particles.
Materials and Methods
See the Supporting Material for complete details. References to the corresponding sections are provided in the end of each paragraph.
Structure preparation summary, simulation protocols
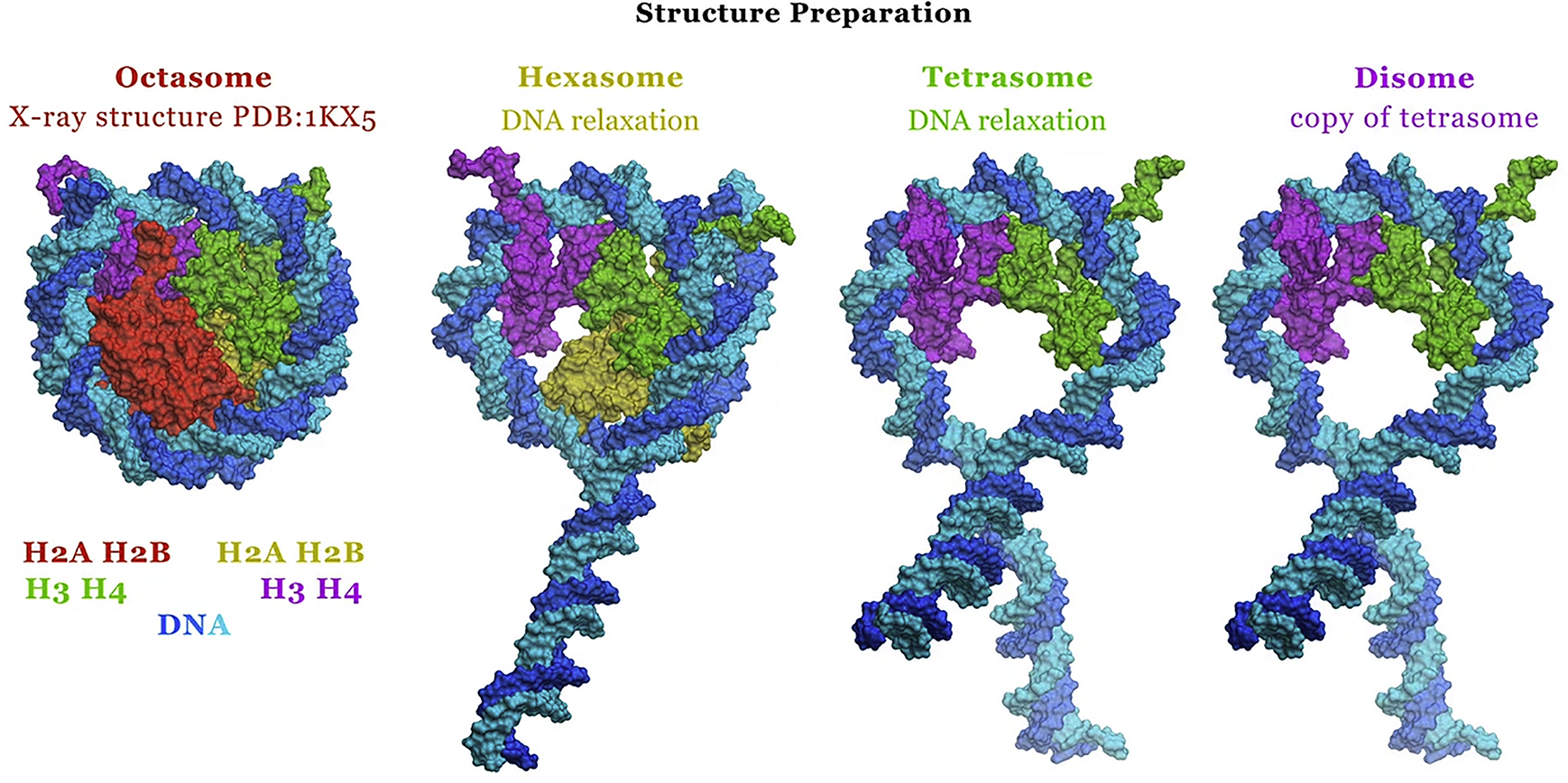
The preparation of the atomistic models of the PANS included three key stages: 1) Appropriate histone proteins were removed from the atomic resolution octasome structure (PDB: 1KX5 (8)) to construct the initial, crude models of the PANS. 2) The structures were equilibrated for 75 ns in the implicit solvent at 300 K. 3) The structures (except for the disome) were refined in the explicit solvent for 100 ns. For implicit solvent calculations we used the AMBER12 (47) package with the ff10 force field and the generalized Born (igb5) solvation model without long-range cutoffs; 0.145 M NaCl was modeled implicitly. Langevin dynamics was used for temperature regulation with a collision frequency (effective viscosity) γ = 0.01 ps–1. The critical advantage of this low effective solvent viscosity regime, successfully used in simulation of nucleic acids, proteins, and their complexes (39, 43, 48, 49), is that it offers ∼100-fold speedup of large conformational transitions relative to the more traditional explicit solvent simulations, for the types of structures considered here (44, 50). For explicit solvent calculations we used the GROMACS package (51), with the AMBER ff10 force field at 300 K for 100 ns in the explicit solvent (PME, TIP3P water (52), with 0.145 M NaCl). Because the disome was too extended to fit in a reasonable solvent box, 75 ns of implicit solvent molecular dynamics (MD) was used instead, for the structure refinement stage 3 described above (see Sections 1.1–1.3 in the Supporting Material).
Robustness of MD protocols
We have verified that key structural characteristics of the PANS—protected DNA length, FRET interdye distances, and histone interdimer distances—are reproducible in independent trajectories, different force fields, and solvent models (see sections 1.4 and 3.2 in the Supporting Material).
MD trajectory analysis
Except for the disome, all of the results presented in this article are based on the last 50 ns of the explicit solvent trajectories. An analysis of the implicit solvent trajectories is provided in sections 1.5 and 3.2 of the Supporting Material.
Protected DNA length. A basepair was considered as protected if minimal distance dmin between atoms of the basepair and the histone core is below a cutoff distance dpro = 15 Å. As the histone tails (as defined in Luger et al. (53)) are very mobile and can tightly attach to the DNA at arbitrary positions, they were excluded from the analysis (see section 1.6 in the Supporting Material).
Calculation of interdye distances to compare with FRET-based data. We mapped positions of the dyes utilized by Gansen et al. (28) onto the 147-bp DNA fragment in the nucleosome structure of PDB: 1KX5 using the primer sequences from Gansen et al. (28). Distances between the phosphorus atoms of these nucleotides were compared to the reported interdye distances. We followed an earlier suggestion that averaging the distance over a range of possible histone core positions provides a better estimate of the expected FRET than using the donor-acceptor distance from a single position (54) (see section 1.7 in the Supporting Material).
Calculation of SAXS spectra to compare with experiment. We used the g_sans GROMACS utility (55). Atom scattering cross sections were adapted for x-ray scattering by the Cromer-Mann method (56). Scattering by DNA component of the sample was simulated by selective application of g_sans to trajectories of DNA atoms only. Experimental I(q) values obtained for 601NP nucleosome particles in varying salt conditions and with contrast variation in 50% sucrose (32) were fitted by a linear weighted combination of the computed intensity curves (see section 1.8 in the Supporting Material).
