

Abstract
CRISPR-Cas systems provide microbes with adaptive immunity by employing short sequences, termed spacers, that guide Cas proteins to cleave foreign DNA1,2. Class 2 CRISPR-Cas systems are streamlined versions in which a single Cas protein bound to RNA recognizes and cleaves targeted sequences3,4. The programmable nature of these minimal systems has enabled their repurposing as a versatile technology that is broadly revolutionizing biological and clinical research5. However, current CRISPR-Cas technologies are based solely on systems from isolated bacteria, leaving untapped the vast majority of enzymes from organisms that have not been cultured. Metagenomics, the sequencing of DNA extracted from natural microbial communities, provides access to the genetic material of a huge array of uncultivated organisms6,7. Here, using genome-resolved metagenomics, we identified novel CRISPR-Cas systems, including the first reported Cas9 in the archaeal domain of life. This divergent Cas9 protein was found in little-studied nanoarchaea as part of an active CRISPR-Cas system. In bacteria, we discovered two previously unknown systems, CRISPR-CasX and CRISPR-CasY, which are among the most compact systems yet identified. Notably, all required functional components were identified by metagenomics, enabling validation of robust in vivo RNA-guided DNA interference activity in E. coli. Interrogation of environmental microbial communities combined with in vivo experiments allows access to an unprecedented diversity of genomes whose content will expand the repertoire of microbe-based biotechnologies.
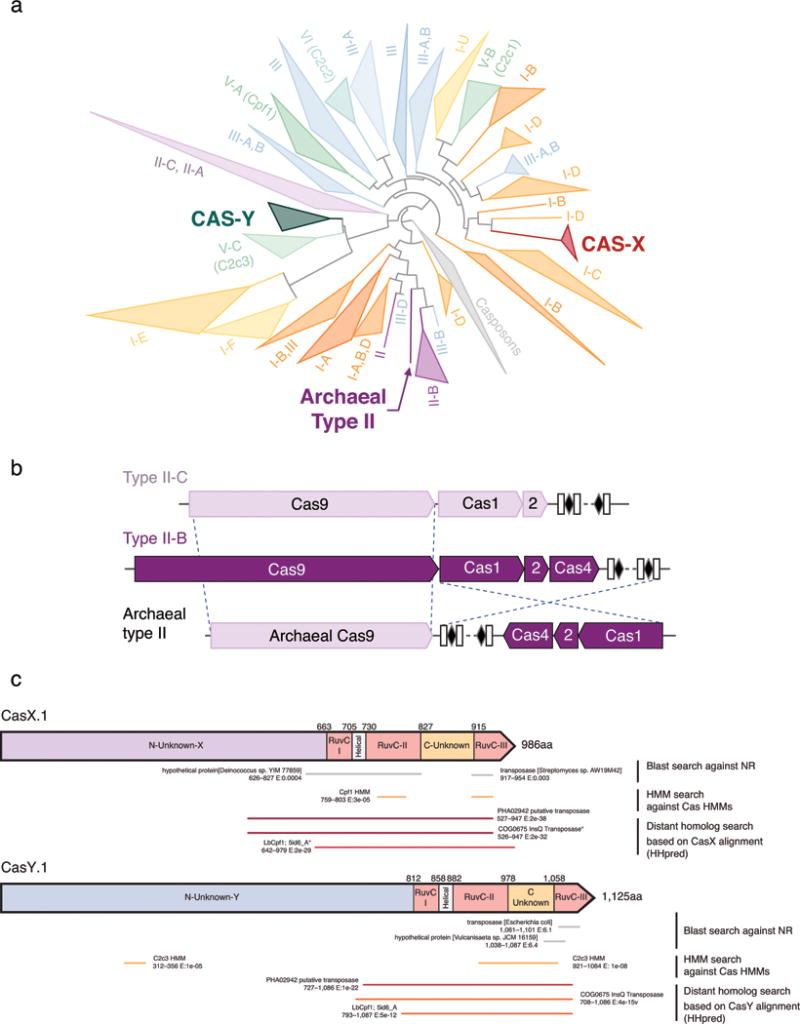
We sought to identify previously unknown class 2 CRISPR-Cas systems in terabase-scale metagenomic datasets from groundwater, sediment, acid mine drainage biofilms, soil, infant gut, and other microbial communities. Our analyses targeted large uncharacterized genes proximal to a CRISPR array and cas1, the universal CRISPR integrase8–10. Among the 155 million protein-coding genes analyzed, we identified the first Cas9 proteins in domain Archaea, and discovered in uncultivated bacteria two new CRISPR-Cas systems, which we refer to as CRISPR-CasX and CRISPR-CasY (Fig. 1). Both the archaeal Cas9 and CasY are encoded exclusively in the genomes of organisms from lineages with no known isolated representatives.
Figure 1. CRISPR-Cas systems identified in uncultivated organisms.
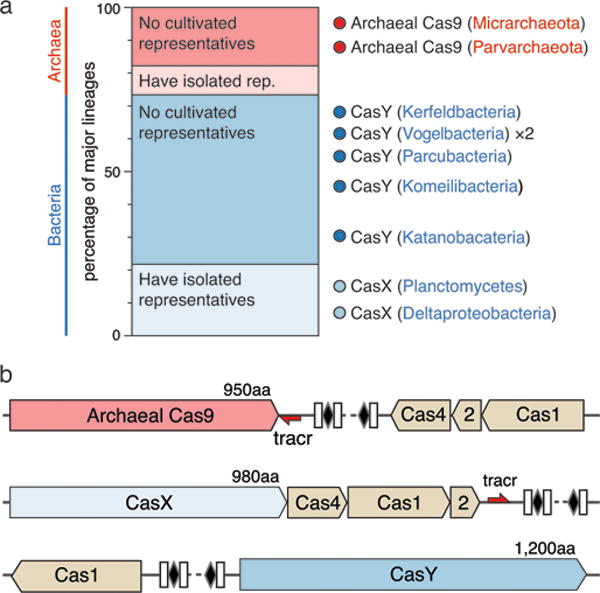
a, Ratio of lineages with and without isolated representatives in Bacteria and Archaea, based on 31 major lineages described by Hug et al. (2016)29. The results highlight the massive scale of as-yet little investigated biology in these domains. Archaeal Cas9 and the novel CRISPR-CasY were found exclusively in lineages with no isolated representatives. b, Locus organization of the discovered CRISPR-Cas systems.
One of the hallmarks of CRISPR-Cas9 (type II) systems was their presumed presence only in the bacterial domain3,11. We were therefore surprised to discover Cas9 proteins encoded in genomes of the nanoarchaea ARMAN-1 (Candidatus Micrarchaeum acidiphilum ARMAN-1) and ARMAN-4 (Candidatus Parvarchaeum acidiphilum ARMAN-4)12,13 in acid-mine drainage (AMD) metagenomic datasets (Extended Data Table 1 and Extended Data Fig. 1). These findings expand the occurrence of Cas9-containing CRISPR systems to another domain of life.
