

Abstract
Background and Purpose
AMPA receptor positive allosteric modulators represent a potential therapeutic strategy to improve cognition in people with schizophrenia. These studies collectively constitute the preclinical pharmacology data package used to build confidence in the pharmacology of this molecule and enable a clinical trial application.
Experimental Approach
[N‐[(2S)‐5‐(6‐fluoro‐3‐pyridinyl)‐2,3‐dihydro 1H–inden‐2‐yl]‐2‐propanesulfonamide] (UoS12258) was profiled in a number of in vitro and in vivo studies to highlight its suitability as a novel therapeutic agent.
Key Results
We demonstrated that UoS12258 is a selective, positive allosteric modulator of the AMPA receptor. At rat native hetero‐oligomeric AMPA receptors, UoS12258 displayed a minimum effective concentration of approximately 10 nM in vitro and enhanced AMPA receptor‐mediated synaptic transmission at an estimated free brain concentration of approximately 15 nM in vivo. UoS12258 reversed a delay‐induced deficit in novel object recognition in rats after both acute and sub‐chronic dosing. Sub‐chronic dosing reduced the minimum effective dose from 0.3 to 0.03 mg·kg−1. UoS12258 was also effective at improving performance in two other cognition models, passive avoidance in scopolamine‐impaired rats and water maze learning and retention in aged rats. In side‐effect profiling studies, UoS12258 did not produce significant changes in the maximal electroshock threshold test at doses below 10 mg·kg−1.
Conclusion and Implications
We conclude that UoS12258 is a potent and selective AMPA receptor modulator exhibiting cognition enhancing properties in several rat behavioural models superior to other molecules that have previously entered clinical evaluation.
Abbreviations
- MEST
maximal electroshock seizure threshold test
- NOR
novel object recognition
Tables of Links
| TARGETS | |
|---|---|
| AMPA receptors | GRIK1 |
| GluN1 receptor | Kainate receptors |
| GluN2A receptor | NMDA receptors |
| GluN2B receptor |
| LIGANDS | |
|---|---|
| AMPA | Phencyclidine |
| Cyclothiazide | Picrotoxin |
| Glutamate | SB‐399885 |
| Kainate | Scopolamine |
| Ketamine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Many lines of evidence support the premise that enhancing AMPA receptor function should be a viable route to treating a variety of disorders that are underpinned by a hypoglutamatergic state, such as schizophrenia (Ozawa, 1998, Goff and Coyle, 2001, Sanacora et al., 2008, Zarate and Manji, 2008). The AMPA receptor itself is a family of tetrameric ionotropic receptors arising from four genes that encode four distinct subunits, named GluA1–4, or previously GluR1–4/A‐D (Collingridge et al., 2009). This diversity of subunits gives rise to heterogeneity of AMPA receptor composition in native brain states (Beneyto 2004), further complicated by both the presence of splice variants and post‐translational editing (Sommer et al., 1990; Seeburg et al., 2001). Two main sub‐populations arise from the editing of a Ca2+‐permeable glutamine residue in the re‐entrant M2 loop of the GluA2 subunit to a Ca2+‐impermeable arginine, which renders the edited receptors permeable to just Na+ and K+ (Gouaux, 2004). Other than this re‐entrant M2 loop, each subunit also comprises three transmembrane domains (M1, 3 and 4), an intra‐cellular C‐terminal domain, key for regulation of trafficking, as well as an extensive extracellular N‐terminal domain containing both the glutamate‐binding site domain (commonly referred to as the ligand‐binding or agonist‐binding domain) and an N‐terminal domain (Traynelis et al., 2010). Functionally active AMPA receptors can be formed from any combination of the GluA subunits, although different combinations of different subunits display different biophysical and kinetic properties as well as distinct regional distributions and trafficking potential (Herguedas et al., 2016). Specifically for distribution, GluA1, GluA2 and GluA3 subunits are found at high levels in the hippocampus, basal ganglia, amygdala, lateral septum and cerebellum in rat and macaque, whereas GluA4 is most highly expressed in the cerebellum (Boulter et al., 1990, Keinänen et al., 1990, Beneyto and Meador‐Woodruff, 2004). Glutamate is the major excitatory neurotransmitter, and AMPA receptors are responsible for mediating the majority of fast, excitatory synaptic transmission, and as such, offer a great opportunity for modulation for therapeutic benefit. Repetitive AMPA receptor stimulation leads to activation of another group of ionotropic glutamate receptors, NMDA receptors, by voltage‐mediated release of magnesium ion block. This enables greater influx of calcium ions into the post synaptic neuron, and onward synaptic transmission and processes required for memory deposition and learning, which are further strengthened by increased trafficking of AMPA receptors to the membrane (Lynch, 2002).
Glutamate receptors and schizophrenia
The considerable body of data linking glutamate receptors to schizophrenia comprises both empirical observations and genetic associations. Studies in schizophrenic patients have identified various supporting lines of data, including reduced glutamate concentrations in the CSF (Kim et al., 1980), reduced hippocampal glutamate from post mortem studies (Harrison, 2000), reduced glutamate neurotransmission, reduced carboxypeptidase II (Tsai et al., 1995) and increased N‐acetyl‐aspartyl glutamate (endogenous iGlu receptor antagonist) (Jessen et al., 2013). Additionally, evidence from immunocytochemical studies on different brain regions of patients as well as analysis of mRNA transcript level changes in hippocampal regions of schizophrenia brains indicates reductions in AMPA receptor levels in prefrontal and temporal cortical and thalamic regions that are associated with the performance of cognitive tasks (Akbarian et al., 1995, Wright and Vissel, 2012). Wider evidence comes from the NMDA receptor, for which it is clear that administration of an NMDA receptor blocker, such as phencyclidine or ketamine, is able to both amplify the symptoms of schizophrenia in schizophrenic patients (Lahti et al., 1995), and also induce a ‘schizophrenia‐like’ state including psychosis and disrupted cognition in healthy volunteers (Lahti et al., 2001). These data are more compelling for NMDA receptor blocking agents than for other agents, such as dopamine‐releasers such as the amphetamines (Le Pen et al., 2003).
AMPA receptor positive modulators
Given the evidence associating reduced AMPA receptor‐mediated neurotransmission with various disease states, it is not surprising that considerable effort has been expended in trying to identify ways to increase glutamatergic transmission. Direct activation approaches suffer from the loss of spatial and temporal control of activation of endogenous glutamate binding and also will run a significant safety and tolerability risk of generating undesirably high excitatory signalling levels. To overcome both of these liabilities, positive allosteric modulators have been targeted over the recent decades, such that the glutamate signalling will be enhanced if, and only if, both glutamate and positive modulator are bound at the receptor. A range of molecules has been reported, from various groups, which have been studied in different preclinical species as well as clinically. These molecules have generated considerable in vitro data to support their mechanisms of action, and in particular, have been shown to be selective for the AMPA receptor subtype of glutamate receptor over the other iGlu and mGlu receptor subtypes (unlike the majority of orthosteric‐site binding ligands). In addition to effects directly at the receptor, these molecules have demonstrated potentiation in native tissue and hippocampal slice preparations, particularly generating facilitation of polysynaptic responses (Arai et al., 1996, Lynch, 2002), long‐term potentiation, increased BDNF levels and activity in a wide range of animal models of learning and memory (Woolley et al., 2009). Specifically for models of cognition, molecules have demonstrated improved performance in olfactory discrimination, radial arm maze (Staubli et al., 1994), conditioned fear (Rogan et al., 1997), water maze performance (Zivkovic et al., 1995; Quirk and Nisenbaum, 2001), delayed non match to sample (Hampson et al., 1998), novel object recognition (NOR) (Lebrun et al., 2000) and passive avoidance (PA) (Lebrun et al., 2000, Quirk and Nisenbaum, 2001) in rodents, and have also demonstrated encouraging results in non‐human primates (Thompson et al., 1995, Buccafusco et al., 2004, Porrino et al., 2005). In addition to understanding the behavioural pharmacology, the knowledge of the overall structure of the receptor offers the opportunity to exploit structure‐based design opportunities against the extracellular binding domains (Baranovic et al., 2016).
AMPA receptor positive modulators – clinical studies
Although there have been many reports of AMPA receptor positive allosteric modulators in preclinical studies, relatively few have been progressed beyond Phase I into patient studies. The initial clinical evaluation was driven by the set of CX molecules originating from the University of California, subsequently as Cortex, from the pioneering work of Gary Lynch. These molecules initially demonstrated effects in healthy young (Ingvar et al., 1997) and aged volunteers (Lynch et al., 1997), as well as in schizophrenic patients stabilized on the atypical antipsychotic drug clozapine (Goff et al., 2001), but were subsequently ineffective in a later study (Goff et al., 2008). Studies with later compounds have generated effects in attention deficit hyperactivity disorder (ADHD) patients (2006, Weisler, 2007). The second major class of the phenethylamine sulphonamides produced two clinical stage molecules from Lilly, one of which was reported as negative in a Phase II study of cognition in Alzheimer's disease, as well as molecules from GlaxoSmithKline (Ward et al., 2010, 2011) and Pfizer (Shaffer et al., 2015).
Methods
Compound
The chemical structure of UoS12258 is shown in Figure 1. The preparation and preliminary characterization of UoS12258 has been described previously (Ward et al., 2010).
Figure 1.
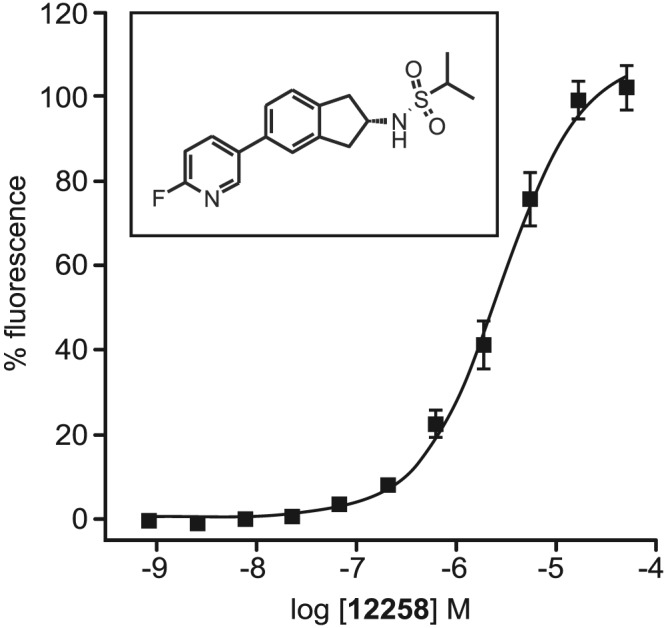
Concentration–response curve and chemical structure for UoS12258 showing its potentiation of glutamate‐induced rises in intracellular calcium in HEK293 cells stably expressing the hGluA2i homomeric AMPA receptor, pEC50 = 5.57. Responses are normalized to 150 μM cyclothiazide. Mean ± SEM, n = 6 independent experiments.