Experiment: AFM visualization and analysis of the PANS
Nucleosome reconstitution was performed (57) using the 353-bp DNA amplified by polymerase chain reaction with plasmid pGem-3Z-601 (58) and human recombinant histones purchased from New England BioLabs (Ipswich, MA). The reconstituted nucleosome samples were homogeneous with uniformly positioned histone octamer on DNA template (Fig. S3 in the Supporting Material), thus additional purification of nucleosomes was not required. APS-treated mica was used for immobilization of nucleosomes on the surface (59). The nucleosome samples were diluted to final concentration 2 nM with 10 mM Tris-HCl buffer, pH 7.5, and 140 mM NaCl, leading to accumulation of PANS, observed at low concentrations of the nucleosomes (28). Samples were deposited on a mica surface, rinsed with Milli-Q Ultrapure water (Millipore, Billerica, MA), and dried under argon flow (see section 2.1 in the Supporting Material).
The samples were scanned in tapping mode with a Nanoscope III system (Veeco, Fremont, CA) and silicon probes. The scanning rate was 1.7 Hz over scan areas of 1 μm. Measurements of contour length and cross-section analysis were performed using Femtoscan software (Advanced Technologies Center, Moscow, Russia) (see section 2.2. in the Supporting Material).
Wrapped DNA length, L, was calculated by subtraction of two DNA arm lengths from the total template DNA length. Volume, V, of the protein-DNA complex was calculated using the equation proposed in Henderson et al. (60). Only well-defined single particles with intact DNA were selected for further analysis. Measured L values were processed with kernel density analysis using the package for R (61). Local minima of the density distribution of L values were used to separate the nucleosome structures into groups (see sections 2.3 and 2.4 in the Supporting Material).
Results and Discussion
The combination of atomistic simulation with AFM experimental measurements has yielded morphological and dynamical characteristics of the subnucleosomal particles including the hexasome, the tetrasome, and the disome. While atomistic modeling is arguably (62, 63) the only method available at the moment that can deliver the transient PANS at full atomic resolution, the method (64) cannot yet probe timescales of seconds relevant to key biological processes such as transcription (65), which depend critically on the DNA packing in the nucleosome. Here, the difficulty is overcome by verifying experimentally that the key geometric characteristics of the modeled structures, including the length of protected DNA, remain close to the observed values on the AFM timescales of minutes. While the reported MD simulations may not be long enough to properly sample the entire configurational landscape of the PANS, the noteworthy quantitative consistency with multiple experiments (AFM imaging, FRET, and SAXS spectra analysis) gave us confidence that the proposed PANS structures are nevertheless meaningful approximations to reality.
Partially assembled nucleosome structures
One of our key results is that the specific PANS investigated here—the hexasome, the tetrasome, and the disome—are characterized by well-defined structures (see Figs. 1 and 2 and the Supporting Material in PDB format). Each of these structures contains roughly 18 bp of protected DNA per histone protein (Table 2).
Figure 1.

Overall structures and protected DNA regions in the PANS and the octasome. Distributions of the minimal distance dmin from the DNA basepair to the histone core are shown for the disome (A), tetrasome (B), hexasome (C), and octasome (D) structures obtained in the MD simulations. The basepairs are classified as follows: protected (dmin < dpro = 15 Å for all the snapshots); accessible (dmin > dpro for all the snapshots); and transient. Here dpro is a minimal distance separating histones atoms from the DNA atoms in a realistic complex of the nucleosome with a nuclear factor. Minimal, median, and maximal dmin values are shown by solid lines, 25th and 75th percentiles are shown by dashed lines. Black marks with labels indicate basepair positions. Colored lines connect centers of mass of the histone heterodimers (purple dots); the distances are specified in Å; the color scheme is the same for all the structures. Additional images (Fig. S4; Movie S1) and representative snapshots (in PDB format) are provided in the Supporting Material. To see this figure in color, go online.
Figure 2.

Representative images from four subpopulations of nucleosomal structures observed in the AFM experiment under the conditions (very low nucleosome concentration) when the equilibrium is shifted toward the PANS shown in Fig. 1. (Circles) Approximate extent of the atomistic models on the same scale. The height of the particle above the mica plane is indicated by the color intensity: progressive disassembly of the nucleosome particle leads to the decreasing size and height of the particle core. Additional AFM images (Figs. S7 and S8) are provided in the Supporting Material. To see this figure in color, go online.
Table 2.
Protected DNA Length (bp) in the PANS, from Atomistic MD Simulations and Experiment
| MD Simulations |
AFM Dataa |
MNase Protection |
|||
|---|---|---|---|---|---|
| Median | Range | Median | Rangeb | Peaksa,c | |
| Disome | 43 | 14 | 36 | 8 | 15–45 |
| Tetrasome | 69 | 10 | 74 | 16 | 46–75 |
| Hexasome | 110 | 7 | 109 | 18 | 76–120 |
| Octasome | 147 | 0d | 148 | 6 | 140–150 |
Obtained from analysis of the experimental data representing a mix of PANS.
Interquartile range.
Data from Teves and Henikoff (31) (see section Interpretation of genome-wide mapping data).
Modeled octasome structure does not contain linker DNA.
The overall shapes of the disome, the tetrasome, and the hexasome resemble Greek letters ζ, υ, and σ, respectively, and are very different from the octasome shape that resembles an o, when viewed down the DNA superhelical axis (Fig. 1). In contrast to the often used schematics, the subnucleosomal particles are highly nonplanar structures, especially the disome and the tetrasome. The nonplanar layout naturally helps mitigate the energetically unfavorable interaction between the arms of the highly charged DNA, contributing to the stability of the histone-DNA contacts. Progressive removal of the histone dimers results in an increase of the distances between the dimers remaining in the core (Fig. 1).
In the PANS, the free DNA not in direct contact with the histones is flexible, experiencing considerable occasional fluctuations. However, large swings are relatively rare: most of the time DNA arms fluctuate near their average positions (see Movie S1). For the tetrasome structure, the asymmetry of the fluctuations seen in the simulations (Fig. 1) is determined by the asymmetry in the structure of the histone tails in the original octasome and stochastic behavior of histones tails annealing to DNA.
DNA accessibility in the PANS
Despite large-scale fluctuations of DNA arms seen in the simulations, the histone-DNA contacts are rather stable; these define DNA regions protected by histones from contacts with hypothetical nuclear factors (Table 2). The key result of biological significance is the specific pattern of accessible and inaccessible (protected) DNA in the PANS (Fig. 1). Each H2A·H2B dimer protects ∼40 bp in the hexasome and the octasome; and each H3·H4 dimer protects ∼35 bp in the tetrasome and 43 bp in the disome. Critically, the DNA protection pattern of the four nucleosomal structures inferred from the atomistic models is in good agreement with corresponding experimental values from the AFM measurements (Table 2).