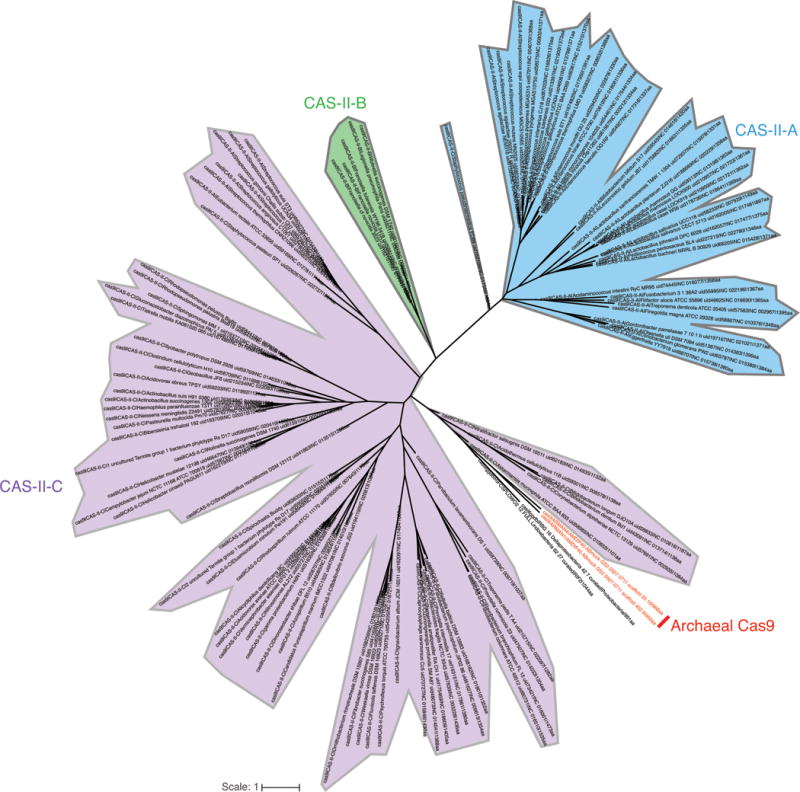
The CRISPR-Cas locus in ARMAN-1 includes large CRISPR arrays adjacent to cas1, cas2, cas4 and cas9 genes. This system was found on highly similar contigs (average nucleotide identity of 99.7% outside of the CRISPR array) reconstructed independently from 16 different samples. We reconstructed numerous alternative ARMAN-1 CRISPR arrays with a largely conserved end (likely comprised of the oldest spacers) and a variable region into which many distinct spacers have been incorporated (Fig. 2a, Extended Data Fig. 2, and Supplementary Table 1). Given the polarity of the array, we predict that the ~200 bp region between the end of the Cas9 gene and the variable end of the array likely contains the leader sequence and transcriptional start site. Based on the hypervariability in spacer content, we conclude that the ARMAN-1 CRISPR-Cas9 system is active in the sampled populations. Phylogenetic analysis of Cas1 (Extended Data Fig. 3a) suggests that this archaeal CRISPR-Cas system does not clearly fall into any existing type II subtype. The presence of cas4, affiliate it with type II-B systems3,11, yet the Cas9 sequence is more similar to type II-C proteins (Extended Data Fig. 4, Supplementary Data 3). Thus, the archaeal type II system may have arisen as a fusion of type II-C and II-B systems (Extended Data Fig. 3b).
Figure 2. ARMAN-1 CRISPR array diversity and identification of the ARMAN-1 Cas9 PAM sequence.
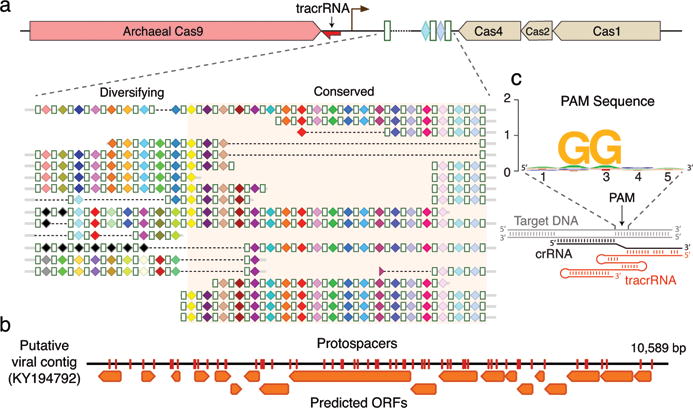
a, CRISPR arrays reconstructed from AMD samples. White boxes indicate repeats and colored diamonds indicate spacers (identical spacers are similarly colored; unique spacers are in black). The conserved region of the array is highlighted. The diversity of recently acquired spacers (on the left) indicates the system is active. Analysis of within-population CRISPR variability is presented in Extended Data Fig. 2. b, A single circular, putatively viral, contig contains 56 protospacers (red vertical bars) from the ARMAN-1 CRISPR arrays. c, Sequence analysis of 240 protospacers (Supplementary Table 1) revealed a conserved ‘NGG’ PAM downstream to the protospacers.
Fifty-six of the spacers of the ARMAN-1 CRISPR-Cas9 system target a 10 kbp circular sequence that encodes mostly short hypothetical proteins, and is likely an ARMAN-1 virus (Fig. 2b). Indeed, cryo-electron tomographic reconstructions often identified viral particles attached to ARMAN cells12,14. ARMAN-1 protospacers also derived from a putative transposon within the genome of ARMAN-2 (another nanoarchaeon13) and a putative mobile element in the genomes of Thermoplasmatales archaea, including that of I-plasma15 from the same ecosystem (Extended Data Fig. 5). Direct cytoplasmic “bridges” were observed between ARMAN and Thermoplasmatales cells, implying a close relationship between them12,14. The ARMAN-1 CRISPR-Cas9 may thus defend against transposon propagation between these organisms, a role that is reminiscent of piRNA-mediated defense against transposition in the eukaryotic germ line16.
Unlike the ARMAN-1 CRISPR-Cas system, the ARMAN-4 cas9 gene has only one adjacent CRISPR repeat-spacer unit and no other cas genes in its vicinity (Extended Data Fig. 6). The lack of a typical CRISPR array and cas1 points to a system with no capacity to acquire additional spacers. No target could be identified for the spacer sequence, but given the conservation of the locus in samples collected over several years, we cannot rule out it is functional as a “single-target” CRISPR-Cas system. Conservation of a single spacer may indicate that the ARMAN-4 Cas9 exerts an alternative role, such as gene regulation17 or involvement in cell-cell interactions18.
Active DNA-targeting CRISPR-Cas systems use 2 to 4 nt protospacer-adjacent motifs (PAMs) located next to target sequences for self versus non-self discrimination19,20. Examining sequences adjacent to the genomic target sequences revealed a strong ‘NGG’ PAM preference in ARMAN-1 (Fig. 2c). Cas9 also employs two separate transcripts, CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA), for RNA-guided DNA cleavage21. We identified a putative tracrRNA in the vicinity of both ARMAN-1 and ARMAN-4 CRISPR-Cas9 systems (Extended Data Fig. 7). Previously, it was suggested that type II CRISPR systems were absent in archaea due to a lack of the host factor, RNase III, responsible for crRNA-tracrRNA guide complex maturation11,22. Notably, no RNase III homologs were identified in the ARMAN-1 genome (estimated to be 95% complete) and no internal promoters are predicted for the CRISPR array23, suggesting an as-yet undetermined mechanism of guide RNA production. Biochemical experiments to test cleavage activity of ARMAN-1 and ARMAN-4 Cas9 proteins purified from both E. coli and yeast and in vivo E. coli targeting assays did not reveal any detectable activity (see Extended Data Table 2 and Extended Data Fig. 7). Given the unique physiology and ecological niche of these nanoarchaea, lack of activity may be due to a post-translational modification or a co-factor absent in the experimental expression systems.
