

Abstract
A hallmark of systemic lupus erythematosus (SLE) is the consistent production of various auto-antibodies by auto-reactive B cells. Interferon-α (IFN-α) signaling is highly activated in SLE B cells and plays a vital role in the antibody response by B cells. Previous studies have shown that CD180-negative B cells, which are dramatically increased in SLE patients, are responsible for the production of auto-antibodies. However, the association between CD180 and IFN-α signaling remains unknown. In the present study, we explored the effect of CD180 on regulating the activation of IFN-α signaling in B cells. We found that the number of CD180-negative B cells was increased in MRL/Mp-Fas(lpr/lpr) lupus-prone mice compared with wild-type mice. Phenotypic analysis showed that CD180-negative B cells comprised CD138+ plasmablast/plasma cells and GL-7+ germinal center (GC) B cells. Notably, ligation of CD180 significantly inhibited the IFN-α-induced phosphorylation of signal transducer and activator of transcription 2 (STAT-2) and expression of IFN-stimulated genes (ISGs) in a Lyn-PI3K-BTK-dependent manner in vitro. Moreover, ligation of CD180 could also inhibit IFN-α-induced ISG expression in B cells in vivo. Furthermore, the Toll-like receptor 7 and Toll-like receptor 9 signaling pathways could significantly downregulate CD180 expression and modulate the inhibitory effect of CD180 signaling on the activation of IFN-α signaling. Collectively, our results highlight the close association between the increased proportion of CD180-negative B cells and the activation of IFN-α signaling in SLE. Our data provide molecular insight into the mechanism of IFN-α signaling activation in SLE B cells and a potential therapeutic approach for SLE treatment.
Keywords: B cells, CD180, IFN-α, SLE
Introduction
Systemic lupus erythematosus (SLE) is a disorder of systemic autoimmunity characterized by the formation of a variety of auto-antibodies by hyper-reactive B cells.1,2 Endogenous DNA- and RNA-associated auto-antigens are recognized by the B-cell antigen receptor (BCR) and then delivered to endosomal Toll-like receptor 9 (TLR9) and Toll-like receptor 7 (TLR7), respectively.3 Activation of B cells through these pathways leads to the production of class-switched DNA- and RNA-reactive auto-antibodies, contributing to an inflammatory amplification loop characteristic of the disease.3
Interferon-α (IFN-α), a central cytokine in the pathogenesis of SLE, can induce and accelerate the SLE symptoms in patients and mice.4 SLE patients have increased serum levels of IFN-α and expression of IFN-stimulated genes (ISGs) in peripheral blood mononuclear cells (PBMCs).5,6,7 More importantly, we and others have found that B cells display an upregulated type I IFN-inducible gene signature in SLE patients,8,9suggesting that IFN-α signaling is highly activated in SLE B cells. As is well known, IFN-α impacts the function of B cells through various mechanisms, including BCR engagement, TLR7 expression, survival, differentiation, and class-switch recombination.10,11,12,13,14 Moreover, IFN-α could also potentiate TLR7 and TLR9 activation, which leads to excessive auto-antibody production.11,15,16,17 Thus, the highly activated IFN-α signaling in SLE B cells plays an important role in the pathogenesis of lupus.
CD180, also known as RP105, is a TLR-like protein that physically associates with MD-1.18,19 The CD180/MD-1 complex is expressed on B lymphocytes, macrophages, and dendritic cells (DCs).20,21 CD180 contains a conserved extracellular leucine-rich repeat domain typical for members of the TLR family but lacks the intracellular Toll/IL-1-receptor- (TIR-) domain.21 However, it has been shown that ligation of CD180 by anti-CD180 antibodies leads to B-cell proliferation, resistance to radio-induced apoptosis, and expression of the costimulatory molecule CD86.22 CD180 ligation leads to Lyn activation and then phosphorylates phosphatidylinositol 3-kinase (PI3K), which recruits Bruton tyrosine kinase (BTK) to the cell membrane and causes Ca2+ mobilization, followed by the activation of protein kinase C β (PKC β), mitogen-activated protein kinases (MAPKs), and nuclear factor-kappa B.23,24
Notably, CD180 was first reported to be involved in SLE in 1999. The number of CD180-negative B cells is significantly increased in PBMCs from SLE patients and is positively associated with disease activity.25 Moreover, only CD180-negative B cells obtained from SLE patients spontaneously produce IgG and IgM in vitro.26 These studies suggest that CD180-negative B cells may be responsible for the production of auto-antibodies. However, the reason for abnormal expression and the function of CD180 in B cells in SLE remain largely elusive.
In the present study, we first found that the number of CD180-negative B cells was increased in B cells from MRL/Mp-Fas(lpr/lpr) (MRL/lpr) lupus-prone mice compared with wild-type mice. Phenotypic analysis showed that CD180-negative B cells include CD138+ plasmablast/plasma cells and GL-7+ GC B cells. Intriguingly, ligation of CD180 by anti-CD180 antibody could inhibit the IFN-α-induced ISG expression in B cells both in vitro and in vivo. Mechanistically, ligation of CD180 inhibited the tyrosine phosphorylation of signal transducer and activator of transcription 2 (STAT-2) induced by IFN-α via a Lyn-PI3K-BTK-dependent pathway. Moreover, TLR7 and TLR9 signaling could downregulate CD180 expression and attenuate the inhibitory effect of anti-CD180 on the activation of IFN-α signaling in B cells. Altogether, our study indicates that CD180 plays an important role in regulating the activation of IFN-α signaling in B cells, thereby providing molecular insight into the mechanism of the enhanced activation of IFN-α signaling in SLE B cells.
Materials and methods
Collection of human blood samples and isolation of CD19+ B cells from human PBMCs
Blood samples from age-matched female SLE patients (n = 10) and female healthy donors (n = 10) were obtained from Jiangsu Province Hospital of Traditional Chinese Medicine (Jiangsu Province, China). PBMCs were separated from the plasma using Ficoll centrifugation (Lymphoprep, Nycomed, Oslo, Norway) according to the standard procedures. B cells were isolated using human CD19+ B-Cell Isolation Beads, as previously described.8 The purity of B cells was consistently above 95%. For in vitro experiments, isolated human CD19+ B cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and stimulated with the TLR7 ligand R848 (1 μg mL−1, Enzo Life Sciences, Farmingdale, NY, USA), the TLR9 ligand CpG-2006S (0.3 μM, Invitrogen, Carlsbad, CA, USA), or human IFN-α (1000 U mL−1, eBioscience, San Diego, CA, USA).