Transiently accessible regions (yellow regions in Fig. 1) between protected and accessible zones span ∼5–7 bp at each DNA arm. In these regions the DNA can bend and transiently unwrap, potentially allowing nuclear factors and nucleases an additional access to the DNA. A reasonable correspondence to the data on chromatin protection from MNase digestion (31) is noteworthy; its biological implications will be discussed below.
Contacts between the DNA and the histone proteins
Dynamics of protein-DNA contacts can be evaluated from the distribution of protein-DNA distance in the protected region (Fig. 1), where clear peaks and troughs are seen: higher peaks indicate looser contacts. Three kinds of contacts can be distinguished by the peak height: tight, medium strength, and loose. In all the PANS, a clear 10-bp periodicity is observed for medium-strength contacts, initially reported by Luger and Richmond (66) for the octasome. Regions of tight histone-DNA contacts are located between the peaks with ∼5-bp periodicity, seen experimentally by mechanical unzipping of the nucleosomal DNA (67) and in MD simulations of forced unwrapping of the DNA from the histone core (68). Importantly, all three subnucleosomal particles mostly inherit the pattern of histone-DNA contacts from the octasome, with only slight alteration of medium contacts in the –20 to +20 bp region. The fine details of the histone-DNA contacts may be important because transcription through the tightly packed nucleosome carried out by RNA polymerase unassisted by chromatin remodeling factors and histone chaperones can depend on specific protein-DNA contacts. In vitro experiments detailing the progression of RNA polymerase (RNAP or Pol II) through nucleosomes show strong pausing at approximately –60 bp and –30 bp from the dyad (69, 70), and a distinct periodicity of ∼10 bp pause sites (71). Thus, positions of polymerase strong pausing sites tend to occur in the transient accessibility regions (Fig. 1) from ±30 to ±40 bp in the PANS.
Enhanced accessibility to PTM sites
Posttranslational modifications (PTMs) of histones play critical roles in many biological processes; aberrant PTMs contribute to disease (72). While most of the PTMs with known in vivo roles are located in the N-terminal tails, mounting evidence suggests that PTMs of core histones are as important (73, 74), particularly to regulation of DNA accessibility (75). A large number of core histone PTMs are known to exist (76), investigation of their functional roles is an active area of research (74). Many of the core PTM sites are expected to be inaccessible in the intact octasome, but may become accessible in the PANS. Here we used atomistic structures of PANS to identify putative PTM sites that become accessible in the hexasome, tetrasome, and the disome (Table S5). Specifically, H4K91 important for DNA replication and H3Y41 involved in the control of transcription (74) become accessible in all the PANS.
One may expect the accessibility of histone residues to always increase in the PANS compared to the intact nucleosome. Counterintuitively, some residues that are solvent accessible in the octasome become completely inaccessible in some PANS, due to structural rearrangements that follow the histone removal. The absolute majority (10 out of 13) of these residues are in H2A/B histones (only in the hexasome; Table S6).
The histone tails
Histone N-terminal tails are known to regulate DNA accessibility and gene transcription and chromatin structure (77), often via charge-altering posttranslational modifications that affect chromatin compaction. N-terminal tails of H3 and H2B pinched between two DNA coils can facilitate maintenance of the superhelical structure of the octasome and the hexasome. The long protruding parts of these histone tails have no regular structure, and were found earlier to be broadly distributed over the DNA surface in the octasome (45, 78). Here we observe a similar trend in the PANS. Further, more detailed investigation of the structure and dynamics of the histone tails in PANS at the fully atomistic level will likely require much longer simulation times (35) than reported here, or/and the use of coarse-grained approaches (79). Comparison with more appropriate experimental observables, beyond those considered in this work, may be needed for a detailed validation of the conformational ensembles representing the histone tails. Atomistic structures of PANS may serve as a starting point for future studies aimed at addressing the potentially rich interplay between the roles of PANS and the histone tails in the chromatin function.
Identification of PANS in the AFM data
Dilution of the nucleosome sample to nanomolar concentrations gives rise to the full repertoire of PANS in a single experiment (28, 80, 81). We used this well-characterized technique to visualize a representative set of naturally occurring PANS by AFM.
To identify subnucleosomal structures registered by the AFM, we separated the observed population into groups based on two main characteristics: 1) the length, L, of DNA region in contact with histone core (i.e., protected DNA length); and 2) the volume, V, of protein-DNA complex (the core of a particle, excluding DNA arms) (Fig. 3). These geometric parameters can be obtained from the AFM images and, independently, calculated from the atomistic models. Remarkably, the density distribution of experimentally measured L values forms four well-defined peaks (Fig. 3). Comparison of the experimental and theoretical (based on the atomistic models) median values of L, and relative values of V (Tables 2 and 3) for each AFM subpopulation, shows that their characteristics are consistent with the disome, the tetrasome, the hexasome, and the octasome, respectively, moving from left to right and bottom-upwards (Fig. 3). The representative AFM images of PANS from each subpopulation are shown in Figs. 2 and S8. Partially unwrapped octasomes and PANS were present in the AFM images (outliers with relatively high volume and low DNA length in Fig. 3). However, these species were much less frequent than well-formed PANS and did not affect median values of L and V.
Figure 3.

Distribution of wrapped DNA length L and particle volume V as measured with AFM for complete nucleosomes and PANS. One-dimensional density distribution for DNA lengths is shown by the black curve (arbitrary units). The L-partition based on the local minima of one-dimensional density distribution is represented by blue vertical lines. Experimental points were classified using the L-partition into four classes (disome, tetrasome, hexasome, and octasome) and are represented by the first letter in blue color (D, T, H, and O, respectively). Median values for each class are marked by black plus-symbols (+). A linear fit to the experimental points is shown by the straight black line. Two-dimensional density distribution of experimental points is shown as yellow-to-red gradient fill for 10–99% levels. Median DNA length (protected only; Table 2) and particle volume values calculated from MD simulations are marked by green cross symbols (×); V values were normalized using median octasome values (Table 3). To see this figure in color, go online.
Table 3.
Volume of the Core Particle: MD Simulations and AFM Experiments
| Volume | Simulation |
Experimenta |
||||
|---|---|---|---|---|---|---|
| Median (nm3) | Ratiob | Range (nm3) | Median (nm3) | Ratiob | Rangec (nm3) | |
| Disome | 56.1 | 0.27 | 8.3 | 51.4 | 0.27 | 13.2 |
| Tetrasome | 101.2 | 0.48 | 5.9 | 95.7 | 0.51 | 26.4 |
| Hexasome | 157.0 | 0.74 | 4.7 | 130.2 | 0.69 | 33.3 |
| Octasome | 210.8 | 1.00 | 2.1 | 187.4 | 1.00 | 31.4 |
Obtained from analysis of the experimental data representing a mix of PANS.
Relative to volume of the octasome.
Interquartile range.
An alternative approach for identification of PANS in AFM images, as well as the influence of Mg2+ ions on the structures, was published recently by Nazarov et al. (82).
Interpretation of the DNA geometry and fluctuations in PANS revealed by FRET
FRET technique has emerged as a powerful method to study structure and dynamics of proteins and nucleic acids, as FRET efficiency between two judiciously placed fluorescent labels has a strong distance dependence. Nevertheless, unambiguous interpretation of observed distance distributions can be complicated by inhomogeneity of molecular structures in the sample, caused by the multisubunit composition of the molecular complexes and multidomain organization of their subunits. Realistic atomistic dynamic models can provide a solid basis for the interpretation, as well as generate further insights from the existing experimental data.