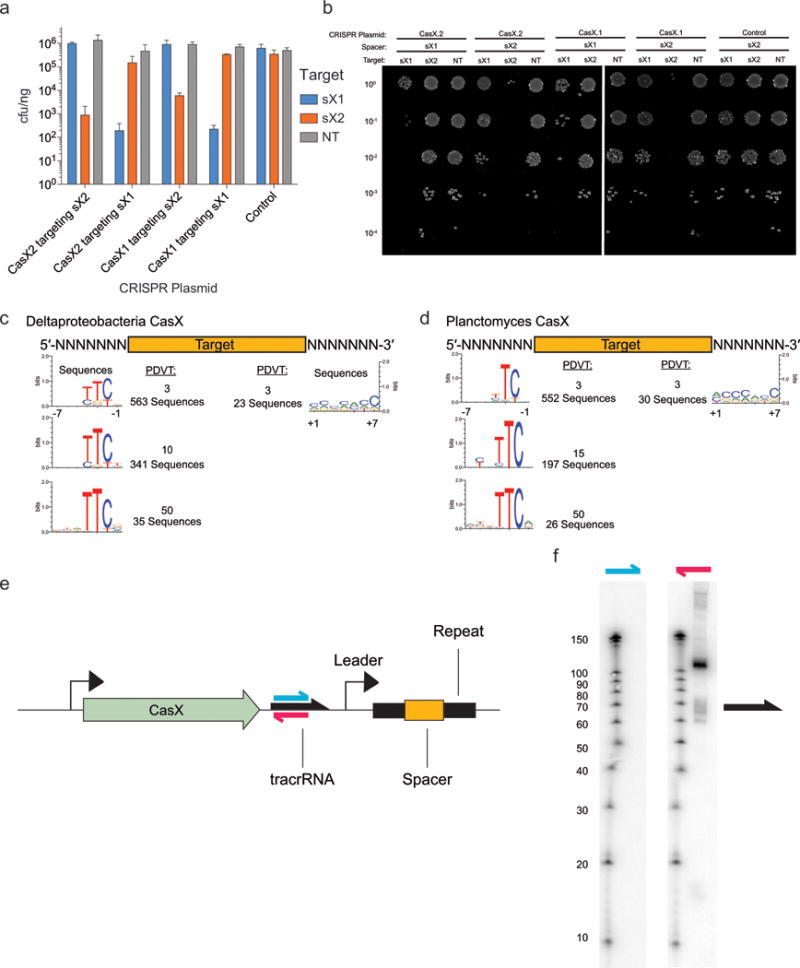
In addition to Cas9, only three families of class 2 Cas effector proteins have been discovered and experimentally validated: Cpf1, C2c1, and C2c24,24,25. Another gene, c2c3, which was identified only on small DNA fragments, has been suggested to also encode such a protein4. We hypothesized that other distinct types of effector proteins might exist within uncultivated microbes whose genomes were reconstructed from our metagenomic datasets. Indeed, a new type of class 2 CRISPR-Cas system was found in the genomes of two bacteria recovered from groundwater and sediment samples26. This system includes Cas1, Cas2, Cas4 and an uncharacterized ~980 aa protein that we refer to as CasX. The high conservation (68% protein sequence identity, Supplementary Data 1) of this protein in two organisms belonging to different phyla, Deltaproteobacteria and Planctomycetes, suggests a recent cross-phyla transfer29. The CRISPR arrays associated with each CasX had highly similar repeats (86% identity) of 37 nt, spacers of 33–34 nt, and a putative tracrRNA between the Cas operon and the CRISPR array (Fig. 1b, Extended Data Table 1). BLAST searches revealed only weak similarity (e-value > 1×10−4) to transposases, with similarity restricted to specific regions of the CasX C-terminus. Distant homology detection and protein modeling identified a RuvC domain near the CasX C-terminal end, with organization reminiscent of that found in type V CRISPR-Cas systems (Extended Data Fig. 3c). The rest of the CasX protein (630 N-terminal amino acids) showed no detectable similarity to any known protein, suggesting this is a novel class 2 effector. The combination of tracrRNA and separate Cas1, Cas2 and Cas4 proteins is unique among type V systems, and phylogenetic analyses indicate that the Cas1 from the CRISPR-CasX system is distant from those of any other known type V (Extended Data Fig. 3a). Further, CasX is considerably smaller than any known type V proteins: 980 aa compared to a typical size of ~1,200 aa for Cpf1, C2c1 and C2c3.
To test whether CasX would be capable of RNA-guided DNA targeting analogous to Cas9 and Cpf1 proteins, we synthesized a plasmid encoding a minimal CRISPR-CasX locus including casX, a short repeat-spacer array and intervening noncoding regions. We found that when expressed in E. coli, this minimal locus blocked transformation by a plasmid bearing a target sequence identified by metagenomic analysis (Fig. 3a–c, Extended Data Fig. 8). Furthermore, interference with transformation occurred only when the spacer sequence in the mini-locus matched the protospacer sequence in the plasmid target.
Figure 3. CRISPR-CasX is a dual-guided system that mediates programmable DNA interference in E. coli.
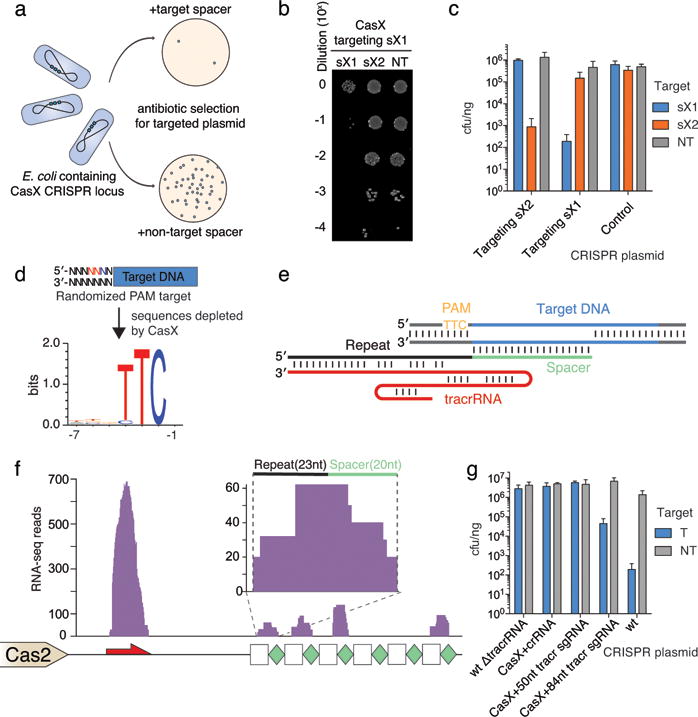
a, Diagram of CasX plasmid interference assays. b, Serial dilution of E. coli expressing the Planctomycetes CasX locus with spacer 1 (sX1) and transformed with the specified target (sX1, CasX protospacer 1; sX2, CasX protospacer 2; NT, non-target). c, Plasmid interference by Deltaproteobacteria CasX, using the same spacers and targets as in (b). d, PAM depletion assays for the Planctomycetes CasX locus expressed in E. coli. Sequence logo was generated from PAM sequences depleted > 30-fold compared to a control library (see also Extended Data Fig. 8). e, Diagram of CasX DNA interference. f, Mapping of environmental RNA sequences to the CasX CRISPR locus (red arrow, putative tracrRNA; white boxes, repeats; green diamonds, spacers); Inset: detailed view of mapping to first repeat and spacer. g, Plasmid interference assays with the putative tracrRNA knocked out of the CasX locus and CasX coexpressed with a crRNA alone, a truncated sgRNA or a full length sgRNA (T, target; NT, non-target). Experiments presented in (c) and (g) were conducted in triplicate and mean ± s.d. is shown.
To identify a PAM sequence for CasX, we repeated the transformation assay in E. coli using a plasmid containing either a 5′ or 3′ randomized sequence adjacent to the target site. This analysis revealed a stringent preference for the sequence ‘TTCN’ located 5′ of the protospacer sequence (Fig. 3d). No 3′ PAM preference was observed (Extended Data Fig. 8). Consistent with this finding, we observed that ‘TTCA’ is the sequence found upstream of the putative Deltaproteobacteria CRISPR-CasX protospacer that was identified in the environmental samples. Notably, both CRISPR-CasX loci share the same PAM sequence, in line with their high degree of protein sequence homology.
Examples of both single-RNA and dual-RNA guided systems exist among type V CRISPR loci. We used environmental RNA (metatranscriptomic) data to determine whether CasX requires a tracrRNA for DNA targeting activity. This analysis revealed a non-coding RNA transcript with sequence complementarity to the CRISPR repeat encoded between the Cas2 open reading frame and the CRISPR array (Fig. 3e). To check for expression of this non-coding RNA in E. coli expressing the CasX locus, Northern blots were conducted against this transcript in both directions (Extended Data Fig. 8). The results showed expression of a transcript of ~110 nt encoded on the same strand as the casX gene, with a more heterogeneous transcript of ~60–70 nt, suggesting that the leader sequence for the CRISPR array lies between the tracrRNA and the array. Transcriptomic mapping further suggests that the CRISPR RNA (crRNA) is processed to include ~23 nt of the repeat and 20 nt of the adjacent spacer, similar to the crRNA processing that occurs in CRISPR-Cas9 systems21,22 (Fig. 3f). To determine the dependence of CasX activity on the putative tracrRNA, we deleted this region from the minimal CRISPR-CasX locus described above, and repeated the plasmid interference assays. Deletion of the putative tracrRNA-encoding sequence from the CasX plasmid abolished robust transformation interference (Fig. 3g). This putative tracrRNA was joined with the processed crRNA using a tetraloop to form a single-guide RNA (sgRNA)21. While expression using a heterologous promoter of the crRNA alone or a shortened version of the sgRNA did not have any significant plasmid interference, expression of the full-length sgRNA conferred resistance to plasmid transformation (Fig. 3g). Together, these results establish CasX as a functional DNA-targeting, dual-RNA guided CRISPR-associated protein.
We identified another new class 2 Cas protein encoded in the genomes of certain candidate phyla radiation (CPR) bacteria6,29 (Fig. 1, Extended Data Table 1). These bacteria typically have small cell sizes, very small genomes and a limited biosynthetic capacity6,30–32, indicating they are most likely symbionts6,30–33. The ~1,200 aa Cas protein, which we named CasY, appears to be part of a minimal CRISPR-Cas system that includes Cas1 and a CRISPR array (Fig. 4a). Most of the CRISPR arrays have unusually short spacers of 17–19 nt, but one system, which lacks Cas1 (CasY.5), has longer spacers (27–29 nt). No predicted tracrRNA was detected in the vicinity of CRISPR-CasY, based on partial complementarity to the repeat sequences; however, we had insufficient metatranscriptomic data mapped to the CasY loci to detect potential tracrRNA sequences. Thus, we cannot exclude the dependence of CasY on a tracrRNA for robust interference from the available data.