Intracellular Ca2+ influx assay
AMPA receptors: HEK293 sticky cells (293 TAg/hsr‐A pCIP4) were grown in a DMEM/F‐12 supplemented with 10% FBS, 2 mM glutamine and 1.5 ug·mL−1 puromycin in a 5% CO2 humidified atmosphere at 37°C. Transient transfection was performed according to the lipofectamine 2000 method. Briefly, a sub‐confluent T‐175 cm2 flask was washed with PBS to remove serum and 20 mL of optimem were added. Separately, in 10 mL optimem, 100 μg DNA were pre‐incubated with 300 μL of lipofectamine for 20 min at room temperature and then added to the flask. After 4 h of incubation at 37°C, 5% CO2, cells were detached with versene and re‐suspended in the post transfection medium (DMEM supplemented with 10% of dialysed FBS). The confluent monolayer for the functional assay was obtained by seeding 7500 cells per well, 2 days before the experiment in 384‐wells, poly‐D‐lysine coated, black with clear bottom plates (Greiner).
The following homomeric AMPA receptor subtypes were studied: human GluA1 flip isoform (hGluA1i), hGluA3i and hGluA4i and rat GluA2i using DNAs (all Q‐unedited in TM2 region to allow Ca2+ permeability) hGRIA1i, 3i and 4i, and rGRIA2i. Human GluA2i was studied using the HEK293‐hGR1A2i Q‐unedited RC17 cell line that was established in‐house.
NMDA receptors: HEK293 sticky cells, grown at 80–95% confluency in 175 cm2 T‐flask, were transduced by addition of 5% NR1A‐BacMam/25% NR2B‐BacMam mixture in 15 mL of medium containing DMEM/F‐12, 10% dialysed FCS, 0.3 mM MgCl2 and 500 μM ketamine hydrochloride (non‐competitive NMDA receptor antagonist). Cells were incubated with the transduction mixture at 37°C in 5% CO2 for 24 h. Then, BacMam was washed away with PBS, and cells were plated onto poly‐D‐lysine coated black/clear plates at a density of 20 000 cells per well, always in presence of 500 μM ketamine hydrochloride. After an additional 24 h, cells were tested in the fluorescent imaging plated reader (FLIPR) assay.
Kainate receptors: Transient transfection was performed according to the lipofectamine 2000 method. Briefly, a sub‐confluent T‐175 cm2 flask was washed with PBS to remove serum and 20 mL of optimem were addded. Separately, in 10 mL optimem, 60 μg pcDNA3.2‐human GRIK1 (Q/unedited) were pre‐incubated with 300 μL of lipofectamine for 20 min at room temperature and then added to the flask. After 4 h incubation at 37°C, 5% CO2, cells were detached with versene and re‐suspended in the post transfection medium (DMEM supplemented with 10% of dialysed FBS). The confluent monolayer for the functional assay was obtained by seeding 7500 cells per well 1 day before the experiment in 384‐wells, poly‐D‐lysine coated, black with clear bottom plates.
Intracellular calcium levels through AMPA receptors were measured. Buffer comprised (mM): 20 HEPES, 145 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 5.5 glucose, pH 7.3 with NaOH. On the day of the experiment, cell plates were washed three times with the assay buffer using the EMBLA instrument (Skatron), and 20 μL of buffer was left in each well. Then 20 μL of Fluo‐4AM (Molecular Probes) buffer was added to each well to give a final concentration of 2 μM, and cells were then incubated at room temperature for 60 min in the dark. After the loading incubation, cell plates were washed three times to remove unloaded Fluo‐4 and 30 μL of buffer was left in each well. Then cell plates were placed into the fluorescent imaging plated reader (FLIPR) instrument with 1 W laser power, 10% gain and 0.4 s exposure, and a dual addition protocol of 10 min fluorescence measurement was used. Compounds were collected in 384‐well plates at a concentration of 10 mM. A serial dilution (1:3) was performed using a Beckman FX. A copy of the serial dilution plates was prepared and diluted with buffer immediately before the experiment. The first 10 μL addition into a well was from a buffer pre‐diluted compound plate, and it was followed by 10 min incubation. AMPA positive modulators did not give rise to detectable calcium increases when applied in the absence of glutamate. Then for the second 10 μL addition, glutamate was applied to give a final concentration of 100 μM. Glutamate caused a dramatic increase in FLIPR counts when a positive modulator was pre‐applied. Cyclothiazide (Sigma Aldrich) 150 μM, a known AMPA receptor positive modulator, was used as a positive control.
Intracellular calcium measurements through NMDA receptors were obtained in a similar manner to that described for AMPA receptors except for the addition of 500 μM ketamine hydrochloride during labelling with FLUO‐4. The method used to obtain calcium influx measurements through kainate receptors was also similar to that described for AMPA receptors except for the addition of concanavalin A (Sigma Aldrich) 0.125 mg·mL−1, which reduced the level of receptor desensitization.
Assay quality was determined by calculating the Z′ value: high (150 μM cyclothiazide and 100 μM glutamate) and low controls (only 100 μM glutamate) were always present within each 384‐well plate. FLIPR kinetics data were transformed to a single endpoint using the peak high of the response. Values were then normalized to the specific % increase from the 100 μM glutamate effect (0%) to the maximal cyclothiazide positive modulator effect (150 μM) in the presence of the 100 μM glutamate stimulation (100%). In general, data from plates were only accepted if Z′ > 0.3 and two out of three pharmacological standards had values that were within 3× the SD of historical values. The pEC50 for UoS12258 was calculated using a non‐linear, four parameter logistic curve‐fitting programme.
Recombinant receptor electrophysiology
AMPA receptors: HEK293‐hGluA2i unedited RC17 cells were grown in a DMEM medium supplemented with 10% FBS, 2 mM glutamine, 10% non‐essential amino acids, 1% penicillin/streptomycin and 500 μg·mL−1 geneticin (G418) in a 5% CO2 humidified atmosphere at 37°C. Cells were plated on glass treated with poly‐D‐lysine (BD BioCoat coverslips, BD Biosciences Cat.No. 354086) at a density of 50 000–75 000 cells·mL−1 (2 mL of cell suspension were applied to 35 × 10 mm petri dish containing the glass coverslips), kept at 30°C and used, respectively, 1 and 2 days after plating. Experiments were carried out at room temperature.
NMDA receptors: HEK293‐MSRII cells (passages 19–23) were seeded onto 12 mm poly‐D‐lysine coated coverslips (100 000 cells per 35 mm petri dish holding three coverslips), and the cells were transfected 24 h after being seeded. The following DNAs: hNR1A (0.55 μg), hNR2B (2.8 μg) and pCMS‐EGFP (0.8 μg) were added to the petri dish in the presence of optimem 1 (0.25 mL; Invitrogen) and lipofectamine 2000 (9 μL) and incubated for 4 h at 37°C in an atmosphere of 5% CO2. After the transfection medium had been rinsed off, the cells were maintained in culture medium containing 0.3 mM MgSO4 plus 200 μM D‐2‐amino‐5‐phosphonovaleric acid (D‐AP5; NMDA receptor antagonist), at 30°C in an atmosphere of 5 or 10% CO2 for 24 h before the electrophysiological recordings were obtained.
The external recording solution comprised (in mM), 140 NaCl, 2 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 12 HEPES, pH 7.35 (with NaOH). The intracellular solution comprised (in mM) 150 CsCl, 10 EGTA, and 10 HEPES, pH 7.3 (with CsOH). For perforated patch‐clamp recordings, intracellular solution containing 240 μg·mL−1 amphotericin B was used to backfill the pipette while intracellular solution alone was used to fill the tip. Glutamate (Sigma Aldrich) was dissolved in H2O to generate a 1 M stock solution. Cyclothiazide (Sigma Aldrich) was dissolved in DMSO to generate a 100 mM stock solution.
Whole‐cell currents were recorded from cells using the perforated patch‐clamp technique (Hamill et al., 1981; Sherman‐Gold, 1993), using the EPC9 or EPC10 patch‐clamp system and the Pulse programme (HEKA). Patch pipettes were pulled from thin‐wall borosilicate glass capillary (1.5 mm outer diameter) using a P‐97 pipette puller (Sutter Instruments) and had resistances of 2–5 MΩ. Cells were discarded unless the seal formation permitted low resistance access within 10 min (series resistance ≤20 MΩ) due to the amphotericin B. Results were not used when the access resistance changed significantly during the experiment. Exchange of solution around the cell was achieved by holding the cell in front of one of 16–48 channels with a constant solution flow and quickly (10–50 ms) moving the chip to place an adjacent channel in front of the cell (Dynaflow).
The cell was clamped at −60 mV. Automatic series resistance and capacity compensation were applied and checked regularly. The current induced by a 1 s application of the agonist was recorded. The agonist application was repeated every 60 s. Control currents elicited by a fixed concentration of agonist (3 mM, approximate EC50) were recorded during the experiment in order to monitor any variations in charge. For the concentration‐response curve, the AUC of the current for the first 500 ms (charge) was measured for the control application of glutamate alone and for glutamate in the presence of increasing concentrations of the positive modulator (five different concentrations from 10 nM up to 100 μM) in the same cell. Cyclothiazide (Sigma Aldrich) 30 μM, a known AMPA receptor positive modulator, was used as positive control.
Concentration‐response curves for positive modulator were fitted to the equation of the form:
Y is the response in % normalized to 30 μM cyclothiazide, Top is the extrapolated or asymptote maximal % of potentiation, X is the logarithm of concentration. Y starts at 0 and goes to the top with a sigmoid shape. Graphs were constructed by averaging the results from all experiments and fitting a single curve to the pooled data.
Native receptor electrophysiology
Neuronal cultures were prepared from embryonic rat brains harvested following the killing of the pregnant female by CO2 inhalation in accordance with GlaxoSmithKline animal welfare guidelines and the U.K. Animals (Scientific Procedures) Act 1986. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). The dissected hippocampi were placed into ice‐cold Hank's balanced salt solution (HBSS) consisting of: pyruvate, 1 mM; penicillin, 100 mg·mL−1; streptomycin, 100 mg·mL−1; HEPES, 10 mM; NaHCO3, 0.035%, Ca2+‐ and Mg2+‐free. The tissue was then trypsinized for 30 min at 37°C in trypsin/EDTA diluted (final concentration 0.05%) in HBSS with sodium pyruvate (Ca2+‐ and Mg2+‐free). Tissue pieces were physically dissociated, and neurons were plated onto poly‐D‐lysine coated coverslips in plating medium consisting of: neurobasal medium +1 mM sodium pyruvate; penicillin, 100 mg·mL−1; streptomycin, 100 mg·mL−1; B27 supplement 1x; L‐glutamine, 1 mM. Half of the culture medium was replaced twice weekly, and the cells were used for recordings after 5 to 13 days in culture.