Mice
Six- to eight-week-old female C57BL/6 (B6) mice were used to isolate splenic B cells and were purchased from the Model Animal Research Center of Nanjing University. Age-matched female C57BL/6 mice (n = 13) and MRL/lpr mice (n = 13) were also purchased from the Model Animal Research Center of Nanjing University and euthanized at 22–24 weeks old. The mice were maintained under specific pathogen-free conditions. All manipulations were in accordance with the institutional guidelines for animal care and used based on the Guide for the Animal Care Committee at Nanjing University.
Purification of murine splenic B cells and in vitro experiment
Splenic lymphocytes were isolated by Ficoll density centrifugation according to standard procedures. B cells were purified from the spleen by positively selecting B220+ B cells using B220 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and the purity of B cells was consistently above 95%. The purified B cells were cultured in RPMI 1640 medium containing 10% FBS and stimulated with the TLR7 ligand R848 (1 μg mL−1, Enzo Life Sciences, Farmingdale, NY, USA), the TLR9 ligand CpG-1826 (0.3 μM, Invitrogen, Carlsbad, CA, USA), mouse IFN-α (1000 U mL−1, eBioscience, San Diego, CA, USA), or anti-CD180 antibody (0.2 μg mL−1, eBioscience, San Diego, CA, USA). For inhibitor studies, the B cells were pretreated with dasatinib (5 μM, Selleck, Houston, TX, USA), ibturinib (1 μM, Selleck, Houston, TX, USA), enzastaurin (1 μM, Selleck, Houston, TX, USA), LY294002 (5 μM, Beyotime, Haimen, China), U0126 (3 μM Beyotime, Haimen, China), SP600126 (2 μM Beyotime, Haimen, China), or SB20358 (1 μM Beyotime, Haimen, China) 1 h prior to stimulation with anti-CD180 or IFN-α.
Anti-CD180 antibody and IFN-α treatment
Female C57BL/6 mice (n = 24), 8–10 weeks old, were purchased from Model Animal Research Center of Nanjing University and were randomly divided into four groups: (a) PBS; (b) anti-CD180; (c) IFN-α; (d) anti-CD180 + IFN-α. The mice were injected with 100 μg anti-CD180 antibody (contained in 200 μl PBS) or 10 000 U IFN-α (contained in 200 μl PBS). All of the injections were conducted intraperitoneally (IP). Six hours later, the mice were euthanized. The spleens, bone marrow, and PBMCs were isolated and used to analyze the expression of IFIT1, MX1, and TLR7. The B220+ B cells were purified from spleen as described above.
RNA isolation and quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. qPCR assays for mRNA were carried out on a StepOne Plus real-time polymerase chain reaction system or an ABI Vii 7 detection system (Applied Biosystems, Foster City, CA, USA) using SYBR Green PCR Master Mix. The 2−ΔΔCt method was used for real-time PCR gene expression analysis. All quantification data are presented as a ratio to the GAPDH level.
Western blot analysis
Proteins were extracted using lysis buffer containing 10 mM Tris-HCl (pH 7.3), 150 mM NaCl, 2 mM Na3VO4, 0.4 mM EDTA, 10 mM NaF, 1 mM PMSF, 5 μg mL−1 aprotinin and leupeptin, and 1% NP-40. The total lysates were resolved by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore Corp, Bedford, MA, USA). After blocking, the membranes were incubated with primary antibodies against GAPDH, phos-JAK1 at tyrosine 1022, total-JAK1, phos-signal transducer and activator of transcription 1 (STAT-1) at tyrosine 701, phos-STAT-1 at serine 727, total STAT-1, phos-STAT-2 at tyrosine 690, total STAT-2 and TLR7 (Cell Signaling Technology, Danvers, MA, USA, all dilutions at 1:1000) overnight at 4 °C. After washing, the membranes were incubated at room temperature with the secondary antibody AffiniPure Goat Anti-Rabbit IgG (H+L) (Beyotime, Haimen, China, dilution at 1:3000). ECL Plus western blotting detection reagents (Millipore Corp) were used to visualize protein expressions. The protein expression level was analyzed by Quantity One v4.62.
Flow cytometry
The cells were washed twice with phosphate-buffered saline (PBS) and stained according to the standard procedure. The antibodies used were as follows: FITC-conjugated anti-human CD19, PE-conjugated anti-human CD180, FITC- and APC-conjugated anti-mouse B220, PE-conjugated anti-mouse CD180, PerCP- and APC-conjugated anti-mouse-IgM, and PerCP-conjugated anti-mouse CD38 were purchased from eBioscience (San Diego, CA, USA); APC-conjugated anti-mouse CD138 and FITC-conjugated GL-7 were obtained from BD Biosciences (San Jose, CA, USA). The cells were analyzed with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). An isotype control was used for each antibody. Data analysis was performed using FlowJo software (TreeStar, San Carlos, CA, USA).
Statistical analysis
All values in the graphs are given as the mean values plus or minus the standard error of the mean (SEM). To assess statistical significance, t-tests or analysis of variance (ANOVA) were performed using GraphPad Prism 5.0 software. Statistical significance was set at p < 0.05. All experiments were repeated at least three times.
Results
CD180-negative B cells are increased in SLE patients and MRL/lpr mice
Because it was reported that the number of CD180-negative B cells is dramatically increased in SLE patients,25 we first tested the number of CD180-negative B cells from SLE patients and healthy donors. Consistently, the number of CD180-negative B cells from SLE patients was increased significantly compared with the healthy donors (Figure 1a). Moreover, the mRNA level of CD180 in CD19+ B cells from SLE patients was significantly lower compared with healthy donors (Figure 1b).
Figure 1.

Increased CD180-negative B cells and decreased overall CD180 expression in B cells from SLE patients and MRL/lpr mice. (a) Flow cytometric analysis of the CD180-negative B-cell population within PBMCs from active SLE patients (n = 10) and healthy donors (n = 10). (b) qPCR analysis of CD180 expression in CD19+ B cells of active SLE patients (n = 10) and healthy donors (n = 10). (c) Flow cytometric analysis of the CD180-negative B-cell population in the spleens of MRL/lpr mice (n = 13) and wild-type B6 mice (n = 13). (d) qPCR analysis of CD180 expression in splenic B cells from MRL/lpr mice (n = 13) and wild type mice (n = 13). Error bars represent the SEM. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by t-test; ns denotes p > 0.05.