Figure 4. Expression of a CasY locus in E. coli is sufficient for DNA interference.
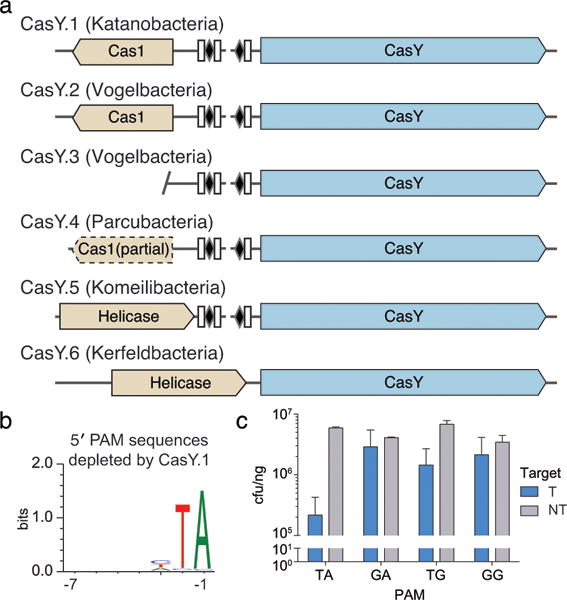
a, Diagrams of CasY loci and neighboring proteins. b, Sequence logo of the 658 5′ PAM sequences depleted greater than 3-fold by CasY relative to a control library. c, Plasmid interference by E. coli expressing CasY.1 and CRISPR array expressed with a heterologous promoter and transformed with targets containing the indicated PAM. Experiments were conducted in triplicate and mean ± s.d. is shown.
The six examples of CasY proteins we identified had no significant sequence similarity to any protein in public databases. A sensitive search using profile Hidden Markov Models (HMMs)34 built from published Cas proteins3,4 indicated that four of the six CasY proteins had local similarities (e-values 4×10−11– 3×10−18) to C2c3 in the C-terminal region overlapping the RuvC domains and a small region (~45 aa) of the N-terminal region (see Extended Data Fig. 3c). The remaining two CasY proteins had no significant similarity to C2c3s, despite sharing significant sequence similarity (best Blast hits: e-values 6×10−85, 7×10−75) with the other CasY proteins (Supplementary Data 2). C2c3 proteins are putative type V Cas effectors4 that were identified on short contigs with no taxonomic affiliation, and have not been validated experimentally. Strikingly, both CRISPR-CasY and C2c3 were found next to arrays with short spacers and within loci lacking Cas2, a protein considered essential for integrating DNA into the CRISPR array9,35. It remains to be seen whether these type V systems are functional for spacer acquisition.
Given the low homology of CRISPR-CasY to any experimentally validated CRISPR loci, we wondered whether this system confers RNA-guided DNA interference, but due to the short spacer length we did not have reliable information about a possible PAM motif that might be required for such activity. To work around this, the entire CRISPR-CasY.1 locus was synthesized with a shortened CRISPR array and introduced into E. coli on a plasmid vector. These cells were then challenged in a transformation assay using a target plasmid with a sequence matching a spacer in the array and containing an adjacent randomized 5′ or 3′ region to identify a possible PAM. Analysis of transformants revealed depletion of sequences containing a 5′ TA directly adjacent to the targeted sequence (Fig. 4b). Based on the identified PAM sequence, the CasY.1 locus was overexpressed using a heterologous promoter and tested against plasmids containing single PAMs. Plasmid interference was strongest in the presence of a target containing the identified 5′ TA PAM sequence (Fig. 4c). Thus, we conclude that CRISPR-CasY has DNA interference activity.
The systems described here are some of the most compact CRISPR-Cas loci identified to date and are found exclusively in metagenomic datasets. The small number of proteins that are required for interference and their relatively short length make these systems especially valuable for the development of genome editing tools. Interestingly, some of these compact loci were identified in organisms with very small genomes. As a consequence of their small genome size, these organisms likely depend on other community members for basic metabolic requirements and thus have remained largely outside the scope of traditional cultivation-based methods. For CasX and CasY, genomic context was critical for predicting functions that would not have been evident from unassembled sequence information. Furthermore, the identification of a putative tracrRNA, as well as targeted sequences uncovered through analysis of the genome-resolved metagenomic data, guided functional testing. Importantly, we show that metagenomic discoveries related to CRISPR-Cas systems are not restricted to in silico observations, but can be introduced into an experimental setting where their activity can be analyzed. Given that virtually all environments where life exists can now be probed by metagenomic methods, we anticipate that the combined computational-experimental approach will greatly expand the diversity of known CRISPR-Cas systems, enabling new technologies for biological research and clinical applications.
METHODS
Metagenomics and metatranscriptomics
Metagenomic samples from three different sites were analyzed: (1) Acid mine drainage (AMD) samples collected between 2006 and 2010 from the Richmond Mine, Iron Mountain, California36,37 (2) Groundwater and sediment samples collected between 2007 and 2013 from the Rifle Integrated Field Research (IFRC) site, adjacent to the Colorado River near Rifle, Colorado6,26. (3) Groundwater collected in 2009 and 2014 from Crystal Geyser, a cold, CO2-driven geyser on the Colorado Plateau in Utah38.
For the AMD data, DNA extraction methods and short read sequencing were reported by Denef and Banfield (2012)36 and Miller et al. (2011)37. For the Rifle data, DNA extraction, sequencing, assembly, and genome reconstruction were described by Anantharaman et al. (2016)26 and Brown et al. (2015)6. For samples from Crystal Geyser, methods follow those described by Probst et al. (2016)38 and Emerson et al. (2016)39. Rifle metatranscriptomic data was used from the data reported by Brown et al. (2015)6.
Briefly, DNA was extracted from samples using the PowerSoil DNA Isolation Kit (MoBio Laboratories Inc., Carlsbad, CA, USA). RNA was extracted from 0.2 μm filters collected from six 2011 Rifle groundwater samples. Following RNA extraction using the Invitrogen TRIzol reagent, DNA removal was done with the Qiagen RNase-Free DNase Set and Qiagen Mini RNeasy kits, and cDNA template library was generated using the Applied Biosystems SOLiD Total RNA-Seq kit. DNA was sequenced on Illumina HiSeq2000 platform, and Metatrancriptomic cDNA on 5500XL SOLiD platform after emulsion clonal bead amplification using the SOLiD EZ Bead system (Life Technologies). For the Crystal Geyser data and reanalysis of the AMD data, sequences were assembled using IDBA-UD40. DNA and RNA (cDNA) read-mapping used to determine sequencing coverage and gene expression, respectively, was performed using Bowtie241. Open reading frames (ORFs) were predicted on assembled scaffolds using Prodigal42. Scaffolds from the Crystal Geyser dataset were binned on the basis of differential coverage abundance patterns using a combination of ABAWACA6, ABAWACA2 (https://github.com/CK7), Maxbin243, and tetranucleotide frequency using Emergent Self-Organizing Maps (ESOM)44. Genomes were manually curated using % GC content, taxonomic affiliation, and genome completeness. Scaffolding errors were corrected using ra2.py (https://github.com/christophertbrown).
CRISPR-Cas computation analyses
The assembled contigs from the various samples were scanned for known Cas proteins using Hidden Markov Model (HMMs) profiles, which were built using the HMMer suite34, based on alignments from Makarova et al. (2015)3 and Shmakov et al. (2015)4. CRISPR arrays were identified using a local version of the CrisprFinder software45. Loci that contained both Cas1 and a CRISPR array were further analyzed if one of the ten ORFs adjacent to the cas1 gene encoded for an uncharacterized protein larger than 800 aa, and no known cas interference genes were identified on the same contig. These large proteins were further analyzed as potential class 2 Cas effectors. The potential effectors were clustered to protein families based on sequence similarities using MCL46. These protein families were expanded by building HMMs representing each of these families, and using them to search the metagenomic datasets for similar Cas proteins. To compare the identified protein families to known proteins, homologs were searched using BLAST47 against NCBI’s non-redundant (nr) and metagenomic (env_nr) protein databases, as well as HMM searches against the UniProt KnowledegeBase34,48. Only proteins with no full-length hits (> 25% of the protein’s length) were considered novel proteins. Distant homology searches of the putative Cas proteins were performed using HHpred from the HH-suite49. High scoring HHpred hits were used to infer domain architecture based on comparison to solved crystal structures50,51, and secondary structure that was predicted by JPred452. Protein modeling was performed using Phyre253. The HMM database, including the newly discovered Cas proteins, is available in Supplementary Data 6.