AMPA receptors: Extracellular solution comprised (in mM) NaCl 145, KCl 2.5, HEPES 10, glucose 10, CaCl2 1.5, MgCl2 1.2; pH 7.3 with NaOH. Intracellular solution comprised (in mM) CsF 80, CsCl 80, HEPES 10, MgATP 14, DiTRIS creatine phosphate 14, creatine phosphokinase 50 U·mL−1; pH 7.3 CsOH. UoS12258 was dissolved in DMSO to generate a 100 mM stock solution. Test solutions were diluted to concentrations between 10 nM and 100 μM from this stock using extracellular solution (maximum final DMSO concentration ≤ 0.1%). AMPA (Tocris‐Cookson) was dissolved in H2O to generate a 10 mM stock. All experiments were performed at 20–21°C. Whole‐cell voltage‐clamp recordings from rat cultured hippocampal neurons were made using standard methods (Hamill et al. 1981). Briefly, using an Axopatch 200B amplifier and pClamp 8.0 software (Axon Instruments) each cell was held at −70 mV throughout the recording and solution exchange was achieved using a fast‐step two‐tube perfusion system (Biologic RSC 160). Control AMPA receptor‐mediated currents were evoked by rapidly moving from normal extracellular solution to an extracellular solution containing 30 μM AMPA, (approximate EC50 as determined previously) for 2 s then returning to normal extracellular solution for 30 s. This cycle of solution changes was repeated continuously throughout the baseline recording period. The lowest test concentration of UoS12258 was added to both perfusion tubes and the solution change cycle repeated until a stable current was measured. Test concentrations were increased sequentially. Inward currents induced by AMPA application were complex in that their magnitude and duration depended on three different mechanisms: (i) opening of the AMPA receptor channel measured in terms of peak amplitude; (ii) desensitization of AMPA receptors measured as the degree of relaxation of the inward current from its initial peak amplitude to a reduced steady state plateau level in the continued presence of AMPA; and (iii) deactivation of AMPA receptors measured as the rate at which the inward current decayed back to baseline following termination of the AMPA application.
Peak current amplitude was measured as the difference between baseline current and the maximum inward current detected following fast perfusion application of either AMPA alone or AMPA + UoS12258. Peak current amplitude, measured at each concentration of UoS12258 in the presence of 30 μM AMPA, was normalized to the peak current amplitude after application of AMPA alone. Desensitization of AMPA receptors is a measure of the reduction from peak inward current amplitude to a steady‐state current influx in the continued presence of AMPA. AMPA receptor deactivation is the rate at which the inward current returns to baseline after agonist removal and is represented by the decay constant, τ, of a mono‐exponential curve fitted to the relaxing inward current after the AMPA application has stopped. The τ value, measured after each concentration of UoS12258 in the presence of 30 μM AMPA, was normalized to the value of τ after application of AMPA alone. Each of these parameters was measured and analysed using Clampfit 8.0 (Axon Instruments), Excel (Microsoft) and Origin (Microcal). The statistical significance of differences in each of the measured parameters described above was assessed using Student's paired t‐test (Excel).
NMDA receptors: Extracellular solution comprised (in mM): NaCl 145, KCl 2.5, HEPES 10, glucose 10, CaCl2 1.5, glycine (30 μM) – (Mg2+ free to prevent NMDA receptor block). Intracellular solution comprised (in mM): K gluconate 140, HEPES 10, NaCl 17, MgATP 4, NaGTP 0.3; pH 7.3 KOH
Baseline whole cell currents were evoked by rapidly exchanging the perfusing medium from extracellular solution to extracellular solution containing 100 μM NMDA for a period of 2 s. In contrast, test currents were evoked by rapidly exchanging the perfusing medium from extracellular solution containing 100 μM UoS12258 with an extracellular solution containing 100 μM UoS12258 plus 100 μM NMDA for a period of 2 s. As with baseline currents, each test pulse was separated from the next by a 30 s interval.
Kainate receptors: Extracellular solution comprised (in mM): NaCl 145, KCl 2.5, HEPES 10, glucose 10, CaCl2 1.5, MgCl2 1.2; pH 7.3 with NaOH. Intracellular solution comprised (in mM): CsF 80, CsCl 80, HEPES 10, MgATP 14, DiTRIS creatine phosphate 14, creatine phosphokinase 50 U·mL−1; pH 7.3 CsOH. Baseline whole cell currents were evoked by rapidly exchanging the perfusate medium from extracellular solution containing 100 μM SYM2206 (a selective non‐competitive AMPA receptor antagonist) with an extracellular solution containing 100 μM SYM2206 plus 100 μM kainate for a period of 2 s. In contrast, test currents were evoked by rapidly exchanging the perfusate medium from extracellular solution containing 100 μM SYM2206 plus 100 μM UoS12258 with an extracellular solution containing 100 μM SYM2206 plus 100 μM UoS12258 plus 100 μM kainate for a period of 2 s. As with baseline currents, each test pulse was separated from the next by a 30 s interval.
In vivo electrophysiology
All animal studies were ethically reviewed and carried out in accordance with Animals (Scientific Procedures) Act 1986 and the GSK Policy on the Care, Welfare and Treatment of Animals. GlaxoSmithKline safety regulations were adhered to at all times.
Adult male Lister Hooded rats (Charles River) weighing 243–340 g were housed under a 12 h light/dark cycle with food (Harlan Maintenance Diet) and water available ad libitum.
For electrophysiological recordings, rats were prepared individually. Specifically, once removed from their home cage, they were placed in an induction chamber and anaesthetized with 5% isoflurane in a 1:1 mix of oxygen and nitrous oxide, which was adjusted to 2.5% isoflurane to maintain anaesthesia throughout the surgical procedure. Surgery involved cannulating the jugular and femoral veins using Portex tubing such that the cannula projected externally from the animal. Each rat was then placed in a stereotaxic frame and secured with atraumatic ear bars. Body temperature was monitored and maintained at appropriate levels by means of a thermostatically‐controlled heated blanket and rectal probe. A midline incision was made to expose the dorsal aspect of the skull and a burr hole made to allow implantation of a multi‐barrelled electrode (Kation Scientific, Hungary) into the CA1 region of the hippocampus (from Bregma: AP −4.2 mm, ML 2.4 mm, depth 1.8–2.2 mm). The multi‐barrelled electrode consisted of a carbon fibre recording electrode and three glass barrels, two of which were filled with the following solutions: one barrel contained 2% w.v‐1 methylene blue dissolved in 0.5 M sodium acetate, which acted as the balance for the current ejection, and the second barrel contained 5 mM AMPA. A silver ground electrode was also positioned under the skin margin. Isoflurane levels were lowered to 1.75% before commencement of the recordings. Neuronal activity was recorded using a Neurolog NL100 AK head stage connected to a NL104A AC preamplifier. The signal was filtered (0.5–5 kHz), passed through a Humbug noise eliminator and fed into an audio amplifier and a CED 1401 digital analogue interface. Spike 2 software (CED) was used to record neuronal activity. AMPA was ejected via iontophoresis (currents of 4–40 nA) for periods of 40 s, repeated at intervals of 40–60 s.
UoS12258 was prepared as a nano‐milled formulation in 1% w.v‐1 HPMC (Pharmacoat 603)/0.1% sodium lauryl sulphate (Sigma‐Aldrich) in purified water. The diluent and vehicle was 1% w.v‐1 HPMC/0.1% sodium lauryl sulphate, and all i.v. doses of UoS12258 were administered as 0.05 mL. 100 g‐1. For p.o. dosing, the same batch of UoS12258 that was used for i.v. dosing was administered (batch number: VNAA\6768\149\4), vehicle was 1% methylcellulose and dose volume was 2 mL·kg−1 in all cases. AMPA (Sigma‐Aldrich) was prepared as a 5 mM stock solution in 0.9% saline (pH 8).
For acute administration, animals were divided into two groups, UoS12258 (n = 4) and saline (n = 3). Once a minimum of five consistent and consecutive responses to AMPA application had been achieved, either UoS12258 or saline was injected. In this respect, rats in group one were injected i.v. dosed with UoS12258 in a cumulative dosing paradigm consisting of 0.5, 1.0 and 1.5 mg·kg−1 doses delivered at 20 min intervals. A similar procedure was used for the second group of rats except that three doses of saline were administered i.v. at 20 min intervals. For sub‐chronic administration of UoS12258 animals were divided into four groups: vehicle–vehicle (n = 5); vehicle–UoS12258 (n = 5); UoS12258–UoS12258 (n = 5) and UoS12258–vehicle (n = 5), where the first treatment term indicates what the animals were treated with for the 7 day period leading up to the test treatment on day 8; this latter treatment being indicated by the second treatment term. The dose of UoS12258 used for sub‐chronic dosing was 0.03 mg·kg−1 (p.o.) in a dose volume of 2 mL·kg−1 and the vehicle was 1% methylcellulose (p.o.). All animals were treated at approximately 09 h every day for 7 days. Animals receiving UoS12258 on the test day were injected i.v. using a cumulative dosing paradigm comprising of 0.1, 0.5 and 1.0 mg·kg−1 at 20 min intervals, whereas animals receiving vehicle on the test day received three doses of saline at 20 min intervals.
Brains and blood samples (50 μL) were collected at the end of each experiment and frozen at −80°C in brain and EDTA tubes, respectively, until analysed. UoS12258 content was determined by LC–MS/MS using a Quattro Premier (Micromass) mass spectrometer with positive‐ion electrospray ionization.
Novel object recognitions (NOR)
Adult male Lister Hooded rats (Charles River) weighing 250–320 g on the day of testing were housed in groups of four in the absence of environmental enrichment (sawdust and bedding only) and maintained on a 12 h light/dark cycle with lights on at 06:00 h. Food (Harlan Maintenance Diet) and water were available ad libitum. For all experiments, rats were habituated to the perspex test arenas (42 cm length × 21 cm width × 20 cm height) for 60 min (am) and 3 min (pm) on the day before the testing. For experiments 1 and 2, rats were sham dosed with vehicle on four occasions before being tested. On the first test day, rats were placed in a test arena for a 3 min habituation period and then presented with two identical objects (T1, 3 min). Twenty‐four hours later rats were placed back in the test arena and presented one of the previous (familiar) objects and a novel object (T2, 3 min). Objects used were of similar size, black plastic ‘kong’ and cylinder shapes (approximately 6.5 cm height × 6 cm width). Objects were cleaned with 70% ethanol between animals to remove any odour traces. The novel object and novel object side (left or right) was randomized equally across all treatment groups. The time spent exploring the objects during T1 and T2 was recorded by an observer blind to the novel object. Rats received UoS12258 or vehicle by oral gavage (p.o.). Treatment group sizes were n = 12 unless stated otherwise. For acute studies, rats were treated 4 h before both T1 and T2 at 0.1, 0.3 and 1 mg·kg−1 p.o. For sub‐chronic studies, rats were treated once daily (08–10 h) for 7 days followed by a single dose 4 h before T1 (day 8) and T2 (day 9) at 0.003, 0.01 and 0.03 mg·kg−1 p.o.
