

Abstract
A single extracellular stimulus can promote diverse behaviors among isogenic cells by differentially regulated signaling networks. We examined Ca2+ signaling in response to VEGF (vascular endothelial growth factor), a growth factor that can stimulate different behaviors in endothelial cells. We found that altering the amount of VEGF signaling in endothelial cells by stimulating them with different VEGF concentrations triggered distinct and mutually exclusive dynamic Ca2+ signaling responses that correlated with different cellular behaviors. These behaviors were cell proliferation involving the transcription factor NFAT (nuclear factor of activated T cells) and cell migration involving MLCK (myosin light chain kinase). Further analysis suggested that this signal decoding was robust to the noisy nature of the signal input. Using probabilistic modeling, we captured both the stochastic and deterministic aspects of Ca2+ signal decoding and accurately predicted cell responses in VEGF gradients, which we used to simulate different amounts of VEGF signaling. Ca2+ signaling patterns associated with proliferation and migration were detected during angiogenesis in developing zebrafish.
INTRODUCTION
Intracellular signaling pathways and networks mediate context-specific decision-making by individual cells and cell ensembles. However, the transfer of information through these molecular systems is subject to uncertainty, and thus, the resulting decision repertoire can be limited (1). Furthermore, there is diversity in both signaling and phenotypic responses to identical stimuli, such as in the regulation of single cell apoptosis or migration (2, 3). Is phenotypic diversity a direct consequence of variability in signal processing among individual cells, or are there additional sources of noise affecting the fidelity of cell responses? Which, if any, aspects of cell phenotype specification are robust to variability in signaling inputs? Can the limited information provided by signaling networks be used to specify cell phenotypes with high fidelity (1)? To address these questions, we explored the vascular endothelial growth factor (VEGF) signaling network, activation of which enables distinct phenotypic responses, such as cell migration or proliferation (4).
VEGF signaling is a key component of vascular sprout formation, a process known as angiogenesis. VEGF stimulates normally quiescent endothelial cells to loosen interconnections and take on individualistic roles as they leave the parent vessel and form a new structure. Throughout angiogenesis, cells at the tip of forming vessels migrate under the guidance of directional cues, such as growth factors, whereas other cells lagging behind divide and eventually form a new vessel wall (5). Growth factors, including VEGF, promote both of these behaviors (6–8), but it remains unclear how genetically identical endothelial cells interpret this signal to elicit distinct roles and whether cell phenotype selection is robust to noise.
VEGF is a pleiotropic signaling ligand that triggers activation of multiple pathways, including those mediated by dynamic Ca2+ responses. Disrupting Ca2+ signaling prevents both tube formation in vitro and angiogenesis in vivo (9). Moreover, modulation of Ca2+ signaling regulates many aspects of cell physiology, including gene transcription (10), cell migration (9), cell proliferation (11, 12), and apoptosis (13). Both experimental (14, 15) and theoretical (14,16) studies suggest that Ca2+ signaling is influenced by stochastic perturbations in cellular Ca2+ regulating components, leading to response variability from isogenic cell populations (17). In addition, experimentally imposed artificial Ca2+ inputs, such as regular oscillations (18) and sustained concentration increases (18,19), activate different transcription factors and gene expression. Thus, Ca2+ signaling may mediate the heterogeneous interpretation of extracellular cues while simultaneously conveying phenotype specificity through signal dynamics.
Whereas previous studies indicate that distinct responses within a single signaling pathway can be used to transmit information from different upstream extracellular stimuli (20), we sought to determine whether noisy signaling responses, triggered by a single stimulus, can be used for robust downstream phenotype selection. We focused on two cell responses that are key components of angiogenesis (21–23): cell migration and the persistent translocation of the transcription factor NFAT (nuclear factor of activated T cells) to the nucleus. In this fashion, we investigated Ca2+ signaling in individual endothelial cells stimulated with VEGF and correlate both stochastic and deterministic response characteristics to the selection of phenotypes associated with angiogenesis.