Spacer sequences were determined from the assembled data using CrisprFinder45. CRASS54 was used to locate additional spacers in short DNA reads of the relevant samples. Spacer targets (protospacers) were then identified by BLAST47 searches (using “-task blastn-short”) against the relevant metagenomic assemblies for hits with ≤ 1 mismatch to spacers. Hits belonging to contigs that contained an associated repeat were filtered out (to avoid identifying CRISPR arrays as protospacers). Protospacer adjacent motifs (PAMs) were identified by aligning regions flanking the protospacers and visualized using WebLogo55. In cases that one spacer had multiple putative protospacers with different composition of flanking nucleotides, each distinct combination of protospacer and downstream nucleotides was taken into account for the logo calculation. RNA structures were predicted using mFold56. Average nucleotide identity was computed with the pyani Python module (https://github.com/widdowquinn/pyani), using the Mummer57 method. CRISPR array diversity was analyzed by manually aligning spacers, repeats and flanking sequences from the assembled data. Manual alignments and contig visualizations were performed with Geneious 9.1.
For the phylogenetic analyses of Cas1 and Cas9 we used proteins of the newly identified systems along with the proteins from Makarova et al. (2015)3 and Shmakov et al. (2015)4. A non-redundant set was compiled by clustering together proteins with ≥ 90% identity using CD-HIT58. Alignments were produced with MAFFT59, and maximum-likelihood phylogenies were constructed using RAxML60 with PROTGAMMALG as the substitution model and 100 bootstrap samplings. Cas1 tree were rooted using the branch leading to casposons. Trees were visualized using FigTree 1.4.1 (http://tree.bio.ed.ac.uk/software/figtree/) and iTOL v361.
Generation of heterologous plasmids
Metagenomic contigs were made into minimal CRISPR interference plasmids by removing proteins associated with acquisition for CRISPR-CasX and reducing the size of the CRISPR array for both CRISPR-CasX and CRISPR-CasY. The minimal locus was synthesized as Gblocks (Integrated DNA Technology). Native promoters were used, with the exception of the overexpression of CasY.1 and expression of the crRNA alone or sgRNA for CasX in figure 3g where the J23119 constitutive promoter was used. The minimal CRISPR loci were assembled using Gibson Assembly62 into a plasmid with a p15A origin of replication and chloramphenicol resistance gene. Detailed plasmid maps are available at the links provided in Supplementary Table 2.
PAM depletion assay
PAM depletion assays were conducted as previously described63 with modification. Plasmid libraries containing randomized PAM sequences were assembled by annealing a DNA oligonucleotide containing a target with a 7 nt randomized PAM region with a primer (Supplementary Table 2) and extended with Klenow Fragment (NEB). The double stranded DNA was digested with EcoRI and NcoI and ligated into a pUC19 backbone. The ligated library was transformed into E. coli DH5α and >108 cells were harvested and the plasmids extracted and purified. 200 ng of the pooled library was transformed into electrocompetent E. coli harboring a CRISPR locus or a control plasmid with no locus. The transformed cells were plated on selective media containing carbenicillin (100 mg L−1) and chloramphenicol (30 mg L−1) for 30 hours at 25°C. Plasmid DNA was extracted and the PAM sequence was amplified with adapters for Illumina sequencing. The 7 nt PAM region was extracted and PAM frequencies calculated for each 7 nt sequence. PAM sequences depleted above the specified threshold were used to generate a sequence logo with WebLogo55.
Plasmid Interference
Putative targets identified from metagenomic sequence analysis or PAM depletion assays were cloned into a pUC19 plasmid. 10 ng of target plasmid were transformed into electrocompetent E. coli (NEB Stable) containing the CRISPR loci plasmid. CasX.1 was used for the plasmid interference assays under control of native promoters or using a strong heterologous promoter (J23119) for sgRNA and crRNA expression. CasY.1 was put under the control of a heterologous promoter (J23119) for these assays. Cells were recovered for 2 hrs at 25°C in Super Optimal Broth (SOB) and an appropriate dilution was plated on selective media. Plates were incubated at 25°C and colony forming units were counted. All plasmid interference experiments were performed in triplicate and electrocompetent cells were prepared independently for each replicate.
Northern Blots
E. coli containing the deltaproteobacteria CasX CRISPR locus was grown to OD600=1 at 25°C in SOB media. RNA was extracted by warm phenol extraction, separated on 10% denaturing polyacrylamide gel and blotted as previously described by Zhang et al. (2013)64.
ARMAN-Cas9 protein expression and purification
Expression constructs for Cas9 from ARMAN-1 (AR1) and ARMAN-4 (AR4) were assembled from gBlocks (Integrated DNA Technologies) that were codon-optimized for E. coli. The assembled genes were cloned into a pET-based expression vector as an N-terminal His6-MBP or His6 fusion protein. Expression vectors were transformed into BL21(DE3) E. coli cells and grown in LB broth at 37°C. For protein expression, cells were induced during mid-log phase with 0.4 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) and incubated overnight at 16°C. All subsequent steps were conducted at 4°C. Cell pellets were resuspended in lysis buffer (50mM Tris-HCl pH 8, 500 mM NaCl, 1 mM TCEP, 10 mM Imidazole 0.5% Triton X-100) and supplemented with Complete protease inhibitor mixture (Roche) before lysis by sonication. Lysate was clarified by centrifugation at 15000g for 40 min and applied to Superflow Ni-NTA agarose (Qiagen) in batch. The resin was washed extensively with Wash Buffer A (50 mM Tris-HCl pH 8, 500 mM NaCl, 1mM TCEP, 10 mM Imidazole) followed by 5 column volumes of Wash Buffer B (50 mM Tris-HCl pH 8, 1M NaCl, 1 mM TCEP, 10 mM Imidazole). Protein was eluted off of Ni-NTA resin with Elution Buffer (50 mM Tris-HCl pH 8, 500 mM NaCl, 1 mM TCEP, 300 mM Imidazole). The His6-MBP tag was removed by TEV protease during overnight dialysis against Wash Buffer A. Cleaved Cas9 was removed from the affinity tag through a second Ni-NTA agarose column. The protein was dialyzed into IEX Buffer A (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 1 mM TCEP, 5% glycerol) before application to a 5 mL Heparin HiTrap column (GE Life Sciences). Cas9 was eluted over a linear NaCl (0.3–1.5 M) gradient. Fractions were pooled and concentrated with a 30 kDa spin concentrator (Thermo Fisher). When applicable, Cas9 was further purified via size-exclusion chromatography on a Superdex 200 pg column (GE Life Sciences) and stored in IEX Buffer A for subsequent cleavage assays. For yeast expression, AR1-Cas9 was cloned into a Gal1/10 His6-MBP TEV Ura S. cerevisiae expression vector (Addgene plasmid # 48305). The vector was transformed into a BY4741 URA3 strain and cultures were grown in CSM media at 30°C. At an OD600 of ~0.6, protein expression was induced with 2% w/v galactose and incubated overnight at 16°C. Protein purification was performed as above.
RNA in vitro transcription and oligonucleotide purification
In vitro transcription reactions were performed as previously described65 using synthetic DNA templates containing a T7 promoter sequence. All in vitro transcribed putative guide RNA sequences and target RNA or DNA were purified via denaturing PAGE. Double-stranded target RNA and DNA were hybridized in 20 mM Tris HCl pH 7.5 and 100 mM NaCl by incubation at 95°C for 1 min, followed by slow-cooling to room temperature. Hybrids were purified by native PAGE. RNA and DNA sequences used in this study are listed in Supplementary Table 2.