In a separate study, male Lister Hooded rats were administered two doses, 24 h apart, of UoS12258 (0.1, 0.3 and 1 mg·kg−1 p.o.) separated by 24 h to simulate the dosing conditions used in acute behavioural studies. Animals were killed 4 h after the final dose following schedule 1 procedures; blood and brain samples were taken for analysis. For sub‐chronic experiments, animals were killed immediately following testing in T2 (n = 3) and blood and brains removed for analysis (as detailed for in vivo electrophysiology).
Water maze
This paradigm is used to investigate spatial learning and memory where learning and subsequent retention of the location of a hidden platform are measured. Aged rats show an inherent deficit in this paradigm such that they do not show an improvement in acquisition of the platform position over the course of training and therefore fail to learn where the platform is located, as assessed during the probe tasks performed at the end of the training. The effects of sub‐chronic administration of UoS12258 (0.3, 1, 3 and 10 mg·kg−1 p.o.) upon age‐induced cognitive deficits were determined in aged (22 months old) male Wistar rats. UoS12258 was administered once daily in 1% methylcellulose for 8 days before the training and then on each training day (3 h prior to each session) and also during the recall period, consistent with previous dosing protocols. Plasma samples were not taken during this study. During each trial, the latency to find the hidden platform (to a maximum of 90 s) was recorded. Animals received five trials per day (inter‐trial interval of 300 s) for 4 days. Recall of platform position was assessed by a probe test at 1, 3 and 7 days following the final training session, in which the platform was removed and animals were allowed to explore the maze for 30 s. The time spent in each quadrant was recorded.
Passive avoidance (PA)
The PA procedure is based on the ability of rats to associate a specific context (dark chamber) with an aversive conditioned stimulus (0.75 mA, 0.5 s scrambled foot shock). This experiment investigated the effects of acute UoS12258 on a scopolamine (0.08 mg·kg−1 i.p.)‐induced deficit in PA in male Wistar rats. UoS12258 (0.1, 1, 3 and 10 mg·kg−1 p.o.) with the 5‐HT6 receptor antagonist, SB‐399885 T (10 mg·kg−1 p.o.) used as a positive control. Test drugs were administered in 1% methylcellulose 3 h before the onset of training. Scopolamine hydrobromide (0.8 mg.kg−1 i.p.) was dissolved in saline and administered 6 h post training. Plasma samples were not taken during this study. On the training day, rats were treated with vehicle or UoS12258 3 h prior to assessment of spontaneous activity in the open field apparatus. Immediately afterwards, the rats were placed in the light compartment and the latency to enter the dark chamber was recorded. Upon entering the dark compartment, rats received a foot shock (0.75 mA, 0.5 s) and returned to the light chamber whereupon they were returned to their home cage. Recall of this inhibitory stimulus was evaluated 24 h post‐training by returning the animal to the light chamber and recording the latency to enter the dark chamber up to a maximum of 600 s. At no point during the test period was a foot shock given.
Microdialysis
Male Lister Hooded rats (250–275 g) were housed in groups of four, kept under a 12 h light/dark schedule, lights on at 07:00 h with food and water available ad libitum.
General anaesthesia was induced using isoflurane. Once deep anaesthesia had been obtained, rats were administered synulox (antibiotic; s.c.) and rimadyl (analgesic; s.c.), and the rat transferred to a stereotaxic frame. The skull was exposed and holes drilled relative to the Bregma, four anchor screws (1.6 × 3 mm stainless steel cheese head screws, Wood and Hughs), and additional holes drilled at 12° to the dura surface for placement of microdialysis guide cannula (CMA 11, CMA, UK) into the dorsal hippocampus (AP: + 3.5 mm, ML −2.0 mm, and DV – 2.0) and anterior cingulate cortex (AP: + 2.7 mm, ML −1.6 mm, and DV – 2.0 mm) according to Paxinos and Watson (1986). Probe and anchor screws are secured in place with dental cement (Poly‐F‐plus, zinc polycarboz cement, Cladius Ashe), and the wound sealed. Postoperative fluids (5 mL saline, 2.5 mL per side; s.c.) and analgesic (Nubain 0.4 mL·100 g−1; s.c.) were administered to aid recovery. The rats were monitored until they regained their righting reflex and received at least 5 days post‐operative care.
Eighteen hours before the start of the dialysis experiment, rats were moved into microdialysis cages (285 mm diameter, 355 mm high; manufactured in‐house) to allow for overnight acclimatization to the dialysis procedure room. On the morning of each experiment, microdialysis probes (CMA 11, 14/02, Linton Instruments; 2 mm active membrane) were perfused with a buffered artificial CSF (aCSF) solution (comprising in mM: 145 NaCl, 2.7 KCl, 1 MgCl2, 1.2 CaCl2 and 2 Na2HPO4) at 1 μL·min−1 via a dual‐channel liquid swivel (Instech 375/D/22QE microdialysis swivel, BAS). Microdialysis probes were inserted into the implanted guide cannulae and perfused with aCSF for 2 h before sampling began, to allow for equilibration of neurotransmitter levels, following which samples were collected every 30 min. Four dialysate samples were collected to generate baseline readings before administration of either test compound. UoS12258 was dissolved in 1% methylcellulose and sonicated prior to administration. Doses of UoS12258, 0.1, 0.3 and 1 mg·kg−1, or vehicle (1% methylcellulose) were administered in a volume of 2 mL·kg−1, p.o. Following administration of compound, a further 10 dialysate samples were collected until the end of sampling. Dialysate samples were analysed for 5‐HT, dopamine (DA) and noradrenaline (NA) using HPLC with electrochemical detection and ACh using mass spectrometry. At the end of each experimental day, animals were returned to their home cage and re‐used in a randomized cross‐over design, allowing at least 7 days drug washout before subsequent uses. After the final experiment, brains were removed and stored in formalin solution for probe placement verification.
All dialysate samples were calculated as a subsequent percentage of the average absolute levels of neurotransmitters from the four baseline samples prior to administration of compound. Significant differences between groups of the same compound were calculated by repeated measures ANOVA followed by Fischer's least significance difference post hoc test where appropriate with significance set at P < 0.05 (Statistica v6.0).
Maximal electroshock threshold test
Adult male Sprague Dawley or Lister Hooded rats (Charles River) weighing 95 to 160 g on the day of testing were housed in groups of six or less and maintained on a 12 h light/dark cycle with lights on at 06:00 h. Food (Harlan Maintenance Diet) and water were available ad libitum. Treatment groups of 12 animals were administered UoS12258 at 10, 30 or 100 mg·kg−1 by p.o. gavage 2 h before testing (expt 1) or 30 mg·kg−1 p.o. 2, 5 or 7 h before testing in Sprague Dawley rats (expt 2) or 1.0, 3.0, 10 mg·kg−1 4 h before testing in Lister Hooded rats (expt 3). In all experiments, the known pro‐convulsant, picrotoxin (2.0 mg·kg−1 as a positive control) was administered i.p. 30 min before testing. After the animals had been treated, they were returned to their home cages. Testing occurred in a separate room and consisted of assessing the induction of a tonic hind limb extensor seizure following a 0.1 s shock administered via corneal electrodes according to the ‘up and down’ method (Kimball et al., 1957). In all experiments, a separate group of animals (n = 3 per dose) was treated and blood and brain samples taken at the appropriate pretreatment time for analysis of compound levels. The threshold for tonic hind limb extensor seizures was determined using a Hugo Sachs Elektronic stimulator, which delivered a constant current of 0.1 s duration, 50 Hz, sine wave form, fully adjustable between 1 to 300 mA, via corneal electrodes. The electrodes were briefly immersed in saline before application of the electroshock in order to achieve good electrical contact.
Results
Functional activity at recombinant human GluA2i homomeric AMPA receptors
The functional activity of UoS12258 at the hGluA2i homomeric AMPA receptor stably expressed in HEK293 cells was assessed using FLIPR/Ca2+ influx methodology. Application of glutamate (100 μM) alone did not elicit a signal due to rapid desensitization of the AMPA receptor. However, in the presence of UoS12258, application of glutamate consistently increased intracellular Ca2+ levels such that an increase in the concentration of UoS12258 produced a progressive potentiation of glutamate‐induced responses. A non‐linear, four‐parameter logistic curve‐fit of the data generated pEC50 and maximum potentiation values (relative to the established AMPA receptor positive modulator cyclothiazide, 150 μM) of 5.57 ± 0.07 and 100.7 ± 3.9% respectively (n = 6; Figure 1). When applied alone, in the absence of glutamate, UoS12258 did not affect intracellular Ca2+ levels indicating that it possessed no intrinsic agonist activity.
Electrophysiological activity at human recombinant GluA2i homomeric AMPA receptors
Using this approach, UoS12258 potentiated glutamate‐induced whole‐cell, AMPA receptor‐mediated currents producing a maximal response that amounted to 112 ± 32% of the maximal response induced by the reference AMPA receptor positive modulator cyclothiazide (30 μM). The pEC50 value (5.19 ± 0.02; n = 4, Figure 2) for this positive modulatory effect of UoS12258 was in close agreement to that generated using the FLIPR/Ca2+ influx assay described above.
Figure 2.
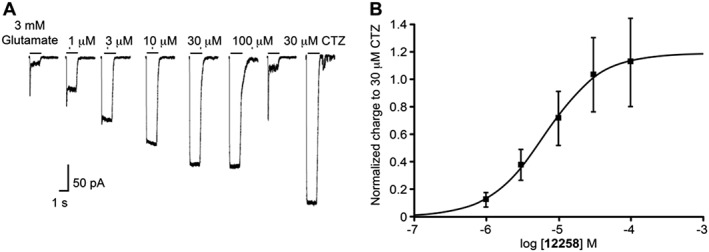
(A) Representative whole cell current traces recorded from a single HEK293 cell expressing hGluA2i homomeric AMPA receptors. UoS12258 (1–100 μM, upper concentration limited by solubility) produced a concentration‐dependent and reversible increase in charge transfer. Positive modulator, cyclothiazide (CTZ) 30 μM, also increased charge transfer following application of glutamate (line 7). (B) Concentration–response curve for the potentiation of hGluA2i‐mediated inward currents by UoS12258, pEC50 = 5.19. Responses are normalised to 30 μM cyclothiazide. n = 4 independent experiments.
Functional activity at recombinant human GluA1i, GluA3i, GluA4i and rat GluA2i homomeric AMPA receptors
To demonstrate (i) the broad spectrum positive modulatory activity of UoS12258 across AMPA receptors formed from different GluA subunits, and (ii) lack of species‐dependent pharmacology, FLIPR‐based assays were used to assess its activity at human AMPA receptors formed by homomeric assembly of GluA1i, 3i or 4i subunits as well as homomeric assembly of rat GluA2i subunits transiently transfected into HEK293 cells (Table 1). There was less than a 10‐fold difference between pEC50 values for all hGluAi homomeric AMPA receptors and between rat and human GluA2i indicating that UoS12258 possesses little specificity for individual GluA subunits or across species (Supporting Information Table 1).