It was also reported that CD180-negative B cells were increased in the spleens from NZB/W F1 mice with age,27 indicating that this phenomenon in SLE patients could also extend to SLE model mice. To further confirm this hypothesis, we then evaluated the population of CD180-negative B cells in the spleens, bone marrow, and PBMCs from MRL/lpr mice. As shown in Figure 1c and Supplementary Figure 1a, the proportion of CD180-negative B cells in the spleen (Figure 1c) and PBMCs (Supplementary Figure 1a) of wild-type mice was minimal, while CD180-negative B cells were significantly increased in MRL/lpr mice. Notably, nearly half of the B cells from the bone marrow of the wild-type mice were CD180-negative; this is consistent with a former study demonstrating that CD180 is not expressed on pre-B cells or immature B cells.22 The proportion of CD180-negative B cells was also increased dramatically in the bone marrow B cells from MRL/lpr mice (Supplementary Figure 1b). We also detected the mRNA level of CD180 in splenic B220+ B cells isolated from MRL/lpr mice and wild-type mice. As expected, the CD180 expression in splenic B cells from MRL/lpr mice was significantly lower compared with wild-type mice (Figure 1d). These data suggest that the proportion of CD180-negative B cells is increased in B cells from SLE patients and MRL/lpr mice.
Phenotypic analysis of CD180-negative B cells in MRL/lpr mice
In SLE patients, the phenotype of CD180-negative B cells resembles the phenotype of activated B cells or germinal center (GC) B cells.25 More recently, a more specific phenotypic analysis of CD180-negative B cells from SLE patients showed that CD180-negative B cells could be divided into at least five subsets of B cells using the plasma cell marker CD138.28 However, the phenotype of CD180-negative B cells in MRL/lpr mice remains unknown. Thus, we used the murine B-cell marker B220 and plasma cell marker CD138 to subdivide CD180-negative B cells from spleen of MRL/lpr mice. As shown in Figure 2a, the CD138+ plasmablasts/plasma cells were CD180-negative. Notably, a substantial proportion of CD180-negative cells were CD138-negative. A previous study showed that MRL/lpr mice have abnormal formation of GC in the spleen,29 so we used the GC B-cell marker GL-7 to further identify whether CD180-negative B cells contain GC B cells. As expected,</emph> the B220+GL-7+ GC B cells were CD180-negative (Figure 2b). These data suggest that CD180-negative B cells consist of CD138+ plasmablasts/plasma cells and B220+GL-7+ GC B cells.
Figure 2.

Phenotypic analysis of CD180-negative B cells in the spleens of MRL/lpr mice. (a) Flow cytometric analysis of CD180 expression on CD138+ plasmablasts/plasma cells in the spleens of MRL/lpr mice and wild-type mice. (b) Flow cytometric analysis of CD180 expression on GL7+ GC B cells in the spleens of MRL/lpr mice and wild-type mice.
We next analyzed the CD180-negative B cells in the bone marrow. As shown in Supplementary Figure 2a and 2b, the proportion of pro- and pre-B cells was increased in the bone marrow of MRL/lpr mice, and these cells were CD180-negative. Notably, we found no differences in the plasma cells between wild-type and MRL/lpr mice (Supplementary Figure 2c). We also found that the memory B cells were CD180-positive (Supplementary Figure 2d). We believe that the increased CD180-negative B cells in the bone marrow of MRL/lpr mice may simply reflect increased numbers of pro- and pre-B cells.
Ligation of CD180 inhibits IFN-α-induced ISG expression in murine B cells in vitro
We next investigated the function of CD180 in B cells. Although previous studies have shown that ligation of CD180 can regulate the TLR7 and TLR9 signaling pathways,30,31 the mechanism of the regulation of IFN-α signaling activation by CD180 remains unknown. To explore whether the ligation of CD180 affects IFN-α-induced ISG expression, murine splenic B cells were treated with anti-CD180 or IFN-α for 4 h and 8 h, and the mRNA levels of MX1, IFIT1, and TLR7 were then measured. As shown in Figure 3a and 3b, anti-CD180 treatment alone could downregulate the basal expression of MX1, IFIT1, and TLR7. Moreover, anti-CD180 could significantly inhibit the IFN-α-induced expression of MX1, IFIT1, and TLR7. To evaluate the protein level, murine B cells were treated with anti-CD180 or IFN-α for 24 h and the protein levels of STAT-1 and TLR7 were measured. Consistently, anti-CD180 could inhibit IFN-α induced STAT-1 and TLR7 expression (Figure 3c). These results demonstrate that ligation of CD180 can indeed inhibit IFN-α-induced ISG expression in B cells.
Figure 3.

Ligation of CD180 inhibits IFN-α-induced expression of ISGs. (a–c) Isolated murine splenic B220+ B cells were stimulated with anti-CD180 antibody (0.2 μg mL−1) or mouse IFN-α (1000 U mL−1). qPCR analysis of the expression of IFIT1, MX1, and TLR7 at 4 h (a) and 8 h (b). Western blot analysis of the protein levels of TLR7 and STAT-1 at 24 h (c). The data shown represent the mean values of three independent experiments, and the error bars represent the SEM. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by ANOVA; ns denotes p > 0.05.
Ligation of CD180 also inhibits the IFN-α-induced tyrosine phosphorylation of STAT-2
We next investigated the mechanism underlying the inhibitory effect of anti-CD180 on the activation of IFN-α signaling. It is known that the activation of IFN-α signaling leads to Janus-activated kinases (JAKs), tyrosine kinase 2 (TYK2), and JAK1 activation, which results in the tyrosine phosphorylation of STAT-2 and STAT-1; IRF9 (IFN-regulatory factor 9) is then recruited to form complexes with STAT-2 and STAT-1, which are known as ISGF3 (IFN-stimulated gene factor 3) complexes.32 These complexes translocate to the nucleus and bind IFN-stimulated response elements (ISREs) in the DNA to initiate ISG transcription.32 Moreover, phosphorylation of STAT-1 at serine 727 is required for the full activation of STAT-1.33,34,35 To determine whether ligation of CD180 could modulate the IFN-α induced phosphorylation of JAK1, STAT-1, and STAT-2, murine B cells were treated with anti-CD180 or IFN-α for 10, 20, or 40 min and then the tyrosine phosphorylation of JAK1 (Y-1022), STAT-1 (Y-701), STAT-2 (Y-690), and serine phosphorylation of STAT-1 (S-727) were measured. Notably, ligation of CD180 had no effect on the IFN-α-induced phosphorylation of JAK1 and STAT-1 (Figure 4a–4c). However, the IFN-α-induced tyrosine phosphorylation of STAT-2 was strongly inhibited by CD180 ligation (Figure 4d). These data suggest that the ligation of CD180 inhibits the activation of IFN-α signaling by inhibiting the tyrosine phosphorylation of STAT-2.