RESULTS
VEGF triggers noisy but distinct Ca2+ waveforms in a dose-dependent manner
To determine whether the regulation of intracellular Ca2+ was similar across a population of endothelial cells exposed to VEGF, we performed live cell microscopy and fluorescence resonance energy transfer (FRET) imaging (Fig. 1A). We found that individual isogenic endothelial cells exposed to the same concentration of VEGF exhibited one of three different dynamic Ca2+ responses or waveforms (Fig. 1B). A fraction of cells displayed a transient single peak of Ca2+. In these cells, the concentration of intracellular Ca2+ often did not completely adapt back to the baseline value, settling at a low persistent amount of signaling (LP waveform). Other cells responded with complex temporal patterns of repeated spikes (RS waveform). Finally, in the remainder of the cells, Ca2+ did not display a detectable response at all, referred to as the null response (NR waveform).
Fig. 1. VEGF promotes multiple Ca2+ waveforms.
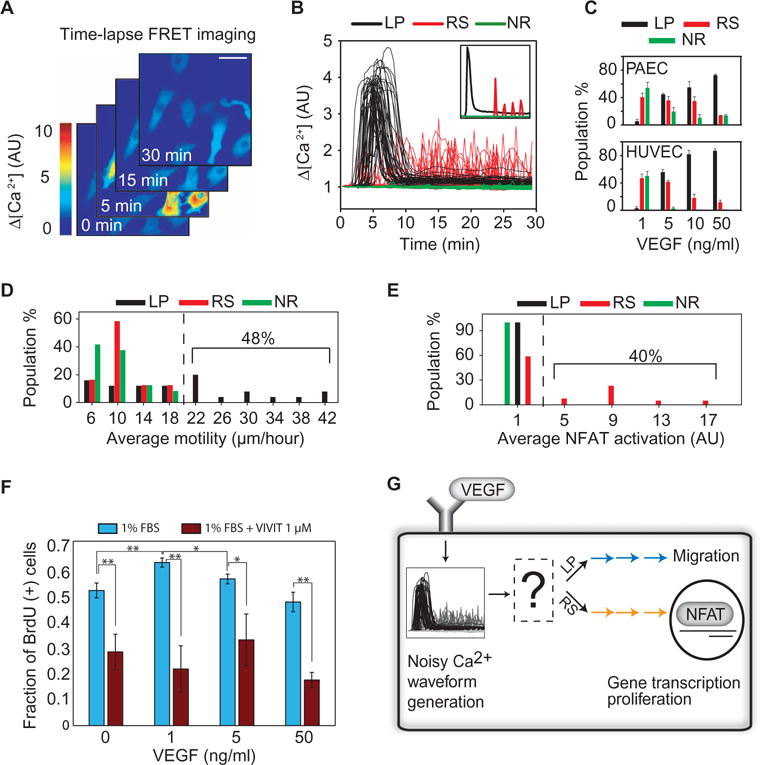
(A) Time series showing YC3.6 FRET detection of cytosolic Ca2+ concentration in individual PAECs after VEGF stimulation. The FRET signal at each time point was normalized to that measured at the starting time. Scale bar, 50 μm. AU, arbitrary unit. (B) Representative traces illustrating different Ca2+ waveforms triggered by VEGF stimulation. Inset shows the three basic waveforms observed (LP, RS, and NR). (C) Distribution of Ca2+ waveforms as a function of VEGF concentration in PAECs and HUVECs. Data are means ± SEM for n = 3 independent experiments with greater than 100 cells for each concentration of VEGF. (D) Distribution of migration distances as a function of waveform after stimulation with VEGF for 1 hour (n = 92 cells from n = 3 independent experiments). (E) The distribution of time-averaged NFAT activation as a function of Ca2+ waveform in PAECs after stimulation with VEGF for 3 hours (n = 76 cells from n = 4 independent experiments). NFAT activation was determined as the ratio of nuclear to cytoplasm NFAT intensity. (F) HUVECs were starved overnight and exposed to VEGF and the NFAT inhibitor VIVIT for 24 hours. Nuclear BrdU (5-bromo-2′-deoxyuridine)incorporation was used to track cell cycle progression (Student’s ttest, **P <0.05, *P < 0.085, n = 3 independent experiments). (G) Schematic summarizing the role of Ca2+ signaling in phenotype selection. LP and RS signals are interpreted differently by cells resulting in either migration or NFAT signaling, which can promote cell proliferation.