Table 1.
Effect of UoS12258 (10, 30, 100 mg·kg−1, p.o., 2 h pretreatment) in CD rats in the pro‐convulsant MEST test
| UoS12258 (mg·kg−1) | CC50 (mA) | SEM | Change compared to vehicle (%) | Mean blood levels (ng·mL−1) | Mean brain levels (ng·g−1) |
|---|---|---|---|---|---|
| Saline | 58.5 | 3.4 | – | – | – |
| Picrotoxin | 25.8 | 1.9 | −56 * | – | – |
| 1% MeCell | 55.5 | 3.7 | – | – | – |
| 10 | 50.0 | 4.9 | −10 | 1285 | 2283 |
| 30 | 32.5 | 3.2 | −41 * | 2136 | 3390 |
| 100 | 23.5 | 2.1 | −58 * | 2151 | 4961 |
P < 0.05, significant difference compared to vehicle treated animals.
Potentiation of rat native AMPA receptor‐mediated responses
Repeated (2 s duration, every 30 s) applications of 30 μM AMPA in rat cultured hippocampal neurons produced inward currents, the peak amplitude and waveform of which were consistent in magnitude between successive applications. Measurements taken reflected the three different mechanisms which occurred during this period: (i) opening of the AMPA receptor channel measured in terms of peak amplitude; (ii) desensitization of AMPA receptors measured as the degree of relaxation of the inward current from its initial peak amplitude to a reduced steady state plateau level in the continued presence of AMPA; and (iii) deactivation of AMPA receptors measured as the rate at which the inward current decayed back to baseline following termination of the AMPA application.
While UoS12258 alone did not evoke a whole‐cell inward current at any concentration tested (10 nM–00 μM), application of 30 μM AMPA in the presence of UoS12258 produced inward currents that exhibited larger peak amplitudes, reduced desensitization and slowed deactivation, which all contributed to an increase in charge transfer (area under the inward current), relative to control responses in the absence of UoS12258. The effect of UoS12258 on each of these electrophysiological parameters was clearly concentration‐dependent and was first evident in the nM concentration range.
Thus, although the pEC50 and maximal potentiation of charge transfer induced by UoS12258 were increased by 5.1 and a 4.1 ± 0.97‐fold, respectively, 10 nM UoS12258 produced a statistically significant 1.4‐fold increase in charge transfer when compared to AMPA alone ( n = 8; P < 0.05, Student's paired t‐test, Figure 3A).
Figure 3.
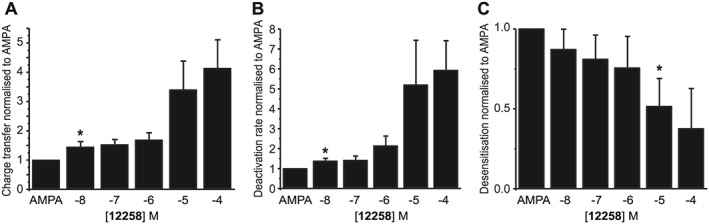
Rat native neuronal data. (A) Concentration‐response plot for UoS12258‐mediated potentiation of charge transfer. (B) Concentration‐response plot for UoS12258‐mediated potentiation of AMPA receptor deactivation. (C) Concentration–response plot for the inhibition of AMPA receptor desensitization induced by UoS12258. Responses were normalized to 30 μM AMPA control. Lowest concentration to cause a significant (*P < 0.05) difference.
UoS12258 produced a concentration‐dependent increase in the rate constant of AMPA receptor deactivation (pEC50 5.3) such that at 100 μM the rate of deactivation was 5.9 ± 1.5‐fold greater than that produced by AMPA application alone. As with charge transfer UoS12258 also potentiated this component of the AMPA receptor‐mediated response at nM concentrations such that at 10 nM it produced a 44 ± 6% decrease in the rate of deactivation (P < 0.05, Student's paired t‐test n = 7; Figure 3B).
In contrast to the significant increases in both charge transfer and rate of deactivation, inhibition of AMPA receptor desensitization by UoS12258 was more variable and only statistically significant at concentrations of 10 μM and above where a greater than 52 ± 17% reduction in AMPA receptor desensitization was observed (n = 7, P < 0.05, Student's paired t‐test, Figure 3C).
Notably, since the minimum effective concentration of UoS12258 on desensitization was 10 μM, the previously described potentiation of charge transfer at 10 nM reflects UoS12258‐mediated potentiation due to changes in peak amplitude and deactivation (Supporting Information Table S2).
Selectivity versus recombinant NMDA and kainate receptors
In the NMDA receptor FLIPR assay, UoS12258 (tested up to 50 μM, n = 4) did not show any activity either as an opener or blocker at human GluN1/2B receptors. Similarly, in the electrophysiology experiments, UoS12258 did not potentiate GluN1/2B receptor‐mediated activity at either 10 nM, the minimum concentration (paired t‐test, n = 5) or 1 μM, 100 times the minimum concentration (paired t‐test, n = 5) shown to potentiate AMPA‐mediated currents; although there was a partial inhibition (18 ± 1% at 100 μM, P < 0.05, paired t‐test, n = 5) when tested at 100 μM.
In FLIPR experiments on transiently expressed GluK1 (Q‐edited) homomeric kainate receptors, UoS12258 (up to 50 μM, n = 3) did not show any activity either as an agonist, antagonist or positive modulator. Identical results were obtained using electrophysiological analysis.
Functional selectivity versus rat native NMDA and kainate receptors
In separate neurons, repeated applications of either NMDA, in the presence of NMDA receptor co‐agonist glycine, or kainate, in the presence of the selective AMPA receptor antagonist SYM2206, generated reproducible inward currents that were mediated by activation of NMDA receptors and kainate receptors, respectively, and that were not significantly affected by UoS12258 (100 μM) (n = 4 for both; paired t‐test). There were also no significant changes in NMDA receptor‐mediated charge transfer with UoS12258 at either 10 nM, the minimum concentration (paired t‐test, n = 4) or 1 μM, 100 times the concentration (paired t‐test, n = 4) shown to potentiate AMPA‐mediated currents.
Potentiation of AMPA receptor‐mediated synaptic responses in vivo
The effect of UoS12258 was evaluated in vivo in an electrophysiological model where synaptic connectivity is intact, specifically the potentiation of electrically evoked AMPA receptor‐mediated synaptic potentials were recorded from the dentate gyrus of the anaesthetized rat. Electrical stimulation of the medial perforant pathway evoked a population spike recorded in the hippocampal dentate gyrus granule cell layer. UoS12258 (0.1 mg·kg−1 i.v.) significantly increased population spike amplitude by 18 ± 3% compared to vehicle (Figure 4, P < 0.05, n = 4 ANCOVA, univariate test of significance. Analysis of brain samples taken immediately after the experiment generated a mean UoS12258 concentration of 198 ± 40 ng·g−1). Brain tissue binding for UoS12258 was measured as 97.4% resulting in an estimated mean unbound concentration of 5.1 ng·g−1, which equates to an approximate free concentration of 15 nM, which is in close agreement with the MEC (10 nM) for UoS12258 induced potentiation of AMPA receptor‐mediated charge transfer and deactivation in rat hippocampal neurons.
Figure 4.
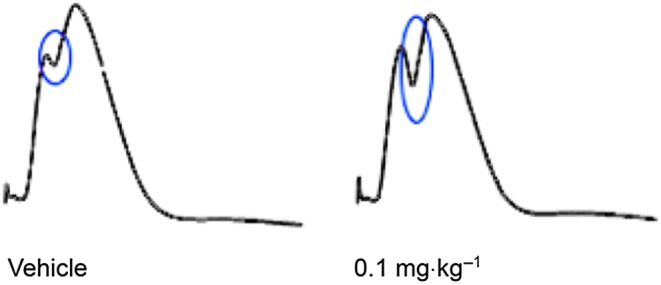
Representative traces recorded from the dentate gyrus of the anaesthetized rat following vehicle (left trace) and 0.1 mg·kg−1 i.v. administration of UoS12258 (right trace). Synaptic traces are an average of 10 consecutive responses; the population spike is ringed in blue.
NOR test
The NOR test is a two‐trial recognition memory test of the ability of rats to discriminate between novel and familiar objects. NOR can be impaired by inserting a time delay (24 h) between presentations of the objects.
Data are expressed both as total time spent exploring the novel and familiar objects in T2 and as the d2 index (proportion of time exploring the novel object in T2). In the first study, acute UoS12258 (0.1, 1, 10 mg·kg−1) produced a bell‐shaped dose‐response curve (i.e. improvement in object recognition at 1 mg·kg−1 with a loss of effect at 10 mg·kg−1). At the higher dose 1 rat out of 12 had a seizure and was excluded from the test. Based on an efficacious dose of 1 mg·kg−1, a follow up experiment was conducted to assess a lower dose range (i.e. 0.1–1 mg·kg−1 p.o.). Following acute administration, UoS12258 significantly increased novel object exploration compared to vehicle‐treated rats at 0.3 (P < 0.05) and 1 mg·kg−1 (P < 0.05, Figure 5A) and the d2 index (P < 0.05, Figure 5A). In a separate study, mean blood and brain levels of UoS12258 were determined in rats dosed under identical conditions (n = 3) and are shown in Supporting Information Table S3.
Figure 5.
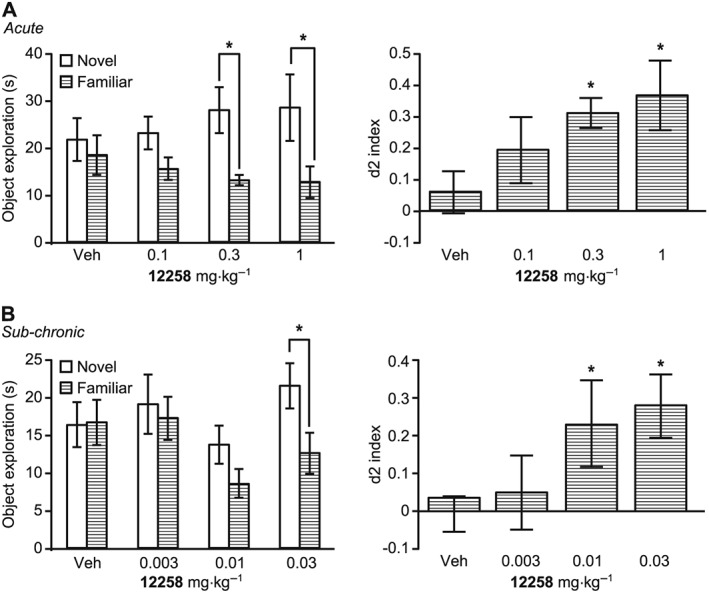
(A) Effects of UoS12258 on NOR in rats after acute administration (0.1–1 mg·kg−1 p.o., 4 h prior to T1 and T2); (B) Effects of UoS12258 on NOR in rats after 7 day sub‐chronic administration (0.003–0.03 mg·kg−1 p.o. dosed daily for 7 days and then 4 h prior to T1 and T2). Statistical analysis was performed using ANOVA, followed by planned comparisons (*P < 0.05) to vehicle‐treated animals.