Figure 4.
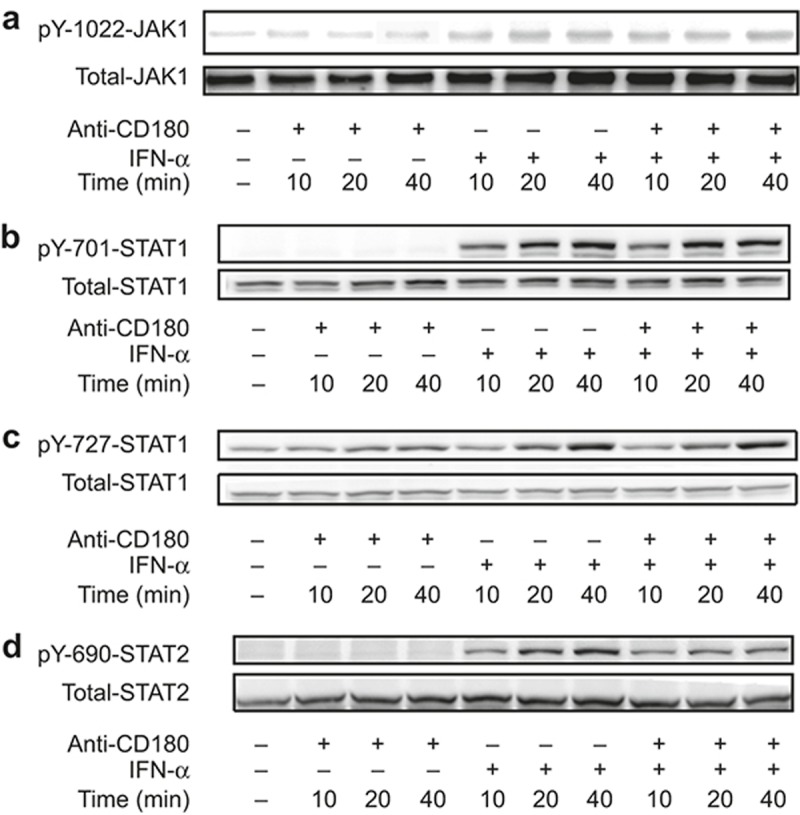
Ligation of CD180 inhibits the IFN-α-induced phosphorylation of STAT-2. (a–c) Isolated murine splenic B220+ B cells were stimulated with anti-CD180 (0.2 μg mL−1) or mouse IFN-α (1000 U mL−1) for 10 min, 20 min, and 40 min. Western blot analysis of the tyrosine phosphorylation of JAK1 (Y-1022) (a), tyrosine phosphorylation of STAT-1 (Y-701) (b), serine phosphorylation of STAT-1 (S-727), (c) and tyrosine phosphorylation STAT-2 (Y-690) (d).
CD180 ligation-mediated inhibition of IFN-α signaling requires Lyn-PI3K-BTK signaling
Previous studies have shown that many kinases participate in regulating the activation of IFN-α signaling.36,37 Notably, ligation of CD180 can trigger the activation of various kinases such as Lyn, PI3K, BTK, PKC β, and MAPKs.23,24 To elucidate which kinases participate in the CD180 ligation-mediated inhibition of IFN-α signaling, murine B cells were pretreated with dasatinib (Lyn inhibitor), LY294002 (PI3K inhibitor), ibturinib (BTK inhibitor), enzastaurin (PKC β inhibitor), U0126 (ERK inhibitor), SP600126 (JNK inhibitor), or SB203580 (p38 inhibitor) for 1 h prior to stimulation with anti-CD180 or IFN-α for 4 h. As shown in Figure 5a and 5b, pretreatment of dasatinib, LY294002, and ebturinib, rather than enzastaurin, U0126, SP600126, SB203580, could totally reverse the inhibitory effect of anti-CD180 on the IFN-α-induced expression of IFIT1 and MX1. To determine whether blockage of Lyn, PI3K, and BTK could reverse the inhibition of tyrosine phosphorylation of STAT-2 by anti-CD180, murine B cells were pretreated with dasatinib, LY294002, or ibturinib for 1 h prior to stimulation with anti-CD180 and IFN-α for 40 min. As expected, the inhibition of IFN-α-induced tyrosine phosphorylation of STAT-2 by CD180 ligation was reversed by dasatinib, LY294002, or ibturinib (Figure 5c). These results suggest that Lyn-PI3K-BTK signaling is required for the inhibitory effect of CD180 ligation on the activation of IFN-α signaling.
Figure 5.

Lyn-PI3K-BTK signaling is required for the inhibitory effect of anti-CD180 on the activation of IFN-α signaling. (a and b) Isolated murine splenic B220+ B cells were pretreated with dasatinib (5 μM), a Lyn inhibitor; LY294002 (5 μM), a PI3K inhibitor; ibturinib (1 μM), a BTK inhibitor; enzastaurin (1 μM), a PKC β inhibitor; U0126 (3 μM), an ERK inhibitor; SP600126 (2 μM), a JNK inhibitor or SB20358 (1 μM), a p38 inhibitor for 1 h, followed by stimulation with anti-CD180 antibody (0.2 μg mL−1) or mouse IFN-α (1000 U mL−1) for 4 h. qPCR analysis of the expression of IFIT1 (a) and MX1 (b). (c) Isolated murine splenic B220+ B cells were pretreated with dasatinib (5 μM), LY294002 (5 μM), and ibturinib (1 μM) for 1 h, followed by stimulation with anti-CD180 antibody (0.2 μg mL−1) or mouse IFN-α (1000 U mL−1) for 40 min. Western blot analysis of the phosphorylation level of STAT-2. The data shown represent the mean values of three independent experiments, and the error bars represent the SEM. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by ANOVA; ns denotes p > 0.05.