In 2009, Gansen et al. (28) reported several average distance characteristics of incomplete nucleosomal species using single-molecule FRET measurements on reconstituted nucleosomes containing a 170-bp strong positioning sequence 601 (83) labeled with donor and acceptor fluorophores, separated by 93 bp (Fig. 4 B). Measured FRET efficiencies were divided into three distinct subpopulations (Low FRET and DOnly (LF); Mid-FRET (MF); and High-FRET (HF)). For these subpopulations, FRET efficiencies were converted to interdye distances using probability distribution analysis and taking into account dye linker flexibility (28) (Table 4). As experimental data were obtained in highly diluted solution (picomolar to nanomolar concentrations of nucleosome particles), the whole set of PANS was expected to contribute to interdye distance distributions.
Figure 4.

(A) Comparison of interdye distances, calculated for the ensemble of atomic PANS (curves) and obtained by FRET in Gansen et al. (28) (gray areas). Gray areas represent three subpopulations observed in the FRET experiment: Low FRET and DOnly (LF); Middle FRET (MF); and High FRET (HF) species (see Table 4). Calculated distributions are in the form of a stacked density plot, assuming equal contribution of the octasome, hexasome, and tetrasome. Median values are indicated by dashed lines. The three interdye distance values for the hexasome model shown in (B) are indicated by red dots. (B) Different positions of the dyes, contributing to distinct FRET subpopulations, are illustrated on the hexasome structure. Phosphorus atoms (to which the dyes are connected) are indicated by red markers with labels showing basepair positions relative to the dyad. Interdye distances (Å) are represented by line segments. Color scheme used for the histones: H2A, yellow; H2B, red; H3, blue; and H4, green. To see this figure in color, go online.
Table 4.
Measured FRET Distances vs Calculated from MD Simulations
| Simulationa |
Experimentb |
||||
|---|---|---|---|---|---|
| Median |
Rangec |
Median |
Ranged |
||
| (Å) | (Å) | (Å) | (Å) | ||
| Tetrasome | 91.1 | 20.6 | 119.0 | 31.0 | Low FRET |
| Hexasome | 66.5 | 13.6 | 63.1 | 3.2 | Middle FRET |
| Octasome | 56.2 | 3.8 | 54.0 | 3.0 | High FRET |
Disome median distance of 203 Å is too large to be reliably detected by FRET.
Data from Table S1 of Gansen et al. (28).
Interquartile range.
Half-width of Gaussian distribution.
In the absence of atomistic models, Gansen et al. (28) suggested an assignment of the experimental HF, MF and LF populations to the octasome, the hexasome, and the tetrasome, respectively (Table 4). The assignment was based on schematic coarse-grained models and the assumption that each of the PANS produces a single distinct Gaussian peak in the FRET efficiency distribution. However, our atomistic models suggest a more nuanced interpretation and further insights from the same experimental data.
Specifically, each FRET band (Fig. 4 A) is not formed by a single particle; instead, it contains significant contributions from other species. The wide range of the observed interdye distances in PANS cannot be explained by the flexibility of the rather short dye linkers (see the Supporting Material for details). While the interdye distance distribution for the octasome has a relatively narrow peak predominantly corresponding to the HF band, the tetrasome has a broad distribution spanning through the three FRET bands with the maximum in the LF band, which reflects large fluctuations of the free DNA. Particular attention should be paid to the hexasome that has irregularly shaped distance distribution with three maxima, contributing to each of three FRET bands. Differing extents of DNA unwrapping results in shifting of the major peak positions near HF and MF bands, while taking into account the possible asymmetric repositioning of the histone core in reconstituted nucleosome particles (54) giving rise to a broad peak in the LF zone (Fig. 4 B). Consequently, the hexasome can be misinterpreted not only as the octasome, but more importantly, as a more disassembled particle—the tetrasome—even at low salt concentrations. The dynamic models of PANS can provide the basis for quantitative analysis of particle mixtures once appropriate experimental data on interdye distance distributions become available.
Interpretation of SAXS spectra of subnucleosomal particles
Recently, salt-induced nucleosome disassembly was investigated using SAXS with contrast variation (32). SAXS is a label-free technique that provides information about the average composition, size, and shape of macromolecules. Interpretation of SAXS experimental results in biology is most reliable for monodisperse systems (84); given the diversity and flexibility of PANS, the corresponding SAXS experimental data may be challenging to interpret. A detailed comparison between experimental and theoretical curves based on atomistic models of PANS can both validate the all-atom models and provide a more detailed interpretation to SAXS measurements.
Here we compared modeled SAXS spectra of the PANS structures to experimental spectra of the nucleosomes obtained (32) in 50% sucrose, which reveals primarily the DNA part of the system. This concentration of sucrose has virtually no effect on the salt-dependent equilibrium of the nucleosome particle (32). The standard approaches of calculating SAXS curves based on static structural models (85) appear to have only a limited utility in this case due to the presence of relatively large and mobile fragments of free DNA. Instead, we calculated I(q) spectra directly, as averages over MD trajectory of each PANS species. Specifically, we fit the experimental scattering intensities by a linear weighted combination of the modeled I(q) functions. The weights of the best-fit solutions represent the unknown fractions of each species.
The results (Fig. 5 A) clearly indicate that nucleosomal DNA unwrapping observed under elevated salt conditions may be explained by the formation of disassembled nucleosome species, which also occur under physiologically relevant conditions. This interpretation of the SAXS data implies that with increasing salt concentration the composition of the nucleosome particles changes in favor of the more dissociated species, from the mixture of hexasomes (∼45%) with tetrasomes (∼27%) and octasomes (∼23%) in 1 M NaCl to almost exclusively (>90%) disomes in 1.9 M NaCl (Fig. 5 B). In the physiological and the relatively low-salt regime (from 0.2 up to 0.5 M), the octasomes remain the predominant species as expected. The small amount (∼20%) of the dissociated states at low salt is most likely due to the presence of intact octasomes with partially unpeeled DNA (that structure was not modeled).
Figure 5.

(A) Least-square fit (red lines) of the SAXS scattering data (symbols) by the linear combination of the simulated I(q) for the different PANS in Fig. 1; vertical offset (y axis) for clarity. Experimental equilibrium I(q) are for 601NP nucleosomes in 50% sucrose reported earlier (32) for five different NaCl concentrations: 0.2, 0.5, 1, 1.5, and 2.0 M. Best fits to experiment are shown as solid lines; χ2 were 1.56, 2.07, 0.83, 0.87, and 1.14, respectively. (B) Composition of the best fit mixture of the modeled PANS as a function of salt concentration. To see this figure in color, go online.