These responses, which were present under conditions with limited cell-cell contact (Fig. 1A) and with substantial cell-cell contact (movies S1 and S2), were observed in porcine aortic endothelial cells (PAECs), in which only VEGF receptor 2 (VEGFR2) is present, and in primary human umbilical vein endothelial cells (HUVECs), in which multiple VEGFR isoforms are present. Moreover, by testing several different concentrations of VEGF, we found that the fraction of cells exhibiting each waveform was dependent on the amount of VEGF signaling in both PAECs (Fig. 1C, top) and HUVECs (Fig. 1C, bottom). At a low concentration of VEGF (1 ng/ml), most cells had either NR or RS waveforms. When the concentration was increased to 5 ng/ml, approximately the dissociation constant of VEGFR2 (24), most cells exhibited either an RS or an LP waveform. As the concentration of VEGF approached saturating amounts, the LP response was dominant. These observations raised the possibility that Ca2+ signals in endothelial cells can relay differences in VEGF signaling in vitro, and possibly in vivo, in the form of dynamically distinct profiles. These Ca2+ waveforms, if decoded by distinct signaling pathways, could then translate into distinct phenotypic outcomes, such as enhanced cell proliferation or enhanced migration.
Distinct noisy temporal Ca2+ dynamics are specifically converted into either enhanced cell migration or increased NFAT signaling
VEGF-dependent Ca2+ signaling stimulates cell motility. We simultaneously measured Ca2+-dependent FRET and the path length of cell migration in response to VEGF stimulation. We found that cells with LP waveforms were more motile (Fig. 1D) than those with NR or RS waveforms. To assess proliferative capacity, we examined signaling mediated by the transcription factor NFAT, which is a terminal node of the central pathway that links intracellular Ca2+ to cell proliferation (25–29) and survival (30). NFAT isoforms can show differential activation in response to dynamic Ca2+ stimulation (31). We thus examined NFAT2 (also known as NFATC1, henceforth referred to as NFAT), which promotes cell proliferation (25, 30). Because dephosphorylated NFAT translocates into the nucleus (31), we used changes in the ratio of nuclear to cytoplasmic NFAT to estimate changes in NFAT activation. Simultaneous imaging of Ca2+ waveforms and phenotypic responses revealed that only cells with RS waveforms exhibited enhanced NFAT activation (Fig. 1E) compared to those with NR or LP waveforms. Furthermore, we found that the dependence of HUVEC proliferation on VEGF concentration was similar to that of RS waveforms, with proliferation suppressed at all VEGF concentrations by an NFAT inhibitor (Fig. 1F).
How do signaling pathways controlling NFAT activation and cell migration discriminate between LP and RS waveforms? Individual signaling pathways could respond to the average amount of Ca2+ over a given amount of time to reduce noise and discriminate between waveforms. Because the distribution of Ca2+ concentration for LP and RS waveforms, averaged for 30 min after VEGF stimulation, completely overlapped (fig. S1), time averaging was an unlikely mechanism of signal discrimination. This finding was consistent with biological constraints that prevent sustained high concentrations of Ca2+ for prolonged periods (32). Therefore, the mechanisms of waveform discrimination likely rely on information encoded by the dynamic, time-dependent attributes of different Ca2+ waveforms (Fig. 1G). We thus explored how distinct downstream signaling pathways might enable robust dynamic decoding of distinct noisy Ca2+ inputs.