Determination of the correlation between CD180 signaling and the IFN-α-induced expression of ISGs in murine splenic B cells in vivo
To further validate the inhibitory effect of anti-CD180 on the activation of IFN-α signaling, an in vivo study was constructed. Wild-type mice were randomly divided into four groups and injected with PBS, anti-CD180, IFN-α, or anti-CD180 plus IFN-α for 6 h. The mRNA levels of IFIT1, MX1, and TLR7 in the spleens, bone marrow, and PBMCs from these mice were measured. As shown in Figure 5a, the expression of IFIT1, MX1, and TLR7 were significantly lower in the spleens from the anti-CD180- and IFN-α-treated mice compared with the IFN-α-treated mice. However, no significant differences were found in the bone marrow (Figure 6b) or PBMCs (Figure 6c). To further confirm the expression of ISGs in B cells, we isolated B cells from the spleens of these mice and evaluated the levels of IFIT1, MX1, and TLR7. Intriguingly, the expression levels of IFIT1, MX1, and TLR7 were significantly lower in B cells from anti-CD180- and IFN-α-treated mice compared with IFN-α-treated mice (Figure 6d). Additionally, no differences were found in the B-cell-removed splenocytes of these mice (Figure 6e). Consistent with these data, the protein levels of TLR7 and STAT-1 were all decreased in anti-CD180- and IFN-α-treated mice compared with IFN-α-treated mice (Figure 6f). These results indicate that ligation of CD180 inhibits the IFN-α-induced expression of ISGs in B cells in vivo.
Figure 6.

In vivo study of the inhibitory effect of anti-CD180 on the activation of IFN-α signaling. Female C57BL/6 mice (n = 24) were randomly divided into four groups: (a) PBS; (b) anti-CD180; (c) IFN-α; (d) anti-CD180+ IFN-α. The mice were IP injected with 200 μl PBS, 200 μl PBS containing 100 μg anti-CD180 antibody or 10 000 U IFN-α. Six hours later, the mice were euthanized. (a–c) qPCR analysis of the expression of IFIT1, MX1, and TLR7 in spleen (a), bone marrow (b), and PBMCs (c). (d and e) qPCR analysis of the expression of IFIT1, MX1, and TLR7 in purified splenic B220+ B cells (d) and B cell-removed splenocytes (e). (f) Western blot analysis of the protein levels of TLR7 and STAT-1 in purified splenic B220+ B cells. The data shown represent the mean values of three independent experiments, and the error bars represent the SEM. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by ANOVA; ns denotes p > 0.05.
TLR7 and TLR9 signaling downregulate CD180 expression and attenuate the CD180 ligation-mediated inhibition of IFN-α signaling in B cells
We next investigated the molecular events leading to CD180 downregulation. It is known that the TLR7, TLR9, and IFN-α signaling pathways play vital roles in B cell abnormality in SLE;3,10,11,12,13,14,15,16 we therefore evaluated the effects of the TLR7, TLR9, and IFN-α signaling pathways on CD180 expression in B cells. Isolated CD19+ human B cells were stimulated with the TLR7 agonist R848, the TLR9 agonist CpG2006S or human IFN-α, and the mRNA level of CD180 was detected at 8 h and 16 h. Notably, CD180 was significantly downregulated by R848 and CpG 2006S but not by IFN-α (Figure 7a). To further confirm the effects of the TLR7, TLR9, and IFN-α signaling pathways on CD180 expression, isolated B220+ murine splenic B cells were stimulated with the TLR7 agonist R848, the TLR9 agonist CpG 1826 or mouse IFN-α for different time courses. Consistently, both the mRNA (Figure 7b) and the protein levels (Figure 7c) of CD180 were downregulated by R848 and CpG1826 but not by IFN-α.
Figure 7.

TLR7 and TLR9 signaling downregulates CD180 expression and attenuates the inhibitory effect of anti-CD180 on the activation of IFN-α signaling in B cells. (a) Isolated CD19+ B cells from human PBMCs were stimulated with the TLR7 ligand R848 (1 μg mL−1), the TLR9 ligand CpG2006S (0.3 μM) or human IFN-α (1000 U mL−1). qPCR analysis of the expression of CD180 at 8 h and 16 h. (b and c) Isolated murine splenic B220+ B cells were stimulated with the TLR7 ligand R848 (1 μg mL−1), the TLR9 ligand CpG1826 (0.3 μM), or mouse IFN-α (1000 U mL−1). qPCR analysis of the mRNA level of CD180 at 8 h and 16 h (b); flow cytometric analysis of the protein level of CD180 at 24 h and 36 h (c). (d and e) Isolated murine splenic B cells were pretreated with R848 (1 μg mL−1) or CpG1826 (0.3 μM) for 24 h prior to stimulation with anti-CD180 or IFN-α for 4 h. qPCR analysis of the expression of IFIT1 (d) and MX1 (e). The data shown represent the mean values of three independent experiments, and the error bars represent the SEM. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by ANOVA; ns denotes p > 0.05.
The above results prompted us to ask whether TLR7 and TLR9 signaling can alter the inhibitory effect of anti-CD180 on the activation of IFN-α signaling. To confirm this hypothesis, murine B cells were treated with R848 or CpG1826 for 24 h prior to stimulation with anti-CD180 and IFN-α for 4 h. As shown in Figure 7d and 7e, R848 and CpG1826 pretreatment could significantly attenuate, despite failing to totally reverse, the inhibitory effect of anti-CD180 on the IFN-α-induced expression of IFIT1 (Figure 7d) and MX1 (Figure 7e). Taken together, these data demonstrate that TLR7 and TLR9 signaling could downregulate CD180 expression and attenuate the inhibitory effect of anti-CD180 on the activation of IFN-α signaling in B cells.
Discussion
In this report, we first demonstrated that CD180-negative B cells in MRL/lpr lupus-prone mice were increased and that the mRNA level of CD180 was decreased. Phenotypic analysis showed that CD180-negative B cells comprise CD138+ plasmablasts/plasma cells and GL-7+ GC B cells. Furthermore, we showed a novel role for CD180 in inhibiting the activation of IFN-α signaling. Finally, we demonstrated that TLR7 and TLR9 signaling could downregulate CD180 expression and attenuate the inhibitory effect of anti-CD180 on the activation of IFN-α signaling.
Although CD180-negative B cells were shown to be significantly increased in SLE patients,25 studies have not yet addressed whether CD180-negative B cells were also increased in SLE model mice. In this study, we demonstrated for the first time that CD180-negative B cells were also significantly increased in MRL/lpr mice. Our results, in conjunction with the former study that CD180-negative B cells were increased in the spleens of NZB/W F1 mouse with age,27 make it reasonable to speculate that increased CD180-negative B cells could also be found in other SLE models. A recent study showed that serum soluble MD-1 levels are increased in MRL/lpr mice;38 since MD-1 can form a complex with CD180 on the surface of B cells,19 the increased CD180-negative B cells in MRL/lpr mice may account for the increased serum soluble MD-1 levels.