Interpretation of genome-wide mapping data
A key result of this work is a quantitative characterization of DNA structural protection in various PANS (Fig. 1; Table 2). Atomistic models of the disome, the tetrasome, and the hexasome developed here provide a precise and unambiguous characterization of areas of unprotected DNA in the subnucleosomal particles, which can be used for interpretation of genome-wide data (9). For example, a 2011 study (31) used nuclease protection assays to identify a peculiar pattern of lengths of protected DNA fragments in active genes of Drosophila (right column of Table 2). In addition to the expected peak at ∼147 bp, corresponding to the intact nucleosome, three additional, lower intensity satellite peaks were observed (31). The oscillatory profile of DNA protection for fragments shorter than 147 bp was in contrast to the flat profiles that characterize inactive genes. Teves and Henikoff (31) interpreted the first two peaks as intact and partially unwrapped nucleosome, while the peaks at 65 and 35 bp were interpreted as corresponding to areas protected by transcription factors or machinery. We propose that the specific number of the peaks and their peculiar positions observed by Teves and Henikoff (31), can be explained, at least to an extent, by the specific lengths of protected DNA identified in the PANS (Table 2).
Thus, we suggest that the PANS are present in a substantial amount in actively transcribing regions and in promoter areas of actively transcribing genes. This conclusion is supported by other recent in vivo works. A genomic-scale study in yeast (23) detected widespread subnucleosomal structures in dynamic chromatin, including what appear to be half-nucleosomes (consisting of one copy of each histone), hexasomes, and tetrasomes. Moreover, it was shown that in actively transcribing genes and their enhancers (which are also the sites of very active histone exchange), preexisting (H3·H4)2 tetramers might split into dimers, resulting in mixed nucleosomes composed of old and new histones (86, 87). The enhanced presence of mixed tetramers at sites of active transcription suggests that nucleosome perturbation by Pol II involves a transfer of parental H3·H4 as dimers. In summary, in vivo evidence strongly suggest the presence of hexasomes, tetrasomes, and disomes in chromatin of living cells. All-atom models of PANS developed here provide a number of necessary building blocks for determining the landscape of genome-wide nucleosome occupation.
Relevance to DNA accessibility control
Access to regions of structurally protected DNA is expected to be severely impeded for cellular machinery responsible for key processes that require access to the DNA information content, in particular transcription. Indeed, the wrapping of the nucleosomal DNA around the intact core histones is extremely tight—the free energy cost of complete DNA unwrapping is ∼40 kcal/mol (75), which is much larger than a stability of a typical protein (88, 89). It is therefore not surprising that the question of how Pol II machinery overcomes this barrier to access the DNA has remained a fundamental open question in biology (25, 90). In general, the specifics of the structural mechanism through which access to nucleosomal DNA is granted may depend on the desired rate of the access dictated by the required transcription speed. Several lines of evidence (25) suggest that rather than actively unwrapping the DNA, the transcription machinery waits for spontaneous unwrapping fluctuations large enough for the polymerase to be able to transcribe through (91), with the partially unwrapped states possibly stabilized by bound proteins. However, while short DNA fragments near the entry/exit region wrap and rewrap with high frequency, providing rapid and efficient access to regulatory DNA target sites located there, the spontaneous DNA unwrapping rate decreases dramatically with distance inside the nucleosome (10). In fact, characteristic timescales for spontaneous DNA unwrapping deep inside the nucleosome can reach 10 min, and possibly even longer for sites further away from the entry/exit (10). This very slow unwrapping kinetics undoubtedly reduces the speed of Pol II machinery considerably—unimpeded, the latter can transcribe the entire nucleosomal DNA in seconds (65).
While a number of mechanisms may help Pol II approach the unimpeded speed limit of transcription in vivo, one can imagine a scenario in which the nucleosome itself becomes partially, or even completely, disassembled. Such a disassembly can be caused by, for example, a positive torsional stress accompanying the polymerase movement (92). In this scenario, Pol II would be transcribing through PANS rather than through the intact nucleosome (octasome), which would reduce the accessibility barriers dramatically. Note that the number of histone-DNA contacts, and thus the strength of the association, weakens progressively from the hexasome to the tetrasome to the disome. The corresponding timescales for the DNA wrapping/rewrapping are also expected to decrease dramatically. For example, for the disome in which only ∼40 bp are in contact with the positively charged core, one might expect the characteristic DNA unwrapping times to be less than hundreds of milliseconds—unwrapping time of the outer fragment of similar length in the nucleosome (10). The presence of the PANS would, therefore, speed up transcription appreciably. Indeed, widespread presence of subnucleosomal particles in dynamic chromatin of actively transcribing genes (23, 25) suggests that the existence of PANS may not only be a consequence, but also a prerequisite, for fast transcription in vivo.
Conclusions
The nucleosome is the fundamental unit of DNA compaction in cells; its structure is a key for the understanding of how access to the genomic information is regulated when needed by vital processes such as DNA transcription and replication. Recent evidence highlights the critical role of partially assembled states of the nucleosome, in which the DNA is more exposed than in the fully assembled nucleosome. In this work we have presented for the first time, to our knowledge, atomic-resolution models of several PANS—namely the disome, the tetrasome, and the hexasome—that naturally occur on the nucleosome assembly pathways, and are believed to be important for key biological processes related to processing and maintenance of genetic information carried by the DNA. One unexpected conclusion is that these PANS, even those (the tetrasome or the disome) that describe a significantly disassembled nucleosome, retain many key features of protein-DNA interaction of the intact nucleosome. At the same time, noticeable structural rearrangements are observed, in some cases leading to counterintuitive loss of accessibility to some histone residues compared to the intact nucleosome. Significant structural fluctuations are present and PANS structures are definitely highly dynamic, much more so than the intact nucleosome.
The PANS structures considered here can be natural precursors for the full nucleosome assembly that does not require slow rearrangements of misassembled intermediates, even if their stoichiometry is right. Drawing an analogy with the critical importance of atomic resolution structure of the nucleosome for interpretation of experiment and generation of novel, critical insight, we believe that the proposed atomistic models of PANS will become useful in many areas—in vivo, in vitro, and in silico.
We have demonstrated the utility of the atomistic models by proposing an alternative interpretation of a nontrivial pattern of lengths of protected DNA fragments in active genes of Drosophila; we have shown that the specific number of the DNA protection peaks and their peculiar positions observed in vivo, can be explained by the specific lengths of protected DNA identified in the PANS. Based on the insight, we have made a suggestion that the hexasome, the tetrasome, and the disome are present in actively transcribing regions in a substantial amount.
We have also illustrated utility of the atomistic models of subnucleosomal states for in vitro studies, including interpretation of AFM, FRET, and SAXS experiments. Here, the models help in species assignment, and provide a nuanced, detailed quantitative interpretation that would be challenging without fully atomistic, dynamical models.
Our contribution to methodology is, to our knowledge, a novel approach—combining AFM with molecular modeling to build realistic atomic resolution models of transient structural states, otherwise unavailable experimentally at this level of detail. In that sense, our approach bears an analogy with the use of low-resolution Cryo-EM in combination with atomistic modeling to produce fully atomistic models of large structures (93), including chromatin components (94, 95). Unlike Cryo-EM, our approach can be used at ambient conditions.