In vitro cleavage assays
Purified DNA and RNA oligonucleotides were radiolabeled using T4 polynucleotide kinase (NEB) and [γ-32P] ATP (Perkin-Elmer) in 1× PNK buffer for 30 min at 37°C. PNK was heat inactivated at 65°C for 20 min and free ATP was removed from the labeling reactions using illustra Microspin G-25 columns (GE Life Sciences). CrRNA and tracrRNA were mixed in equimolar quantities in 1× refolding buffer (50 mM Tris HCl pH 7.5, 300 mM NaCl, 1 mM TCEP, 5% glycerol) and incubated at 70°C for 5 min and then slow-cooled to room temperature. The reactions were supplemented to 1 mM final metal concentration and subsequently heated at 50°C for 5 min. After slow-cooling to room temperature, refolded guides were placed on ice. Unless noted for buffer or salt concentration, Cas9 was reconstituted with an equimolar amount of guide in 1× cleavage buffer (50 mM Tris HCl pH 7.5, 300 mM NaCl, 1 mM TCEP, 5% glycerol, 5 mM divalent metal) at 37°C for 10 min. Cleavage reactions were conducted in 1× cleavage buffer with a 10× excess of Cas9-guide complex over radiolabeled target at 37°C or the indicated temperature. Reactions were quenched in an equal volume of gel loading buffer supplemented with 50mM EDTA. Cleavage products were resolved on 10% denaturing PAGE and visualized by phosphorimaging.
In vivo E. coli interference assays
E. coli transformation assays for ARMAN-1 Cas9 and ARMAN-4 Cas9 were conducted as previously published66. Briefly, E. coli transformed with plasmids expressing guide RNA sequences were made electrocompetent. Cells were then transformed with 9 fmol of plasmid encoding wild-type or catalytically inactive Cas9 (dCas9). A dilution series of recovered cells was plated on LB plates with selective antibiotics. Colonies were counted after 16 hr at 37°C.
Data Availability
All the sequences reported in this study for the first time have been deposited in NCBI database under BioProject PRJNA349044. The NCBI Nucleotide database accession and coordinates of each locus are specified in Extended Data Table 1. The BioSample and Sequence Reads Archive (SRA) accessions for the ARMAN-1 spacers and protospacers are detailed in Supplementary Table 1. The HMMs used in this study are provided in Supplementary Data 6.
Extended Data
Extended Data Figure 1. Multiple sequences alignment of newly described Cas9 proteins.
Alignment of Cas9 proteins from ARMAN-1 and ARMAN-4, as well as two closely related Cas9 proteins from uncultivated bacteria, to the Actinomyces naeslundii Cas9, whose structure has been solved67.
Extended Data Figure 2. Within-population variability of ARMAN-1 CRISPR arrays.
Variability of reconstructed CRISPR arrays, including the most well represented (and thus assembled) sequences (Fig. 2) and array segments representing locus variants that were reconstructed from the short DNA reads. Variability is due to spacers that were present in only a subset of archaeal cells in the population, as well as spacers whose context differed due to spacer loss (indicated by black lines). White boxes indicate repeats and colored arrows indicate CRISPR spacers (spacers with different colors have different sequences, except for unique spacers that are black). In CRISPR systems, spacers are typically added unidirectionally, so the high variety of spacers on the left side is attributed to recent acquisition.
Extended Data Figure 3. Novelty of the reported CRISPR-Cas systems.
a, Simplified phylogenetic tree of the universal Cas1 protein. CRISPR types of known systems are noted on the wedges and branches; the newly described systems are in bold. Detailed Cas1 phylogeny is provided in Supplementary Data 4. b, Proposed evolutionary scenario that gave rise to the archaeal type II system as a result of a recombination between type II-B and type II-C loci. c, Similarity of CasX and CasY to known proteins based on the following searches: (1) Blast search against the non-redundant (NR) protein database of NCBI, (2) HMM search against an HMM database of known Cas proteins and (3) distant homology search using HHpred49 (E, e-value).
Extended Data Figure 4. Evolutionary tree of Cas9 homologs.
Maximum-likelihood phylogenic tree of Cas9 proteins, showing the previously described systems colored based on their type: II-A in blue, II-B in green and II-C in purple. The archaeal Cas9 (in red), cluster with type II-C CRISPR-Cas systems, together with two newly described bacterial Cas9 from uncultivated bacteria. Detailed tree is provided in Supplementary Data 5.
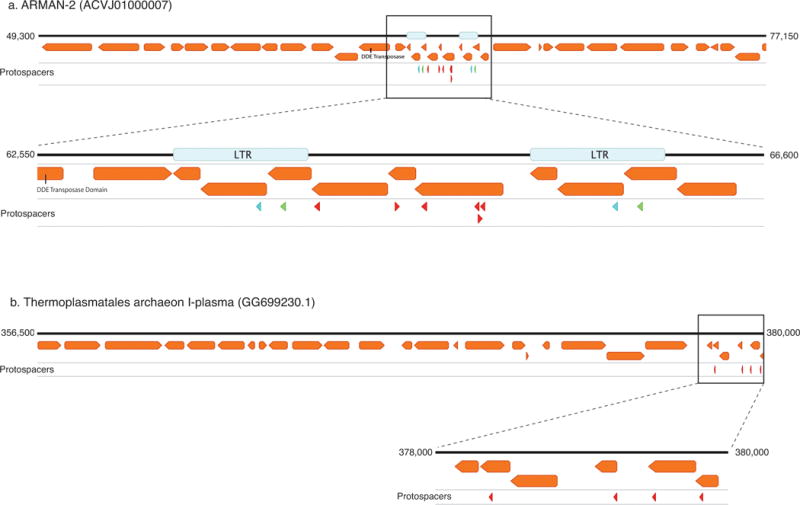
Extended Data Figure 5. ARMAN-1 spacers map to genomes of archaeal community members.

a, Protospacers from ARMAN-1 map to the genome of ARMAN-2, a nanoarchaeon from the same environment. Six protospacers (red arrowheads) map uniquely to a portion of the genome flanked by two long-terminal repeats (LTRs), and two additional protospacers match perfectly within the LTRs (blue and green arrowheads). This region is likely a transposon, suggesting the CRISPR-Cas system of ARMAN-1 plays a role in suppressing mobilization of this element. b, Protospacers also map to a Thermoplasmatales archaeon (I-plasma), another member of the Richmond Mine ecosystem that is found in the same samples as ARMAN organisms. The protospacers cluster within a region of the genome encoding short, hypothetical proteins, suggesting this might also represent a mobile element. NCBI accessions are provided in parenthesis.
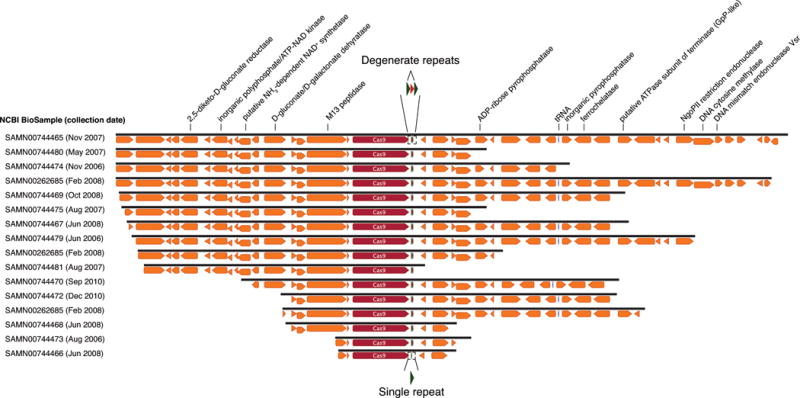
Extended Data Figure 6. Archaeal Cas9 from ARMAN-4 with a degenerate CRISPR array is found on numerous contigs.

Cas9 from ARMAN-4 is highlighted in dark red on 16 nearly identical contigs from different samples. Proteins with putative domains or functions are labeled whereas hypothetical proteins are unlabeled. Fifteen of the contigs contain two degenerate direct repeats (36 nt long with one mismatch) and a single conserved spacer of 36 nt. The remaining contig contains only one direct repeat. Unlike ARMAN-1, no additional Cas proteins are found adjacent to Cas9 in ARMAN-4.