Both SLE patients and lupus-prone mice have alterations in their B-cell subsets, such as an increased proportion of plasmablasts and/or plasma cells, GCs, etc.,39,40,41,42,43 which reflects the disturbed differentiation of B cells in SLE patients and lupus-prone mice. In the present study, we found that compared with wild-type mice, CD138+ plasmablasts/plasma cells and B220+GL-7+ GC B cells are increased in MRL/lpr mice and are CD180-negative. Because the terminal differentiation of GC B cells results in plasma cells,43 and memory B cells are CD180-positive, we deduced that the downregulation of CD180 may participate in the differentiation of B cells into plasma cells but not to memory B cells. Recently, Cheng et al.44 showed that the adoptive transfer of CD138+ antibody-secreting cells (ASCs) from the spleen of NZB/W mice into Rag1-deficient mice could result in elevated serum auto-antibody levels, immune complex nephritis, and a reduced survival rate. Moreover, transplantation of lipogranulomas, which are rich in anti-RNP auto-ASCs, leads to auto-antibody production in recipients.45 We proposed that CD180-negative B cells from MRL/lpr mice may have similar pathogenic roles.
We explored the function of CD180 in B cells and found that the ligation of CD180 inhibited IFN-α signaling and downregulated IFN-α-induced ISG expression both in vitro and in vivo. Since patients undergoing IFN-α therapy for hepatitis C infection or various malignancies often develop lupus-like symptoms46,47 and treatment of exogenous type I IFNs leads to more severe disease activity in NZB and NZB/W lupus-prone mice,48,49 IFN-α is considered an appropriate therapeutic target in SLE.50Recently, a phase I trial of an anti-interferon monoclonal antibody in SLE showed that blocking IFN-α inhibits the IFN-signature in PBMCs and relieves skin symptoms in certain lupus patients,51 which further proved the feasibility of treating SLE by targeting IFN-α. In our present study, we demonstrated that anti-CD180 antibody could significantly inhibit the activation of IFN-α signaling and IFN-α-induced expression of ISGs, including TLR7, in B cells in vivo. Because a moderate chronic upregulation of TLR7 in B cell is sufficient to drive kidney pathogenesis in the lupus-susceptible strain,52 anti-CD180 antibody may be considered as a candidate for SLE treatment.
Ligation of CD180 initiates a BCR-like signaling cascade.53 Notably, BCR signaling enhances the IFN-γ-induced phosphorylation of STAT-1 on serine 727 through calcium mobilization and activation of multiple serine kinase pathways, such as PKC and p38.54 In the present study, we found that the ligation of CD180 inhibited IFN-α-induced tyrosine phosphorylation of STAT-2, while it had no effect on the IFN-α-induced phosphorylation of JAK1 and STAT-1. We propose that this discrepancy may be attributed to the differences between CD180 signaling and BCR signaling. For example, BCR signaling causes a rapid rise in the cytosolic Ca2+ concentration, while the ligation of CD180 causes a very slow and gradual increase in the cytosolic Ca2+ concentration.23 Moreover, BCR-meditated MAP kinase activation is negatively controlled by Lyn, which is essential for CD180-induced MAP kinase activation.23,55 We also found that Lyn-PI3K-BTK signaling is indispensable for the inhibitory effect of CD180 ligation on the activation of IFN-α signaling. Since BTK is required for calcium mobilization in CD180 signaling, it indicates that calcium mobilization and the subsequently activated kinases may contribute to the inhibition of the phosphorylation of STAT-2 induced by CD180 ligation. More studies are still needed to investigate the specific mechanism.
Abnormal expression of CD180 on B cells exists not only in SLE but also in Sjögren's syndrome and marginal zone lymphoma.56,57 However, little is known regarding the regulatory mechanism of CD180 expression. Here, we found that downregulation of CD180 in SLE B cells can be the result of activation of the TLR7 and TLR9 signaling pathways. Recently, our colleagues demonstrated that mice undergoing long-term exposure to imiquimod (a TLR7 agonist) developed lupus-like symptoms.58 Notably, these mice also showed decreased CD180 expression on B cells (data not shown), which is consistent with our current study. In SLE B cells, activation of TLR7 and TLR9 signaling is not only responsible for auto-antibody production3 but also affects B cell function by up- or downregulating the expression of various genes. We previously showed that co-activation of the TLR7 and BCR pathways promotes necroptosis of B cells by elevating the expression of necroptosis-related genes (RIPK1, RIPK3, and PARP1).8 We also showed that activation of TLR7 and TLR9 signaling could upregulate STS-1 expression, which augments the activation of JAK-STAT signaling induced by IFN-α, thus leading to increased B cell autophagy in SLE.59 It will be of interest to explore whether the activation of TLR7 and TLR9 signaling could regulate CD180 expression and affect the activation of TLR4 signaling in macrophages or DCs, in which CD180 negatively regulates TLR4 signaling.20
IFN-α can potentiate the activation of TLR7 and TLR9 signaling and promote auto-antibody production. However, it remains unclear how TLR7 and TLR9 regulate the activation of IFN-α signaling. In the present study, we revealed that TLR7 and TLR9 signaling could alter the inhibitory effect of CD180 ligation on the activation of IFN-α signaling. Our results indicated that TLR7, TLR9, and IFN-α signaling may form a positive feedback loop whereby TLR7 and TLR9 signaling could enhance the activation of IFN-α signaling by modulating the expression of multiple genes (such as CD180 and STS-1), and enhanced activation of IFN-α signaling further boosts the activation of TLR7 and TLR9 signaling. This positive feedback model may account for abnormal B-cell hyperactivation in SLE.
It has been shown that only CD180-negative B cells from SLE patients can produce IgG and IgM spontaneously in vitro.26 Our present study indicates that CD180 ligation could inhibit IFN-α signaling activation in B cells, suggesting that CD180 signaling may play a protective role in the pathogenesis of SLE. However, Toshihiko Kobayashi et al.60 demonstrated previously that CD180−/− MRL/lpr mice showed relieved disease activity and enhanced survival rate compared to MRL/lpr mice, suggesting that CD180 could exacerbate SLE progress. However, it should be noted that CD180 is not exclusively expressed on B cells, and the functions of CD180 in B cells and other cells (such as DCs and monocytes) are also different.20 Thus, the specific role of CD180 in SLE might be complex, and more studies should be conducted to elucidate it.