It is likely that the atomistic structures we constructed can be used as building blocks for in silico modeling of complexes formed by chromatin-interacting factors with PANS during processes of transcription, histone exchange, and histone modifications. An extension of our modeling approach to other biologically relevant subnucleosomal particles (for example, half-nucleosomes), combined with an analysis of genome-wide mapping data, can become useful in determination of genomic landscapes of alternative nucleosome structures and their biological roles.
Author Contributions
G.N.R. performed MD simulations and analyzed MD trajectories; A.V.I. analyzed MD trajectories and AFM data; I.B.N. performed AFM experiments and analyzed AFM data; A.V.S. performed MD simulations and calculated SAXS spectra; D.V.L. analyzed SAXS spectra; A.Y.K. analyzed biological implications of the PANS; V.V.I.-I. analyzed SAXS spectra; A.V.O. performed MD simulations; V.V.I.-I. and A.V.O. designed and supervised the study; all of the authors contributed to discussing the results and writing the article; and G.N.R. and A.V.I. made an equal contribution to this work.
Acknowledgments
Calculations and analysis of MD trajectories were performed using computational resources of federal center for collective usage at NRC Kurchatov Institute (http://computing.kiae.ru/). We thank Alexander Yakimov for technical support. We thank Dr. Suren Felekyan and Professor Claus Seidel for a fruitful discussion of the FRET data. We thank Professor Lois Pollack, Joshua Tokuda, and Yujie Chen for providing SAXS spectra from Chen et al. (32). A.V.O. thanks Professor Y. Lyubchenko for a useful discussion of the AFM data.
This work was supported by the Russian Foundation for Basic Research (obr-i 14-24-01103) and by the Ministry of Science and Education of the Russian Federation (8482 07.09.2012). A.V.O. acknowledges partial support from the National Institutes of Health (R01 GM099450 and GM076121 to A.V.O.); AFM nucleosome image processing was carried out at the Institute of Cytology, Russian Academy of Sciences, and was supported by the Russian Science Foundation (14-50-00068) and the Federal Agency of Scientific Organizations of the Russian Federation.
Editor: Tamar Schlick.
Footnotes
Georgy N. Rychkov and Andrey V. Ilatovskiy contributed equally to this article.
Molecular structures of PANS in PDB format, eight figures, six tables, and one movie are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(16)31044-X.
Supporting Citations
References (96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108) appear in the Supporting Material.
Supporting Material
{kind=link}
References
- 1.Lorch Y., LaPointe J.W., Kornberg R.D. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell. 1987;49:203–210. doi: 10.1016/0092-8674(87)90561-7. [DOI] [PubMed] [Google Scholar]
- 2.Gibney E.R., Nolan C.M. Epigenetics and gene expression. Heredity (Edinb) 2010;105:4–13. doi: 10.1038/hdy.2010.54. [DOI] [PubMed] [Google Scholar]
- 3.Olins A.L., Olins D.E. Spheroid chromatin units (ν bodies) Science. 1974;183:330–332. doi: 10.1126/science.183.4122.330. [DOI] [PubMed] [Google Scholar]
- 4.Woodcock C. Ultrastructure of inactive chromatin. J. Cell Biol. 1973;59:A368. [Google Scholar]
- 5.Kornberg R.D. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 6.Richmond T.J., Finch J.T., Klug A. Structure of the nucleosome core particle at 7 Å resolution. Nature. 1984;311:532–537. doi: 10.1038/311532a0. [DOI] [PubMed] [Google Scholar]
- 7.Luger K., Mäder A.W., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 8.Davey C.A., Sargent D.F., Richmond T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 9.Luger K., Dechassa M.L., Tremethick D.J. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012;13:436–447. doi: 10.1038/nrm3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tims H.S., Gurunathan K., Widom J. Dynamics of nucleosome invasion by DNA binding proteins. J. Mol. Biol. 2011;411:430–448. doi: 10.1016/j.jmb.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das C., Tyler J.K. Histone exchange and histone modifications during transcription and aging. Biochim. Biophys. Acta. 2013;1819:332–342. doi: 10.1016/j.bbagrm.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrews A.J., Luger K. Nucleosome structure(s) and stability: variations on a theme. Annu. Rev. Biophys. 2011;40:99–117. doi: 10.1146/annurev-biophys-042910-155329. [DOI] [PubMed] [Google Scholar]
- 13.Li G., Levitus M., Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 14.Böhm V., Hieb A.R., Langowski J. Nucleosome accessibility governed by the dimer/tetramer interface. Nucleic Acids Res. 2011;39 doi: 10.1093/nar/gkq1279. 3093–31029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donham D.C., 2nd, Scorgie J.K., Churchill M.E. The activity of the histone chaperone yeast Asf1 in the assembly and disassembly of histone H3/H4-DNA complexes. Nucleic Acids Res. 2011;39:5449–5458. doi: 10.1093/nar/gkr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zlatanova J., Bishop T.C., van Holde K. The nucleosome family: dynamic and growing. Structure. 2009;17:160–171. doi: 10.1016/j.str.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 17.Hutcheon T., Dixon G.H., Levy-Wilson B. Transcriptionally active mononucleosomes from trout testis are heterogeneous in composition. J. Biol. Chem. 1980;255:681–685. [PubMed] [Google Scholar]
- 18.Venkatesh S., Workman J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015;16:178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- 19.Barboro P., Pasini A., Patrone E. Chromatin changes in cell transformation: progressive unfolding of the higher-order structure during the evolution of rat hepatocyte nodules. A differential scanning calorimetry study. Biophys. J. 1993;65:1690–1699. doi: 10.1016/S0006-3495(93)81212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isaev-Ivanov V., Lebedev D., Filatov M. Comparative analysis of the nucleosome structure of cell nuclei by small-angle neutron scattering. Phys. Solid State. 2010;52:1063–1073. [Google Scholar]
- 21.Kent N.A., Adams S., Paszkiewicz K. Chromatin particle spectrum analysis: a method for comparative chromatin structure analysis using paired-end mode next-generation DNA sequencing. Nucleic Acids Res. 2011;39:e26. doi: 10.1093/nar/gkq1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henikoff J.G., Belsky J.A., Henikoff S. Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. USA. 2011;108:18318–18323. doi: 10.1073/pnas.1110731108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhee H., Bataille A., Pugh B. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell. 2014;159:1377–1388. doi: 10.1016/j.cell.2014.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bancaud A., Wagner G., Prunell A. Nucleosome chiral transition under positive torsional stress in single chromatin fibers. Mol. Cell. 2007;27:135–147. doi: 10.1016/j.molcel.2007.05.037. [DOI] [PubMed] [Google Scholar]
- 25.Teves S.S., Weber C.M., Henikoff S. Transcribing through the nucleosome. Trends Biochem. Sci. 2014;39:577–586. doi: 10.1016/j.tibs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Lusser A., Kadonaga J.T. Strategies for the reconstitution of chromatin. Nat. Methods. 2004;1:19–26. doi: 10.1038/nmeth709. [DOI] [PubMed] [Google Scholar]