Following 7 day sub‐chronic dosing plus treatment 4 h before T1 and T2, UoS12258 significantly increased novel object exploration compared to vehicle treated rats at 0.03 mg·kg−1 (P < 0.05, Figure 5B). The d2 index was significantly increased at both 0.01 and 0.03 mg·kg−1 (P < 0.05, Figure 5B). The sub‐chronic MED was determined as 0.03 mg·kg−1 based on significant changes in both measures. Mean blood and brain levels of UoS12258 in rats sampled immediately after testing are presented in Supporting Information Table S4 (n = 3). A previous sub‐chronic NOR study with UoS12258 (0.03, 0.1, 0.3 mg·kg−1) also exhibited a bell‐shaped dose–response curve with efficacy at 0.03 and 0.1 mg·kg−1 and no effect at 0.3 mg·kg−1. Thus, two studies confirm that sub‐chronic dosing with UoS12258 results in increased potency to facilitate NOR learning, in accordance with data reported in the literature for compounds of this class.
Passive avoidance
Neither UoS12258 nor SB‐399885T (5‐HT6 receptor antagonist) had any effect on spontaneous open field activity (data not shown), suggesting a lack of confounding motor effects. Scopolamine impaired passive avoidance (P < 0.05, Figure 6). UoS12258 (10 mg·kg−1) did not impair performance when given alone, and prevented the scopolamine‐induced impairment in a dose‐dependent manner (P < 0.05), with significant effects observed at 3 and 10 mg·kg−1 (P < 0.05). The positive control SB‐399885T also prevented the scopolamine‐induced memory deficit (P < 0.05).
Figure 6.
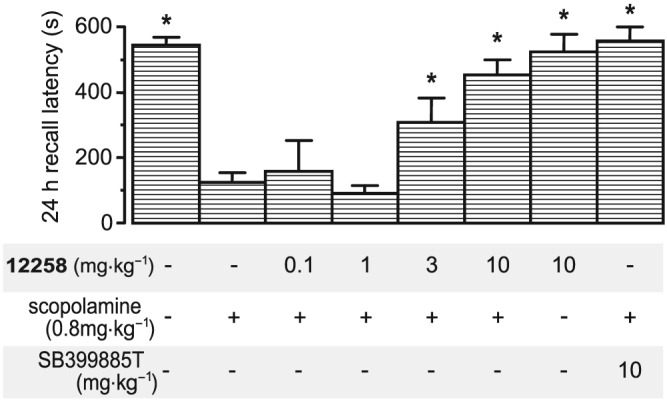
Effects of acute UoS12258 and SB‐399885T on scopolamine‐induced impairment of a passive avoidance response in adult Wistar rats. Data represent mean ± SEM avoidance latency at the 24 h recall time. Significant difference (*P < 0.05) compared to scopolamine‐treated animals (Mann–Whitney U‐test, n = 6 per group).
Morris water maze in aged rats
UoS12258 significantly reduced escape latencies following 3 and 10 mg·kg−1 (Figure 7; F[1180] = 14.1 and 5.9, P < 0.05 for 3 and 10 mg·kg−1 vs. vehicle‐treated controls).
Figure 7.
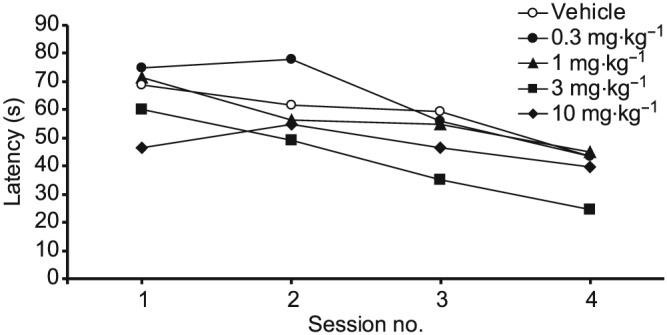
Effect of sub‐chronic UoS12258 on acquisition of a water maze spatial learning task by aged Wistar rats. Data represent mean ± SEM escape latency (s) averaged over the five trials of each training session. By session 3 there was a significant, P < 0.05, difference between groups administered either 3 or 10 mg·kg−1 compared to vehicle treated animals (n = 6 per group).
UoS12258 significantly reduced swim angle, the angle between start position and the position of the hidden platform in the water maze, over the four training sessions (F[1,36] = 19.7 and 5.1; P < 0.05 for 3 and 10 mg·kg−1 vs. vehicle‐treated), suggesting an improved search strategy (data not shown). UoS12258 had variable effects upon swim speed, that is, no consistent increase in swim speed at doses shown to be efficacious at improving acquisition, which therefore does not explain the reduced latency to find the hidden platform.
Recall of the hidden platform position was assessed by probe trials carried out 1, 3 and 7 days following the final training session. UoS12258 (3 mg·kg−1) improved recall of the task in all three probe trials (Figure 7, P < 0.05, Mann–Whitney U‐test). UoS12258 (10 mg·kg−1) significantly improved recall in the 1 day post‐training probe trial only (Figure 8).
Figure 8.
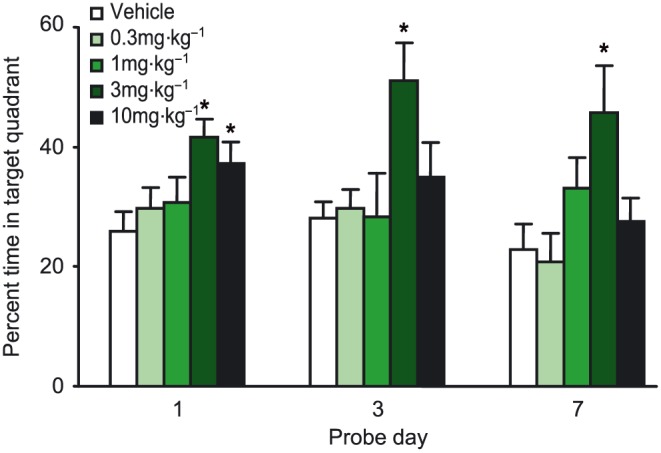
Effect of sub‐chronic UoS12258 on recall of a water maze spatial task by aged rats. Data represent mean ± SEM time (s) swimming in the target quadrant of the water maze during probe trials 1, 3 and 7 days post‐training. Significant difference (*P < 0.05) compared to vehicle treated animals.
Microdialysis
UoS12258 significantly (F3,23 = 5.94, P < 0.05) increased extracellular levels of ACh in the anterior cingulate cortex at all three doses and induced a significant (F3,22 = 5.076, P < 0.05) increase in the dorsal hippocampus again at all three doses (Figure 9).
Figure 9.
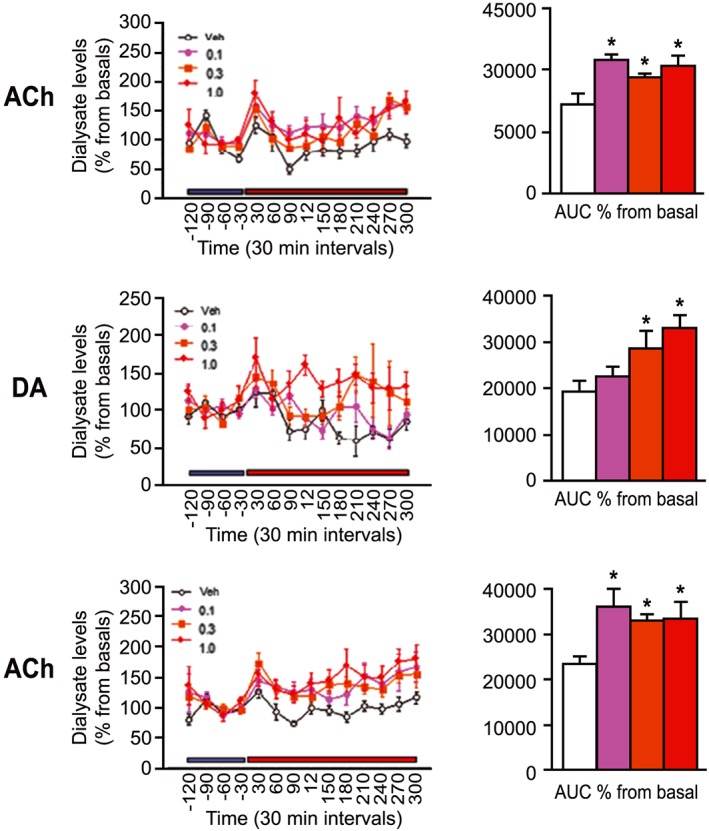
Changes in levels of ACh and dopamine (DA) expressed as % changes from basal (line graphs) and total efflux (bar chart – calculated as AUC of % changes from basal levels) in the anterior cingulate cortex (left panel) and dorsal hippocampus (right panel) following administration of UoS12258 (0.1, 0.3 and 1.0 mg·kg−1, p.o). *P < 0.05, significant differences compared to vehicle treated animals.
UoS12258 also induced significant (F3,24 = 4.47, P < 0.05) increases in extracellular dopamine levels at both 0.3 mg·kg−1 (P < 0.05) and 1 mg·kg−1(P < 0.05) (Figure 9). There was no significant effect of AMPA positive modulation on extracellular levels of 5‐HT or NA in any of the brain structures studied.
Side effect profiling using maximal electroshock seizure threshold test (MEST)
In order to assess the potency of UoS12258 to potentiate induced seizure activity, rats were pretreated with the compound prior to induction of seizure. Corneal application of an electrical current (CC50 approximately 60–70 mA, 0.1 ms duration) in the rat induces tonic and full tonic–clonic seizures. In separate experiments, UoS12258 was administered 10, 30, 100 mg·kg−1, p.o., 2 h pretreatment; 30 mg·kg−1, p.o., 2, 5, 7 h pretreatment to SD rats; 1.0, 3.0 10 mg·kg−1, p.o., 4 h pretreatment to LH rats before testing for pro‐convulsant activity. LH rats were chosen to provide a comparison with the NOR potency data.
In SD rats, UoS12258 significantly decreased seizure threshold at 30 and 100 mg·kg−1 by −41 (3390 ng·g−1 in brain) and −58% (4961 ng·g−1 in brain), respectively, 2 h post dose (Table 1). A subsequent DMPK study showed that the concentration was equivalent at 2 and 5 h. Therefore, in a repeat study, UoS12258 (30 mg·kg−1 p.o.) was tested 2, 5 and 7 h post dose. Seizure threshold compared to vehicle (5 h pretreatment) was reduced at all time points by −54, −43 and −57 (2, 5 and 7 h pretreatment respectively). At 2 h 1/13 animals and at 7 h 1/15 animals showed spontaneous convulsions and were killed just before testing. In LH rats, UoS12258 significantly reduced seizure threshold by −38% at 10 mg·kg−1 (Supporting Information Table S5). Both rat strains show similar brain exposure (2446 ng·g−1 in LH vs. 2283 ng·g−1 SD at 10 mg·kg−1).