In conclusion, our present study extended a human SLE phenomenon to a mouse model, demonstrating that CD180-negative B cells are increased in MRL/lpr mice. We also demonstrated a novel role of CD180 in regulating the activation of IFN-α signaling in B cells both in vitro and in vivo. More importantly, our study may provide new insight into the treatment of SLE by anti-CD180 antibody.
Acknowledgments
This work was supported by a grant from National Natural Science Foundation of China (Project number: 31370899).
Footnotes
Supplementary information of this article can be found on Cellular & Molecular Immunology website: http://www.nature.com/cmi.
The authors declare that they have no conflict of interest.
Supplementary Information
References
- Kotzin BL. Systemic lupus erythematosus. Cell 1996; 85: 303–306. [DOI] [PubMed] [Google Scholar]
- Dorner T, Giesecke C, Lipsky PE. Mechanisms of B cell autoimmunity in SLE. Arthritis Res Ther 2011; 13: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos AM, Busconi L, Marshak-Rothstein A. Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity 2010; 43: 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum 2005; 52: 1491–1503. [DOI] [PubMed] [Google Scholar]
- Kim T, Kanayama Y, Negoro N, Okamura M, Takeda T, Inoue T. Serum levels of interferons in patients with systemic lupus erythematosus. Clin Exp Immunol 1987; 70: 562–569. [PMC free article] [PubMed] [Google Scholar]
- Ytterberg SR, Schnitzer TJ. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum 1982; 25: 401–406. [DOI] [PubMed] [Google Scholar]
- Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA 2003; 100: 2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Liu F, Dong G, Ren D, Xu Y, Dou J et al. Activation-induced necroptosis contributes to B-cell lymphopenia in active systemic lupus erythematosus. Cell Death Dis 2014; 5: e1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB et al. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS One 2013; 8: e67003.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun D, Caramalho I, Demengeot J. IFN-alpha/beta enhances BCR-dependent B cell responses. Int Immunol 2002; 14: 411–419. [DOI] [PubMed] [Google Scholar]
- Bekeredjian-Ding IB, Wagner M, Hornung V, Hornung V, Giese T, Schnurret M et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type-I IFN. J Immunol 2005; 174: 4043–4050. [DOI] [PubMed] [Google Scholar]
- Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity 2004; 20: 785–798. [DOI] [PubMed] [Google Scholar]
- Mathian A, Gallegos M, Pascual V, Banchereau J, Koutouzov S. Interferon-alpha induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZB×NZW) F1 mice but not in BALB/c mice. Eur J Immunol 2011; 41: 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uccellini MB, Busconi L, Green NM, Busto P, Christensen SR, Shlomchik MJ et al. Autoreactive B cells discriminate CpG-Rich and CpG-Poor DNA and this response is modulated by IFN-alpha. J Immunol 2008; 181: 5875–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med 2005; 202: 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green NM, Laws A, Kiefer K, Busconi L, Kim YM, Brinkmann MM et al. Murine B cell response to TLR7 ligands depends on an IFN-beta feedback loop. J Immunol 2009; 183: 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oganesyan G, Saha SK, Pietras EM, Guo B, Miyahira AK, Zarnegar B et al. IRF3-dependent type I interferon response in B cells regulates CpG-mediated antibody production. J Biol Chem 2008; 283: 802–808. [DOI] [PubMed] [Google Scholar]
- Miyake K, Shimazu R, Kondo J, Niki T, Akashi S, Ogata H et al. Mouse MD-1, a molecule that is physically associated with RP105 and positively regulates its expression. J Immunol 1998; 161: 1348–1353. [PubMed] [Google Scholar]
- Miura Y, Shimazu R, Miyake K, Akashi S, Ogata H, Yamashita Y et al. RP105 is associated with MD-1 and transmits an activation signal in human B cells. Blood 1998; 92: 2815–2822. [PubMed] [Google Scholar]
- Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A et al. Negative regulation of toll-like receptor 4 signaling by the toll-like receptor homolog RP105. Nat Immunol 2005; 6: 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K, Yamashita Y, Ogata M, Sudo T, Kimoto M. RP105, a novel B cell surface molecule implicated in B cell activation, is a member of the leucine-rich repeat protein family. J Immunol 1995; 154: 3333–3340. [PubMed] [Google Scholar]
- Miyake K, Yamashita Y, Hitoshi Y, Takatsu K, Kimoto M. Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD molecule unresponsiveness of X-linked immunodeficient B cells. J Exp Med 1994; 180: 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan VW, Mecklenbräuker I, Su I, Texido G, Leitges M, Carsetti R et al. The molecular mechanism of B cell activation by toll-like receptor protein RP-105. J Exp Med 1998; 188: 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa N, Fujimoto M, Sato S, Miyake K, Asano N, Nagai Y et al. CD19 regulates innate immunity by the toll-like receptor RP105 signaling in B lymphocytes. Blood 2003; 102: 1374–1380. [DOI] [PubMed] [Google Scholar]
- Koarada S, Tada Y, Ushiyama O, Morito F, Suzuki N, Ohta A. B cells lacking RP105, a novel B cell antigen, in systemic lupus erythematosus. Arthritis Rheum 1999; 42: 2593–2600. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, Koarada S, Tada Y, Ushiyama O, Morito F, Suzuki N et al. RP105-lacking B cells from lupus patients are responsible for the production of immunoglobulins and autoantibodies. Arthritis Rheum 2002; 46: 3259–3265. [DOI] [PubMed] [Google Scholar]
- Fujita K, Akasaka Y, Kuwabara T, Wang B, Tanaka K, Kamata I et al. Pathogenesis of lupus-like nephritis through autoimmune antibody produced by CD180-negative B lymphocytes in NZB/W F1 mouse. Immunol Lett 2012; 144: 1–6. [DOI] [PubMed] [Google Scholar]
- Koarada S, Tada Y, Suematsu R, Soejima S, Inoue H, Ohta A et al. Phenotyping of RP105-negative B cell subsets in patients with systemic lupus erythematosus. Clin Dev Immunol 2012; 2012: 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzina IG, Atamas SP, Storrer CE, da Silva LC, Kelsoe G, Papadimitriou JC et al. Spontaneous formation of germinal centers in autoimmune mice. J Leukoc Biol 2001; 70: 578–584. [PubMed] [Google Scholar]