- 27.Wilhelm F.X., Wilhelm M.L., Duane M.P. Reconstitution of chromatin: assembly of the nucleosome. Nucleic Acids Res. 1978;5:505–521. doi: 10.1093/nar/5.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gansen A., Valeri A., Seidel C.A. Nucleosome disassembly intermediates characterized by single-molecule FRET. Proc. Natl. Acad. Sci. USA. 2009;106:15308–15313. doi: 10.1073/pnas.0903005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrews A.J., Chen X., Luger K. The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol. Cell. 2010;37:834–842. doi: 10.1016/j.molcel.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henikoff S., Ramachandran S., Henikoff J.G. The budding yeast Centromere DNA Element II wraps a stable Cse4 hemisome in either orientation in vivo. eLife. 2014;3:e01861. doi: 10.7554/eLife.01861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teves S.S., Henikoff S. Heat shock reduces stalled RNA polymerase II and nucleosome turnover genome-wide. Genes Dev. 2011;25:2387–2397. doi: 10.1101/gad.177675.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y., Tokuda J.M., Pollack L. Revealing transient structures of nucleosomes as DNA unwinds. Nucleic Acids Res. 2014;42:8767–8776. doi: 10.1093/nar/gku562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lankaš F., Lavery R., Maddocks J.H. Kinking occurs during molecular dynamics simulations of small DNA minicircles. Structure. 2006;14:1527–1534. doi: 10.1016/j.str.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 34.Adcock S.A., McCammon J.A. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. 2006;106:1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaytan A.K., Armeev G.A., Panchenko A.R. Coupling between histone conformations and DNA geometry in nucleosomes on a microsecond timescale: atomistic insights into nucleosome functions. J. Mol. Biol. 2016;428:221–237. doi: 10.1016/j.jmb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wells D.B., Aksimentiev A. Mechanical properties of a complete microtubule revealed through molecular dynamics simulation. Biophys. J. 2010;99:629–637. doi: 10.1016/j.bpj.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shvetsov A.V., Lebedev D.V., Isaev-Ivanov V.V. Structure of RecX protein complex with the presynaptic RecA filament: molecular dynamics simulations and small angle neutron scattering. FEBS Lett. 2014;588:948–955. doi: 10.1016/j.febslet.2014.01.053. [DOI] [PubMed] [Google Scholar]
- 38.Bishop T.C. Molecular dynamics simulations of a nucleosome and free DNA. J. Biomol. Struct. Dyn. 2005;22:673–686. doi: 10.1080/07391102.2005.10507034. [DOI] [PubMed] [Google Scholar]
- 39.Ruscio J.Z., Onufriev A. A computational study of nucleosomal DNA flexibility. Biophys. J. 2006;91:4121–4132. doi: 10.1529/biophysj.106.082099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roccatano D., Barthel A., Zacharias M. Structural flexibility of the nucleosome core particle at atomic resolution studied by molecular dynamics simulation. Biopolymers. 2007;85:407–421. doi: 10.1002/bip.20690. [DOI] [PubMed] [Google Scholar]
- 41.Materese C.K., Savelyev A., Papoian G.A. Counterion atmosphere and hydration patterns near a nucleosome core particle. J. Am. Chem. Soc. 2009;131:15005–15013. doi: 10.1021/ja905376q. [DOI] [PubMed] [Google Scholar]
- 42.Biswas M., Langowski J., Bishop T. Atomistic simulations of nucleosomes. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013;3:378–392. [Google Scholar]
- 43.Nguyen H., Maier J., Simmerling C. Folding simulations for proteins with diverse topologies are accessible in days with a physics-based force field and implicit solvent. J. Am. Chem. Soc. 2014;136:13959–13962. doi: 10.1021/ja5032776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anandakrishnan R., Drozdetski A., Onufriev A.V. Speed of conformational change: comparing explicit and implicit solvent molecular dynamics simulations. Biophys. J. 2015;108:1153–1164. doi: 10.1016/j.bpj.2014.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erler J., Zhang R., Langowski J. The role of histone tails in the nucleosome: a computational study. Biophys. J. 2014;107:2911–2922. doi: 10.1016/j.bpj.2014.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Götz A.W., Williamson M.J., Walker R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Case D.A., Cheatham T.E., 3rd, Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsui V., Case D. Molecular dynamics simulations of nucleic acids using a generalized Born solvation model. J. Am. Chem. Soc. 2000;122:2489–2498. [Google Scholar]
- 49.Chocholousová J., Feig M. Implicit solvent simulations of DNA and DNA-protein complexes: agreement with explicit solvent vs experiment. J. Phys. Chem. B. 2006;110:17240–17251. doi: 10.1021/jp0627675. [DOI] [PubMed] [Google Scholar]
- 50.Onufriev A. Continuum electrostatics solvent modeling with the generalized Born model. In: Feig M., editor. Modeling Solvent Environments. Wiley; Weinheim, Germany: 2010. pp. 127–165. [Google Scholar]
- 51.Pronk S., Páll S., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jorgensen W., Chandrasekhar J., Klein M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 53.Luger K., Rechsteiner T.J., Richmond T.J. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 54.Gansen A., Hauger F., Langowski J. Single-pair fluorescence resonance energy transfer of nucleosomes in free diffusion: optimizing stability and resolution of subpopulations. Anal. Biochem. 2007;368:193–204. doi: 10.1016/j.ab.2007.04.047. [DOI] [PubMed] [Google Scholar]
- 55.Shvetsov A., Schmidt A., Isaev-Ivanov V. Method for calculating small-angle neutron scattering spectra using all-atom molecular dynamics trajectories. J. Surf. Invest. X-ray, Synchrotron Neutron Tech. 2013;7:1124–1127. [Google Scholar]
- 56.Cromer D., Mann J. X-ray scattering factors computed from numerical Hartree-Fock wave functions. Acta Crystallogr. A. 1968;24:321–324. [Google Scholar]
- 57.Dyer P.N., Edayathumangalam R.S., Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- 58.Lowary P.T., Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 59.Lyubchenko Y., Shlyakhtenko L., Gall A. DNA-Protein Interactions. Humana Press; Totowa, NJ: 2009. Atomic force microscopy imaging and probing of DNA, proteins, and protein-DNA complexes: silatrane surface chemistry. [DOI] [PubMed] [Google Scholar]
- 60.Henderson R.M., Schneider S., Oberleithner H. Imaging ROMK1 inwardly rectifying ATP-sensitive K+ channel protein using atomic force microscopy. Proc. Natl. Acad. Sci. USA. 1996;93:8756–8760. doi: 10.1073/pnas.93.16.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duong T. ks: Kernel density estimation and kernel discriminant analysis for multivariate data in R. Roy. J. Stat. Soft. 2007;21:7. [Google Scholar]
- 62.Henzler-Wildman K.A., Lei M., Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450:913–916. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 63.Ruscio J.Z., Kumar D., Onufriev A.V. Atomic level computational identification of ligand migration pathways between solvent and binding site in myoglobin. Proc. Natl. Acad. Sci. USA. 2008;105:9204–9209. doi: 10.1073/pnas.0710825105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schlick T., Collepardo-Guevara R., Xiao X. Biomolecular modeling and simulation: a field coming of age. Q. Rev. Biophys. 2011;44:191–228. doi: 10.1017/S0033583510000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shermoen A.W., O’Farrell P.H. Progression of the cell cycle through mitosis leads to abortion of nascent transcripts. Cell. 1991;67:303–310. doi: 10.1016/0092-8674(91)90182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luger K., Richmond T.J. DNA binding within the nucleosome core. Curr. Opin. Struct. Biol. 1998;8:33–40. doi: 10.1016/s0959-440x(98)80007-9. [DOI] [PubMed] [Google Scholar]