Discussion
In this article, we present data from experiments that describe the pharmacological properties of UoS12258, a novel and selective AMPA receptor positive modulator. Although, as stated earlier, many molecules have been investigated preclinically and several have progressed into clinical trials, there is limited full disclosure of the enabling preclinical data sets – particularly regarding behavioural and tolerability data. To address this situation, we disclose the data set generated for this molecule, which has progressed into clinical evaluation, including details of the differences observed in behavioural studies between acute and repeat dosing and also the clear side effects seen at elevated concentrations. In addition to the studies reported below, a wider investigation was conducted of UoS12258 in additional behavioural pharmacology models, for which the data were either inconclusive: reversal of ketamine or amphetamine‐induced hyperactivity in rat and reversal of quinelorane (D2/3 receptor agonist)‐induced hypoactivity in rat, or only effective at high doses: mouse forced swim test (10 mg·kg−1 p.o. administered 240 min prior to test gave efficacy of imipramine dosed i.p. 30 min prior to test). Full data for these studies are not included in this manuscript due to space restrictions.
Characterization using recombinant systems
We investigated the effect of UoS12258 on intracellular Ca2+ influx using FLIPR‐based assays, and the results suggest that UoS12258 shows no clear subunit specificity and should act equipotently at heteromeric AMPA receptors with different subunit compositions, which exist in native tissues. Also, given that pEC50 values for UoS12258 were similar at rat and human, there appears to be little evidence of species selectivity. Using the same recombinant system, HEK293‐hGluA2i stable cell line with whole‐cell voltage clamp electrophysiology, the pEC50 for UoS12258 was again similar to the FLIPR values with this AMPA receptor subunit.
Characterization using native systems
Although rates of receptor activation and deactivation are comparable across all homomeric AMPA receptors and are therefore independent of AMPA receptor GluA subunit complement, desensitization kinetics differ depending upon AMPA receptor subunit stoichiometry (see Schlesinger et al., 2005). As whole cell currents in neurons sample activation of a heterogeneous population of AMPA receptors that will differ from neuron to neuron, it is likely that the observed variation in desensitization is due to the heterogeneity of the AMPA receptor populations assessed. Activation (charge transfer) and deactivation of AMPA receptors are measures that define the onset and offset of glutamatergic activity and are therefore directly relevant to the propagation of synaptic transmission and synaptic plasticity. Receptor desensitization, during which the receptor becomes inactive in the continued presence of the ligand, has been more closely associated with excessive or pathological glutamatergic activity. In native hippocampal neurons using whole‐cell voltage clamp electrophysiology, it was possible to measure AMPA receptor‐mediated charge transfer and deactivation which were potentiated significantly by concentrations of UoS12258 as low as 10 nM. It is interesting, however, that the pEC50 of UoS12258 on charge transfer, desensitization and deactivation of AMPA‐mediated currents in native neurons are all in the same range as the pEC50 values in recombinant systems. There are clearly many physiological differences between recombinant and native systems for example increased levels of phosphorylation, the presence of transmembrane AMPA regulatory proteins or increased AMPA receptor expression at the cell membrane, which could explain the observed low concentration‐mediated potentiation of AMPA receptors by UoS12258 in the native system. Perhaps of more interest this concentration is comparable to the concentrations measured in the brain immediately after in vivo electrophysiological studies, in which UoS12258 increased AMPA receptor‐mediated synaptic activity. Together these data mutually support the observation that, at equivalent concentrations, UoS12258 evoked significant potentiation of AMPA receptor‐mediated neuronal activity both in vitro and in vivo.
The selectivity of UoS12258 for AMPA subtype glutamate receptors could however be a desirable property since activation and/or potentiation of other ionotropic glutamate receptor subtypes (i.e. NMDA and kainate) could cause excessive glutamate channel opening, increasing Ca2+ influx and leading to excitotoxic damage. To establish its selectivity for the AMPA receptor, the functional activity (agonism, antagonism and positive modulation) of UoS12258 was assessed at the human heteromeric NMDA receptor GluN1/2B and the human GRIK1 (Q) homomeric kainate receptor using both FLIPR/Ca2+ influx and electrophysiological methods.
UoS12258 did not potentiate NMDA receptor, GluN1/2B‐mediated activity at any concentration tested whether tested by Ca2+ influx or whole‐cell patch clamp. Similarly, in FLIPR experiments on transiently expressed kainite receptors, UoS12258 did not show any activity either as an agonist, antagonist or positive modulator. Identical results were obtained using electrophysiological analysis. UoS12258 also produced no significant potentiation in activity at rat NMDA or kainate receptors suggesting that this compound exhibits selectivity for AMPA compared to other glutamate receptor subtypes.
Cognition
UoS12258 improved performance in three different cognition paradigms using a variety of approaches to cause impairments (i.e. delay, scopolamine and age).
The NOR test was the most sensitive paradigm, particularly following sub‐chronic dosing. UoS12258 reversed a delay‐induced deficit in NOR in rats after both acute dosing and sub‐chronic dosing. However, behavioural studies in the water maze showed that although UoS12258 was effective at improving aged‐induced deficits in learning and retention, the MED, 3 mg·kg−1, was higher than in the NOR study. This difference could be a reflection of young and aged rats' performance in particular cognitive tasks, or it could be due to the underlying mechanisms involved in performing the task. NOR is dependent on the innate curiosity of the rat to explore novelty and might be more sensitive to modulation of AMPA receptor activity than in a model that involves the complex physiological processes, which are altered during cognitive impairment associated with ageing. Similarly the higher MED (3 mg·kg−1) in the passive avoidance cognition model could reflect the increased difficulty in overcoming a scopolamine‐induced impairment compared to a time‐induced deficit used in the NOR model.
Interestingly, a bell‐shaped dose–response relationship (i.e. loss of, or reduced, effect at higher doses) was apparent in both the NOR and water maze paradigms.
In addition to UoS12258 being an AMPA receptor positive modulator, microdialysis studies have shown that it also enhances cholinergic neurotransmission in brain structures, the dorsal hippocampus and anterior cingulate cortex, which are implicated in cognitive functioning.
Safety and tolerability
Although UoS12258 decreased seizure threshold at doses greater than 10 mg·kg−1, it should be noted that brain concentrations in the MEST test were approximately 20‐fold higher than concentrations measured after improved performance of NOR after acute dosing. Further research is ongoing to identify molecules that exhibit a greater margin between efficacious exposures and those which evoke pro‐convulsant activity.
Conclusions
UoS12258 is a selective positive allosteric modulator of AMPA‐type ionotropic glutamate receptors exhibiting equivalent potency at all AMPA receptor subtypes, with no measurable activity on other glutamate receptors. At comparable concentrations (10–15 nM), UoS12258 has been shown to potentiate AMPA receptor‐mediated currents in vitro; enhance synaptic AMPA receptor‐mediated activity in vivo as well as demonstrating an excellent in vivo efficacy profile. UoS12258 improved NOR task performance in the rat at low brain concentrations, which were consistent with its activity on native tissue in vitro; importantly, efficacious concentrations were significantly lower than those required for efficacy with compounds such as CX516 in a similar assay (Damgaard et al., 2010). UoS12258 was also effective at improving rats' performance of water maze and passive avoidance suggestive of enhanced learning and memory, although at higher concentrations. On the basis of the preclinical studies reported in this article, UoS12258 would be expected to be highly efficacious at improving cognitive measures in schizophrenic patients as well as other diseases with cognitive impairments, and is therefore a worthy candidate for clinical evaluation.
Author contributions
S.W. and M.H. conceived the project design and were responsible for the interpretation of all data. M.H., N.C., J.G., L.L., M.H.S.M., B.O., J.P., D.J., L.D., M.W. and K.S. planned, performed, analysed and interpreted individual studies within the manuscript. S.W. and M.H. wrote the manuscript. P.B. revised the document.
Conflict of interest
All authors are or have been employees and shareholders of GlaxoSmithKline. S.W. is grant holder of Wellcome Trust funded project Transforming the treatment of schizophrenia: Design and development of AMPA receptor modulators with a much improved safety profile as novel drugs for treating the cognitive dysfunction associated with schizophrenia and other CNS disorders grant WT‐103096/Z/13/Z. Some authors are currently employed by other pharmaceutical companies: N.C. is currently an employee of Roche, Switzerland; J.C. is currently an employee of Eisai, USA; B.O. is currently an employee of Aptuit, Italy; L.D. is currently an employee of Astex Pharmaceuticals; M.W. is currently an employee of GWPharma; and D.J. is currently an employee of Johnson & Johnson, UK.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Mean ± sem pEC50 values for 1 potentiation of AMPA receptors formed from homomeric combination of different GluA subunits.
Table S2 Summary pEC50 values for 1 on specific AMPA‐mediate current parameters in rat hippocampal neurones.
Table S3 Blood and brain concentrations of 1 after performance of NOR task (dosed 4 h prior to T1 and T2, n = 3).
Table S4 Blood and brain concentrations of 1 following performance of NOR task. Rats were dosed daily for 7 days and then 4 h prior to T1 and T2 (n = 3).
Table S5 (1.0, 3.0, 10 mg/kg, p.o., 4 h ptt) in LH rats in the pro‐convulsant MEST test. Significant differences (* P < 0.05 or ***P < 0.001) compared to vehicle treated animals.
Acknowledgements
We thank and acknowledge the wider team of scientists that were involved with this work at GlaxoSmithKline.
Ward, S. E. , Beswick, P. , Calcinaghi, N. , Dawson, L. A. , Gartlon, J. , Graziani, F. , Jones, D. N. C. , Lacroix, L. , Selina Mok, M. H. , Oliosi, B. , Pardoe, J. , Starr, K. , Woolley, M. L. , and Harries, M. H. (2017) Pharmacological characterization of N‐[(2S)‐5‐(6‐fluoro‐3‐pyridinyl)‐2, 3‐dihydro‐1H‐inden‐2‐yl]‐2‐propanesulfonamide: a novel, clinical AMPA receptor positive allosteric modulator. British Journal of Pharmacology, 174: 370–385. doi: 10.1111/bph.13696.