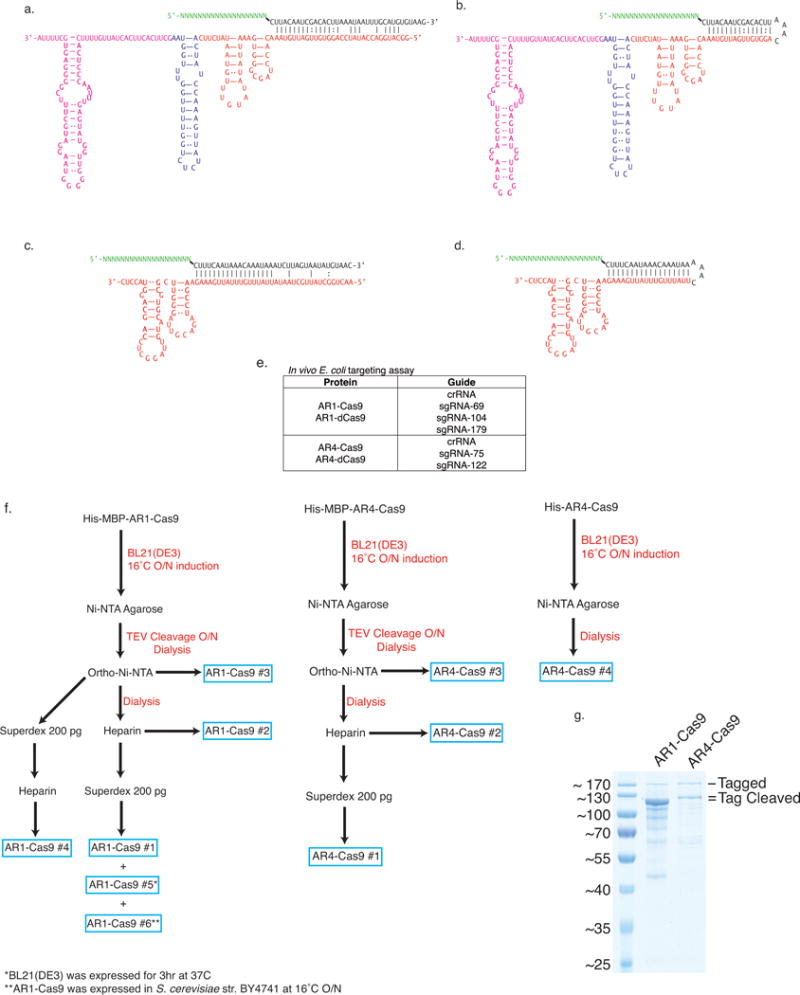
Extended Data Figure 7. Predicted structures of guide RNA and purification schema for in vitro biochemistry studies.
a, The CRISPR repeat and tracrRNA anti-repeat are depicted in black whereas the spacer-derived sequence is shown as a series of green N’s. No clear termination signal can be predicted from the locus, so three different tracrRNA lengths were tested based on their secondary structure – 69, 104, and 179 nt in red, blue, and pink, respectively. b, Engineered single-guide RNA corresponding to dual-guide in (a). c, Dual-guide for ARMAN-4 Cas9 with two different hairpins on 3′ end of tracrRNA (75 and 122 nt). d, Engineered single-guide RNA corresponding to dual-guide in (c). e, Conditions tested in E. coli in vivo targeting assay. f, ARMAN-1 (AR1) and ARMAN-4 (AR4) Cas9 were expressed and purified under a variety of conditions as outlined in the Methods section. Proteins outlined in blue boxes were tested for cleavage activity in vitro. g, Fractions of AR1-Cas9 and AR4-Cas9 purifications were separated on a 10% SDS-PAGE gel.
Extended Data Figure 8. Programmed DNA interference by CasX.
a, Plasmid interference assays for CasX.1 (Deltaproteobacteria) and CasX.2 (Planctomycetes), continued from Figure 3c (sX1, CasX spacer 1; sX2, CasX spacer 2; NT, non-target). Experiments were conducted in triplicate and mean ± s.d. is shown. b, Serial dilution of E. coli expressing a CasX locus and transformed with the specified target, continued from Figure 3b. c, PAM depletion assays for the Deltaproteobacteria CasX and d, Planctomycetes CasX expressed in E. coli. PAM sequences depleted greater than the indicated PAM depletion value threshold (PDVT) compared to a control library were used to generate the sequence logo. e, Diagram depicting the location of Northern blot probes for CasX.1. f, Northern blots for CasX.1 tracrRNA in total RNA extracted from E. coli expressing the CasX.1 locus. The sequences of the probes used are provided in Supplementary Table 2.
Extended Data Table 1. CRISPR-Cas loci identified in this study.
Details regarding the organisms and genomic location in which the CRISPR-Cas system were identified, as well as information on the number and average length of reconstructed spacers, and repeats length (NA, not available). ARMAN-1 spacers were reconstructed from 16 samples, see details in Supplementary Table 1.
| Taxonomic group | Cas effector | NCBI Accession | Coordinates | Repeat length | # spacers | Spacers avg. length |
|---|---|---|---|---|---|---|
| ARMAN-1 | Cas9 | MOEG01000017 | 1827..7130 | 36 | 271 | 34.5 |
| ARMAN-4 | Cas9 | KY040241 | 11779..14900 | 36 | 1 | 36 |
| Deltaproteobacteria | CasX | MGPG01000094 | 4319..9866 | 37 | 5 | 33.6 |
| Planctomycetes | CasX | MHYZ01000150 | 1..5586 | 37 | 7 | 32.3 |
| Candidatus Katanobacteria | CasY.1 | MOEH01000029 | 459..5716 | 26 | 14 | 17.1 |
| Candidatus Vogelbacteria | CasY.2 | MOEJ01000028 | 7322..13087 | 26 | 18 | 17.3 |
| Candidatus Vogelbacteria | CasY.3 | MOEK01000006 | 1..4657 | 26 | 12 | 17.3 |
| Candidatus Parcubacteria | CasY.4 | KY040242 | 1..5193 | 25 | 13 | 18.4 |
| Candidatus Komeilibacteria | CasY.5 | MOEI01000022 | 2802..7242 | 36 | 8 | 26 |
| Candidatus Kerfeldbacteria | CasY.6 | MHKD01000036 | 11503..15366 | NA | NA | NA |
Extended Data Table 2.
In vitro cleavage conditions assayed for Cas9 from ARMAN-1 and ARMAN-4.
| Protein Purification | Buffer | Salt (mM) | Metal | Guide | Target | Temperature |
|---|---|---|---|---|---|---|
| AR1-Cas9 #1 | Tris pH 7.5 | 300 | Mg2+ Mn2+ Zn2+ |
crRNA cr:69 cr:104 cr:179 |
dsDNA ssDNA DNA Bubble ssRNA dsDNA |
37 |
| AR1-Cas9 #1 | Tris pH 7.5 | 100–500 | Mg2+ | cr:69 cr:104 cr:179 |
dsDNA | 37 |
| AR1-Cas9 #1 | Tris pH 7.5 | 300 | Mg2+ Mn2+ Zn2+ |
cr:69 cr:104 cr:179 |
dsDNA | 30–48 |
| AR1-Cas9 #1 | MOPS: pH 6 pH 6.5 pH 7.0 pH 7.5 |
300 | Mg2+ | cr:69 cr:104 cr:179 |
dsDNA | 37 |
| AR1-Cas9 #1 | Citrate: pH 5 pH 5.5 pH 6 |
300 | Mg2+ | cr:69 cr:104 cr:179 |
dsDNA | 37 |
| AR1-Cas9 #1 | Tris pH 7.5 | 300 | Mg2+ Mn2+ Zn2+ |
cr:69 cr:104 cr:179 |
plasmid | 37–50 |
| AR1-Cas9 #2 | Tris pH 7.5 | 300 | Mg2+ Mn2+ Zn2+ |
cr:69 cr:104 cr:179 |
dsDNA | 37 |
| AR1-Cas9 #3 | Tris pH 7.5 | 300 | Mg2+ Mn2+ Zn2+ |
cr:69 cr:104 cr:179 |
dsDNA | 37 |
| AR1-Cas9 #4 | Tris pH 7.5 | 300 | Mg2+ Mn2+ Zn2+ |
cr:69 cr:104 cr:179 |
dsDNA | 37 |
| AR1-Cas9 #5 | Tris pH 7.5 | 300 | Mg2+ Mn2+ Zn2+ |
cr:69 cr:104 cr:179 |
dsDNA | 37 |
| AR1-Cas9 #6 | Tris pH 7.5 | 300 | Mg2+ Mn2+ Zn2+ |
cr:69 cr:104 cr:179 |
ssDNA dsDNA |
37 |
| AR4-Cas9 #1 | Tris ph7.5 | 300 | Mg2+ Mn2+ Zn2+ |
sgRNA-122 | dsDNA | 37 |
| AR4-Cas9 #2 | Tris ph7.5 | 300 | Mg2+ Mn2+ Zn2+ |
sgRNA-122 | dsDNA | 37 |
| AR4-Cas9 #3 | Tris ph7.5 | 300 | Mg2+ Mn2+ Zn2+ |
sgRNA-122 | dsDNA | 37 |
| AR4-Cas9 #4 | Tris ph7.5 | 300 | Mg2+ Mn2+ Zn2+ |
sgRNA-122 | dsDNA | 37 |
Acknowledgments
We thank Nan Ma, Kaihong Zhou and David McGrath for technical assistance; Chris Brown, Matt Olm, Mitchell O’Connell, Janice Chen and Stephen Floor for critical reading of the manuscript and useful discussions. We thank V. Yu for gift of S. cerevisiae expression strain. D.B. was supported by a long-term EMBO fellowship, L.B.H. by a US National Science Foundation Graduate Research Fellowship, and A.J.P. by a fellowship of the German Science Foundation (DFG PR 1603/1-1). J.A.D. is an Investigator of the Howard Hughes Medical Institute. This work was supported in part by a Frontiers Science award from the Paul Allen Institute to J.A.D. and J.F.B., the National Science Foundation (MCB-1244557 to J.A.D.) and the Lawrence Berkeley National Laboratory’s Sustainable Systems Scientific Focus Area funded by the U.S. Department of Energy, (DE-AC02-05CH11231 to J.F.B.). DNA sequencing was conducted at the DOE Joint Genome Institute, a DOE Office of Science User Facility, via the Community Science Program.