- 67.Hall M.A., Shundrovsky A., Wang M.D. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 2009;16:124–129. doi: 10.1038/nsmb.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ettig R., Kepper N., Rippe K. Dissecting DNA-histone interactions in the nucleosome by molecular dynamics simulations of DNA unwrapping. Biophys. J. 2011;101:1999–2008. doi: 10.1016/j.bpj.2011.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bondarenko V.A., Steele L.M., Studitsky V.M. Nucleosomes can form a polar barrier to transcript elongation by RNA polymerase II. Mol. Cell. 2006;24:469–479. doi: 10.1016/j.molcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 70.Kulaeva O.I., Gaykalova D.A., Studitsky V.M. Mechanism of chromatin remodeling and recovery during passage of RNA polymerase II. Nat. Struct. Mol. Biol. 2009;16:1272–1278. doi: 10.1038/nsmb.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jin J., Bai L., Wang M.D. Synergistic action of RNA polymerases in overcoming the nucleosomal barrier. Nat. Struct. Mol. Biol. 2010;17:745–752. doi: 10.1038/nsmb.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berdasco M., Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev. Cell. 2010;19:698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 73.Cosgrove M.S., Boeke J.D., Wolberger C. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 2004;11:1037–1043. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 74.Tessarz P., Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014;15:703–708. doi: 10.1038/nrm3890. [DOI] [PubMed] [Google Scholar]
- 75.Fenley A.T., Adams D.A., Onufriev A.V. Charge state of the globular histone core controls stability of the nucleosome. Biophys. J. 2010;99:1577–1585. doi: 10.1016/j.bpj.2010.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tan M., Luo H., Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 2008;9:15–26. doi: 10.1038/nrg2206. [DOI] [PubMed] [Google Scholar]
- 78.Arya G., Schlick T. Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci. USA. 2006;103:16236–16241. doi: 10.1073/pnas.0604817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Collepardo-Guevara R., Portella G., Orozco M. Chromatin unfolding by epigenetic modifications explained by dramatic impairment of internucleosome interactions: a multiscale computational study. J. Am. Chem. Soc. 2015;137:10205–10215. doi: 10.1021/jacs.5b04086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thåström A., Gottesfeld J.M., Widom J. Histone-DNA binding free energy cannot be measured in dilution-driven dissociation experiments. Biochemistry. 2004;43:736–741. doi: 10.1021/bi0302043. [DOI] [PubMed] [Google Scholar]
- 81.Claudet C., Angelov D., Bednar J. Histone octamer instability under single molecule experiment conditions. J. Biol. Chem. 2005;280:19958–19965. doi: 10.1074/jbc.M500121200. [DOI] [PubMed] [Google Scholar]
- 82.Nazarov I., Chekliarova I., Tomilin A. AFM studies in diverse ionic environments of nucleosomes reconstituted on the 601 positioning sequence. Biochimie. 2016;121:5–12. doi: 10.1016/j.biochi.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 83.Thåström A., Lowary P.T., Widom J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 84.Feigin L., Svergun D. Plenum Press; New York: 1987. Structure Analysis by Small-angle X-ray and Neutron Scattering. [Google Scholar]
- 85.Svergun D., Barberato C., Koch M. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 86.Katan-Khaykovich Y., Struhl K. Splitting of H3-H4 tetramers at transcriptionally active genes undergoing dynamic histone exchange. Proc. Natl. Acad. Sci. USA. 2011;108:1296–1301. doi: 10.1073/pnas.1018308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang C., Zhang Z., Zhu B. H3.3-H4 tetramer splitting events feature cell-type specific enhancers. PLoS Genet. 2013;9:e1003558. doi: 10.1371/journal.pgen.1003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Onufriev A., Case D.A., Bashford D. Structural details, pathways, and energetics of unfolding apomyoglobin. J. Mol. Biol. 2003;325:555–567. doi: 10.1016/s0022-2836(02)01207-x. [DOI] [PubMed] [Google Scholar]
- 89.Dill K.A., Ozkan S.B., Weikl T.R. The protein folding problem. Annu. Rev. Biophys. 2008;37:289–316. doi: 10.1146/annurev.biophys.37.092707.153558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kulaeva O.I., Hsieh F.-K., Studitsky V.M. Mechanism of transcription through a nucleosome by RNA polymerase II. Biochim. Biophys. Acta. 2013;1829:76–83. doi: 10.1016/j.bbagrm.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hodges C., Bintu L., Bustamante C. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science. 2009;325:626–628. doi: 10.1126/science.1172926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Levchenko V., Jackson B., Jackson V. Histone release during transcription: displacement of the two H2A-H2B dimers in the nucleosome is dependent on different levels of transcription-induced positive stress. Biochemistry. 2005;44:5357–5372. doi: 10.1021/bi047786o. [DOI] [PubMed] [Google Scholar]
- 93.Zhao G., Perilla J.R., Zhang P. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature. 2013;497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Villa E., Sengupta J., Frank J. Ribosome-induced changes in elongation factor Tu conformation control GTP hydrolysis. Proc. Natl. Acad. Sci. USA. 2009;106:1063–1068. doi: 10.1073/pnas.0811370106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wong H., Victor J.-M., Mozziconacci J. An all-atom model of the chromatin fiber containing linker histones reveals a versatile structure tuned by the nucleosomal repeat length. PLoS One. 2007;2:e877. doi: 10.1371/journal.pone.0000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Anandakrishnan R., Aguilar B., Onufriev A.V. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012;40:W537–W541. doi: 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Anandakrishnan R., Daga M., Onufriev A.V. An N log N generalized Born approximation. J. Chem. Theory Comput. 2011;7:544–559. doi: 10.1021/ct100390b. [DOI] [PubMed] [Google Scholar]
- 98.Essmann U., Perera L., Pedersen L. A smooth particle mesh Ewald potential. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 99.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 100.Berendsen H., Postma J., Haak J. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 101.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 102.Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984;52:255–268. [Google Scholar]
- 103.McNally R., Bowman G.D., Kuriyan J. Analysis of the role of PCNA-DNA contacts during clamp loading. BMC Struct. Biol. 2010;10:3. doi: 10.1186/1472-6807-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Putnam C.D., Hammel M., Tainer J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 105.Bernadó P., Mylonas E., Svergun D.I. Structural characterization of flexible proteins using small-angle x-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 106.Hura G.L., Menon A.L., Tainer J.A. Robust, high-throughput solution structural analyses by small angle x-ray scattering (SAXS) Nat. Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miller S., Janin J., Chothia C. Interior and surface of monomeric proteins. J. Mol. Biol. 1987;196:641–656. doi: 10.1016/0022-2836(87)90038-6. [DOI] [PubMed] [Google Scholar]
- 108.Wand M., Jones M. Chapman and Hall/CRC; Boca Raton, FL: 1995. Kernel Smoothing. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.