References
- Akbarian S, Smith MA, Jones EG (1995). Editing for an AMPA receptor subunit RNA in prefrontal cortex and striatum in Alzheimer's disease, Huntington's disease and schizophrenia. Brain Res 699: 297–304. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai A, Kessler M, Ambros‐Ingerson J, Quan A, Yigiter E, Rogers G et al. (1996). Effects of a centrally active benzoylpyrrolidine drug on AMPA receptor kinetics. Neuroscience 75: 573–585. [DOI] [PubMed] [Google Scholar]
- Baranovic J, Chebli M, Salazar H, Carbone AL, Faelber K, Lau AY (2016). Dynamics of the ligand binding domain layer during AMPA receptor activation. Biophys J 110: 896–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beneyto M, Meador‐Woodruff JH (2004). Expression of transcripts encoding AMPA receptor subunits and associated postsynaptic proteins in the macaque brain. J Comp Neurol 468: 530–554. [DOI] [PubMed] [Google Scholar]
- Boulter J, Hollmann M, O'Shea‐Greenfield A, Hartley M, Deneris E, Maron C et al. (1990). Molecular cloning and functional expression of glutamate receptor subunit genes. Science 249: 1033–1037. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Weiser T, Winter K, Klinder K, Terry AV (2004). The effects of IDRA 21, a positive modulator of the AMPA receptor, on delayed matching performance by young and aged rhesus monkeys. Neuropharmacology 46: 10–22. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J, Spedding M (2009). A nomenclature for ligand‐gated ion channels. Neuropharmacology 56: 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard T, Larsen DB, Hansen SL, Grayson B, Neill JC, Plath N (2010). Positive modulation of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptors reverses sub‐chronic PCP‐induced deficits in the novel object recognition task in rats. Behav Brain Res 207: 144–150. [DOI] [PubMed] [Google Scholar]
- Goff DC, Leahy L, Berman I, Posever T, Herz L, Leon AC et al. (2001). A placebo‐controlled pilot study of the ampakine CX516 added to clozapine in schizophrenia. J Clin Psychopharm 21: 484–487. [DOI] [PubMed] [Google Scholar]
- Goff DC, Coyle JT (2001). The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 158: 1367–1377. [DOI] [PubMed] [Google Scholar]
- Goff DC, Lamberti JS, Leon AC, Green MF, Miller AL, Patel J et al. (2008). A placebo‐controlled add‐on trial of the Ampakine, CX516, for cognitive deficits in schizophrenia. Neuropsychopharmacology 33: 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouaux E (2004). Structure and Function of AMPA Receptors. J Physiol 554: 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981). Improved patch‐clamp techniques for high‐resolution current recording from cells and cell‐free membrane patches. Pflügers Archiv 391: 85–100. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Rogers G, Lynch G, Deadwyler SA (1998). Facilitative effects of the ampakine CX516 on short‐term memory in rats: enhancement of delayed‐nonmatch‐to‐sample performance. J Neurosci 18: 2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ (2000). Postmortem studies in schizophrenia. Dialogues Clin Neurosci 2: 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herguedas B, Garcia‐Nafria J, Cais O, Fernandez‐Leiro R, Krieger J et al. (2016). Structure and organization of heteromeric AMPA‐type glutamate receptors. Science 352: aad3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvar M, Ambros‐Ingerson J, Davis M, Granger R, Kessler M, Rogers GA et al. (1997). Enhancement by an ampakine of memory encoding in humans. Exp Neurol 146: 553–559. [DOI] [PubMed] [Google Scholar]
- Jessen F, Fingerhut N, Sprinkart AM, Kühn KU, Petrovsky N, Maier W et al. (2013). N‐acetylaspartylglutamate (NAAG) and N‐acetylaspartate (NAA) in patients with schizophrenia. Schizophr Bull 39: 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinänen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA et al. (1990). A family of AMPA‐selective glutamate receptors. Science 249: 556–560. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Kornhuber HH, Schmid‐Burgk W, Holzmüller B (1980). Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett 20: 379–382. [DOI] [PubMed] [Google Scholar]
- Kimball AW, Burnett WT, Doherty DG (1957). Chemical protection against ionizing radiation. I. Sampling methods for screening compounds in radiation protection studies with mice. Radiat Res 7: 1–12. [PubMed] [Google Scholar]
- Lahti AC, Koffel B, LaPorte D, Tamminga CA (1995). Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13: 9–19. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Weiler MA, Tamara M, Parwani A, Tamminga CA (2001). Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 25: 455–467. [DOI] [PubMed] [Google Scholar]
- Lebrun C, Piliere E, Lestage P (2000). Effects of S18986‐1, a novel cognitive enhancer, on memory performance in an object recognition task in rats. Eur J Pharmacol 401: 205–212. [DOI] [PubMed] [Google Scholar]
- Le Pen G, Grottick AJ, Higgins GA, Moreau J‐L (2003). Phencyclidine exacerbates attentional deficits in a neurodevelopmental rat model of schizophrenia. Neuropsychopharmacology 28: 1799–1809. [DOI] [PubMed] [Google Scholar]
- Lynch G, Granger R, Ambros‐Ingerson J, Davis M, Kessler M, Schehr R (1997). Evidence that a positive modulator of AMPA‐type glutamate receptors improves delayed recall in aged humans. Exp Neurol 145: 89–92. [DOI] [PubMed] [Google Scholar]
- Lynch G (2002). Memory enhancement: the search for mechanism‐based drugs. Nat Neurosci 5: 1035–1038. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E, (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa S (1998). Glutamate receptors in the mammalian central nervous system. Prog Neurobiol 54: 581–618. [DOI] [PubMed] [Google Scholar]
- Porrino LJ, Daunais JB, Rogers GA, Hampson RE, Deadwyler SA (2005). Facilitation of task performance and removal of the effects of sleep deprivation by an ampakine (CX717) in nonhuman primates. PLoS Biol 3: e299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk JC, Nisenbaum ES (2001). LY404187: a novel positive allosteric modulator of AMPA receptors CNS. Drug Rev 8: 255–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE (1997). AMPA receptor facilitation accelerates fear learning without altering the level of conditioned fear acquired. J Neurosci 17: 5928–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK (2008). Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 7: 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger F, Tammena D, Krampfl K, Bufler J (2005). Desensitization and resensitisation are independently regulated in human recombinant GluR subunit co assemblies. Synapse 55: 176–182. [DOI] [PubMed] [Google Scholar]
- Seeburg PH, Single F, Kuner T, Higuchi M, Sprengel R (2001). Genetic manipulation of key determinants of ion flow in glutamate receptor channels in the mouse. Brain Res 907: 233–243. [DOI] [PubMed] [Google Scholar]
- Shaffer CL, Patel NC, Schwarz J, Scialis RJ, Wei Y, Hou XJ et al. (2015). The discovery and characterization of the α‐Amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic Acid (AMPA) receptor potentiator N‐{(3S,4S)‐4‐[4‐(5‐Cyano‐2‐thienyl)phenoxy]tetrahydrofuran‐3‐yl}propane‐2‐sulfonamide (PF‐04958242). J Med Chem 58: 4291–4308. [DOI] [PubMed] [Google Scholar]
- Sherman‐Gold R. (ed.) (1993). The Axon Guide for Electrophysiology and Biophysics Laboratory Techniques. Axon Instruments, Foster City, CA. [Google Scholar]
- Sommer B, Keinanen K, Verdoorn T, Wisden W, Burnashev N, Herb A et al. (1990). Flip and flop: a cell‐specific functional switch in glutamate‐operated channels of the CNS. Science 249: 1580–1585. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubli U, Rogers G, Lynch G (1994). Facilitation of glutamate receptors enhances memory. Proc Natl Acad Sci U S A 91: 777–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DM, Guidotti A, DiBella M, Costa E (1995). 7‐chloro‐3‐methyl‐3,4‐dihydro‐2H‐1,2,4‐benzothiadiazine S,S‐dioxide (IDRA 21), a congener of aniracetam, potently abates pharmacologically induced cognitive impairments in patas monkeys. Proc Natl Acad Sci U S A 92: 7667–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62: 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai G, Passani LA, Slusher BS, Carter R, Baer L, Kleinman JE (1995). Abnormal excitatory neurotransmitter metabolism in schizophrenic brains. Arch Gen Psychiatry 52: 829–836. [DOI] [PubMed] [Google Scholar]
- Ward SE, Harries MH, Aldegheri L, Andreotti D, Ballantine S, Bax BD et al. (2010). Discovery of N‐[(2S)‐5‐(6‐fluoro‐3‐pyridinyl)‐2,3‐dihydro‐1H‐inden‐2‐yl]‐2‐propanesulfonamide, a novel clinical AMPA receptor positive modulator. J Med Chem 53: 5801–5812. [DOI] [PubMed] [Google Scholar]
- Ward SE, Harries M, Aldegheri L, Austin NE, Ballantine S, Ballini E et al. (2011). Integration of lead optimization with crystallography for a membrane‐bound ion channel target: discovery of a new class of AMPA receptor positive allosteric modulators. J Med Chem 54: 78–94. [DOI] [PubMed] [Google Scholar]
- Weisler RH (2007). Emerging drugs for attention‐deficit/hyperactivity disorder. Expert Opin Emerg Drugs 12: 423–434. [DOI] [PubMed] [Google Scholar]
- Woolley ML, Waters KA, Gartlon JE, Lacroix LP, Jennings C, Shaughnessy F et al. (2009). Evaluation of the pro‐cognitive effects of the AMPA receptor positive modulator, 5‐(1‐piperidinylcarbonyl)‐2,1,3‐benzoxadiazole (CX691), in the rat. Psychopharmacology (Berl) 202: 343–354. [DOI] [PubMed] [Google Scholar]
- Wright A, Vissel B (2012). The essential role of AMPA receptor GluA2 subunit RNA editing in the normal and diseased brain. Front Mol Neurosci 5: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Manji HK (2008). The role of AMPA receptor modulation in the treatment of neuropsychiatric diseases. Exp Neurol 211: 7–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivkovic I, Thompson DM, Bertolino M, Uzunov D, DiBella M, Costa E et al. (1995). 7‐chloro‐3‐methyl‐3,4‐dihydro‐2H‐1,2,4‐benzothiadiazine S,S‐dioxide (IDRA 21): a benzothidiazine derivative that enhances cognition by attenuating DL‐alpha‐amino‐2,3‐dihydro‐5‐methyl‐3‐oxo‐4‐isoxazolepropanoic acid (AMPA) receptor desensitization. J Pharm Exp Ther 272: 300–309. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Mean ± sem pEC50 values for 1 potentiation of AMPA receptors formed from homomeric combination of different GluA subunits.
Table S2 Summary pEC50 values for 1 on specific AMPA‐mediate current parameters in rat hippocampal neurones.
Table S3 Blood and brain concentrations of 1 after performance of NOR task (dosed 4 h prior to T1 and T2, n = 3).
Table S4 Blood and brain concentrations of 1 following performance of NOR task. Rats were dosed daily for 7 days and then 4 h prior to T1 and T2 (n = 3).
Table S5 (1.0, 3.0, 10 mg/kg, p.o., 4 h ptt) in LH rats in the pro‐convulsant MEST test. Significant differences (* P < 0.05 or ***P < 0.001) compared to vehicle treated animals.