- Chaplin JW, Kasahara S, Clark EA, Ledbetter JA. Anti-CD180 (RP105) activates B cells to rapidly produce polyclonal Ig via a T cell and MyD88-independent pathway. J Immunol 2011; 187: 4199–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki K, Yamazaki T, Taki S, Miyake K, Hayashi T, Ochs HD et al. Potentiation of TLR9 responses for human naive B-cell growth through RP105 signaling. Clin Immunol 2010; 135: 125–136. [DOI] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 2005; 5: 375–386. [DOI] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE Jr. Maximal activation of transcription by Statl and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995; 82: 241–250. [DOI] [PubMed] [Google Scholar]
- Pilz A, Ramsauer K, Heidari H, Leitges M, Kovarik P, Decker T. Phosphorylation of the Stat1 transactivating domain is required for the response to type I interferons. EMBO Rep 2003; 4: 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Ramana CV, Bayes J, Stark GR. Roles of phosphatidylinositol 3-kinase in interferon-γ-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J Biol Chem 2001; 276: 33361–33368. [DOI] [PubMed] [Google Scholar]
- Du Z, Shen Y, Yang W, Mecklenbrauker I, Neel BG, Ivashkiv LB et al. Inhibition of IFN-alpha signaling by a PKC- and protein tyrosine phosphatase SHP-2-dependent pathway. Proc Natl Acad Sci USA 2005; 102: 10267–10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HuangFu WC, Qian J, Liu C, Liu J, Lokshin AE, Baker DP et al. Inflammatory signaling compromises cell responses to interferon alpha. Oncogene 2012; 31, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Nagai Y, Yanagibashi T, Watanabe Y, Ikutani M, Kariyone A et al. Serum soluble MD-1 levels increase with disease progression in autoimmune prone MRL/lpr mice. Mol Immunol 2012; 49: 611–620. [DOI] [PubMed] [Google Scholar]
- Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol 2000; 165: 5970–5979. [DOI] [PubMed] [Google Scholar]
- Odendahl M, Keitzer R, Wahn U, Hiepe F, Radbruch A, Dörner T et al. Perturbations of peripheral B lymphocyte homoeostasis in children with systemic lupus erythematosus. Ann Rheum Dis 2003; 62: 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culton DA, O'Conner BP, Conway KL, Diz R, Rutan J, Vilen BJ et al. Early preplasma cells define a tolerance checkpoint for autoreactive B cells. J Immunol 2006; 176: 790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med 2004; 199: 1577–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol 2009; 9: 845–857. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Mumtaz IM, Khodadadi L, Radbruch A, Hoyer BF, Hiepe F. Autoantibodies from long-lived ‘memory' plasma cells of NZB/W mice drive immune complex nephritis. Ann Rheum Dis 2013; 72: 2011–2017. [DOI] [PubMed] [Google Scholar]
- Weinstein JS, Delano MJ, Xu Y, Kelly-Scumpia KM, Nacionales DC, Li Y et al. Maintenance of anti-Sm/RNP autoantibody production by plasma cells residing in ectopic lymphoid tissue and bone marrow memory B cells. J Immunol 2013; 190: 3916–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson LE, Widman D, Dikman SH, Gorevic PD. Autoimmune disease complicating antiviral therapy for hepatitis C virus infection. Semin Arthritis Rheum 2002; 32: 163–173. [DOI] [PubMed] [Google Scholar]
- Ronnblom LE, Alm GV, Oberg KE. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann Intern Med 1991; 115: 178–183. [DOI] [PubMed] [Google Scholar]
- Adam C, Thoua Y, Ronco P, Verroust P, Tovey M, Morel-Maroger L. The effect of exogenous interferon: acceleration of autoimmune and renal diseases in (NZB/W) F1 mice. Clin Exp Immunol 1980; 40: 373–382. [PMC free article] [PubMed] [Google Scholar]
- Heremans H, Billiau A, Colombatti A, Hilgers J, de Somer P. Interferon treatment of NZB mice: accelerated progression of autoimmune disease. Infect Immun 1978; 21: 925–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow MK. Interferon-alpha: a therapeutic target in systemic lupus erythematosus. Rheum Dis Clin North Am 2010; 36: 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Richman L, Higgs BW, Morehouse CA, de los Reyes M, Brohawn P et al. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum 2009; 60: 1785–1796. [DOI] [PubMed] [Google Scholar]
- Hwang SH, Lee H, Yamamoto M, Jones LA, Dayalan J, Hopkins RB et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol 2012; 189: 5786–5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplin JW, Chappell CP, Clark EA. Targeting antigens to CD180 rapidly induces antigen-specific IgG, affinity maturation, and immunological memory. J Exp Med 2013; 210: 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Nair JS, Malhotra A, Zhang JJ. B cell antigen receptor signaling enhances IFN-gamma-induced Stat1 target gene expression through calcium mobilization and activation of multiple serine kinase pathways. J Interferon Cytokine Res 2005; 25: 113–124. [DOI] [PubMed] [Google Scholar]
- Chan VWF, Meng F, Soriano P, DeFranco AL, Lowell CA. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity 1997; 7: 69–81. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, Koarada S, Nakamura S, Yonemitsu N, Tada Y, Haruta Y et al. Increase of RP105-lacking activated B cells in the peripheral blood and salivary glands in patients with Sjögren's syndrome. Clin Exp Rheumatol 2008; 26: 5–12. [PubMed] [Google Scholar]
- Miguet L, Lennon S, Baseggio L, Traverse-Glehen A, Berger F, Perrusson N et al. Cell-surface expression of the TLR homolog CD180 in circulating cells from splenic and nodal marginal zone lymphomas. Leukemia 2013; 27: 1748–1750. [DOI] [PubMed] [Google Scholar]
- Ren D, Liu F, Dong G, You M, Ji J, Huang Y et al. Activation of TLR7 increases CCND3 expression via the downregulation of miR-15b in B cells of systemic lupus erythematosus. Cell Mol Immunol 2015; 10: in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G, You M, Fan H, Ding L, Sun L, Hou Y. STS-1 promotes IFN-α-induced autophagy by activating the JAK1-STAT1 signaling pathway in B cells. Eur J Immunol 2015; 10: in press. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Takahashi K, Nagai Y, Shibata T, Otani M, Izui S et al. Tonic B cell activation by radioprotective105/MD-1 promotes disease progression in MRL/lpr mice. Int Immunol 2008; 20: 881–891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.