Footnotes
Author Contributions
D.B., L.B.H., S.C.S., J.A.D., and J.F.B. designed the study and wrote the manuscript. A.J.P., K.A., J.F.B., B.T.C., and D.B. assembled the data and reconstructed the genomes. D.B., L.B.H., S.C.S., and J.F.B. computationally analyzed the CRISPR-Cas systems. L.B.H. and D.B. designed and executed experimental work with CRISPR-CasX and CRISPR-CasY. S.C.S. designed and executed the experimental work with ARMAN Cas9. The manuscript that was read, edited, and approved by all authors.
Conflict of Interest
The Regents of the University of California have filed a provisional patent application related to the technology described in this work to the United States Patent and Trademark Office, in which D.B., L.B.H., S.C.S., J.A.D. and J.F.B. are listed as inventors.
References
- 1.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 2.Sorek R, Kunin V, Hugenholtz P. CRISPR — a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6:181–186. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- 3.Makarova KS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shmakov S, et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat Biotechnol. 2016;34:933–941. doi: 10.1038/nbt.3659. [DOI] [PubMed] [Google Scholar]
- 6.Brown CT, et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature. 2015;523:208–211. doi: 10.1038/nature14486. [DOI] [PubMed] [Google Scholar]
- 7.Sharon I, Banfield JF. Genomes from metagenomics. Science. 2013;342:1057–1058. doi: 10.1126/science.1247023. [DOI] [PubMed] [Google Scholar]
- 8.Levy A, et al. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015;520:505–510. doi: 10.1038/nature14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nuñez JK, Lee ASY, Engelman A, Doudna JA. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015;519:193–198. doi: 10.1038/nature14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chylinski K, Makarova KS, Charpentier E, Koonin EV. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:6091–6105. doi: 10.1093/nar/gku241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker BJ, et al. Enigmatic, ultrasmall, uncultivated Archaea. Proc Natl Acad Sci. 2010;107:8806–8811. doi: 10.1073/pnas.0914470107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baker BJ, et al. Lineages of acidophilic archaea revealed by community genomic analysis. Science. 2006;314:1933–1935. doi: 10.1126/science.1132690. [DOI] [PubMed] [Google Scholar]
- 14.Comolli LR, Banfield JF. Inter-species interconnections in acid mine drainage microbial communities. Terr Microbiol. 2014;5:367. doi: 10.3389/fmicb.2014.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yelton AP, et al. Comparative genomics in acid mine drainage biofilm communities reveals metabolic and structural differentiation of co-occurring archaea. BMC Genomics. 2013;14:485. doi: 10.1186/1471-2164-14-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vagin VV, et al. A distinct small RNA pathway silences selfish genetic elements in the germline. Science. 2006;313:320–324. doi: 10.1126/science.1129333. [DOI] [PubMed] [Google Scholar]
- 17.Stern A, Keren L, Wurtzel O, Amitai G, Sorek R. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 2010;26:335–340. doi: 10.1016/j.tig.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zegans ME, et al. Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J Bacteriol. 2009;191:210–219. doi: 10.1128/JB.00797-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah SA, Erdmann S, Mojica FJM, Garrett RA. Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biol. 2013;10:891–899. doi: 10.4161/rna.23764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jinek M, et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deltcheva E, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Rajan R, Seifert HS, Mondragón A, Sontheimer EJ. DNase H activity of Neisseria meningitidis Cas9. Mol Cell. 2015;60:242–255. doi: 10.1016/j.molcel.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zetsche B, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abudayyeh OO, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anantharaman K, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7:13219. doi: 10.1038/ncomms13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Godde JS, Bickerton A. The repetitive DNA elements called CRISPRs and their associated genes: evidence of horizontal transfer among prokaryotes. J Mol Evol. 2006;62:718–729. doi: 10.1007/s00239-005-0223-z. [DOI] [PubMed] [Google Scholar]
- 28.Burstein D, et al. Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat Commun. 2016;7:10613. doi: 10.1038/ncomms10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hug LA, et al. A new view of the tree of life. Nat Microbiol. 2016;1:16048. doi: 10.1038/nmicrobiol.2016.48. [DOI] [PubMed] [Google Scholar]
- 30.Luef B, et al. Diverse uncultivated ultra-small bacterial cells in groundwater. Nat Commun. 2015;6 doi: 10.1038/ncomms7372. [DOI] [PubMed] [Google Scholar]
- 31.Kantor RS, et al. Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. mBio. 2013;4:e00708–13. doi: 10.1128/mBio.00708-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson WC, Stegen JC. The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front Microbiol. 2015;6 doi: 10.3389/fmicb.2015.00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinke C, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 34.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nuñez JK, et al. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat Struct Mol Biol. 2014;21:528–534. doi: 10.1038/nsmb.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denef VJ, Banfield JF. In situ evolutionary rate measurements show ecological success of recently emerged bacterial hybrids. Science. 2012;336:462–466. doi: 10.1126/science.1218389. [DOI] [PubMed] [Google Scholar]
- 37.Miller CS, Baker BJ, Thomas BC, Singer SW, Banfield JF. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011;12:R44. doi: 10.1186/gb-2011-12-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Probst AJ, et al. Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations. Environ Microbiol. 2016 doi: 10.1111/1462-2920.13362. [DOI] [PubMed] [Google Scholar]
- 39.Emerson JB, Thomas BC, Alvarez W, Banfield JF. Metagenomic analysis of a high carbon dioxide subsurface microbial community populated by chemolithoautotrophs and bacteria and archaea from candidate phyla. Environ Microbiol. 2016;18:1686–1703. doi: 10.1111/1462-2920.12817. [DOI] [PubMed] [Google Scholar]
- 40.Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinforma Oxf Engl. 2012;28:1420–8. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 41.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyatt D, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu YW, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–607. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 44.Dick GJ, et al. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009;10:R85. doi: 10.1186/gb-2009-10-8-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Enright AJ, Dongen SV, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Consortium TU. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 50.Dong D, et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature. 2016;532:522–526. doi: 10.1038/nature17944. [DOI] [PubMed] [Google Scholar]
- 51.Yamano T, et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell. 2016;165:949–962. doi: 10.1016/j.cell.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skennerton CT, Imelfort M, Tyson GW. Crass: identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res. 2013;41:e105–e105. doi: 10.1093/nar/gkt183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 63.Esvelt KM, et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, et al. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell. 2013;50:488–503. doi: 10.1016/j.molcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sternberg SH, Haurwitz RE, Doudna JA. Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. RNA. 2012;18:661–672. doi: 10.1261/rna.030882.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oakes BL, et al. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol. 2016;34:646–651. doi: 10.1038/nbt.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jinek M, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343 doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the sequences reported in this study for the first time have been deposited in NCBI database under BioProject PRJNA349044. The NCBI Nucleotide database accession and coordinates of each locus are specified in Extended Data Table 1. The BioSample and Sequence Reads Archive (SRA) accessions for the ARMAN-1 spacers and protospacers are detailed in Supplementary Table 1. The HMMs used in this study are provided in Supplementary Data 6.