Increased NFAT signaling requires temporal integration of frequent Ca2+ spikes
Simultaneous tracking of Ca2+ and NFAT signaling revealed that the naturally occurring interspike intervals of RS waveforms were distributed, such that NFAT activation displayed stepwise increases. These discrete increases in activation correlated with individual Ca2+ spikes (Fig. 2A). Some cells with RS waveforms generated fast and persistent NFAT activation, suggesting that NFAT was able to integrate Ca2+ spikes due to VEGF stimulation [Fig. 2, B (cell I) and C, and movie S3 (cell I)]. LP waveforms did not sustain high NFAT activation as compared to those with RS waveforms [(Fig. 2, B (cell II) and D, and movie S3 (cell II)].
Fig. 2. NFAT signaling is selectively enhanced by the RS Ca2+ waveform.
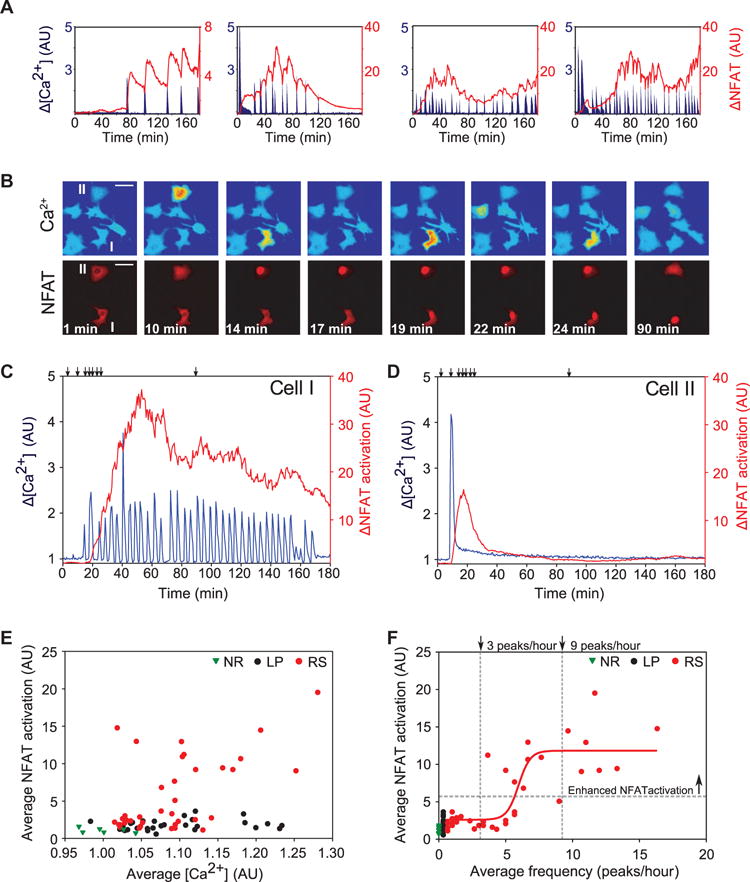
(A) Example traces illustrating “staircase-like” NFAT response to Ca2+ spikes after stimulation with VEGF. (B) Representative images (from movie S3) showing simultaneous Ca2+ (FRET) amplitude and NFAT translocation for cells with RS (I) and LP (II) waveforms. VEGF was added after 1.5 min. Scale bars, 50 μm. (C and D) Ca2+ and NFAT regulation for the cells shown in (B) having RS (C) and LP waveforms (D). Arrows show the times corresponding to the sample images. (E) Comparison of average NFAT activation as a function of average increase in Ca2+ amount for individual cells exhibiting NR, LP, or RS waveforms. (F) Average NFAT activation as a function of the average spike frequency. The horizontal gray line indicates the boundary between the groups with basal and enhanced activation as determined by k-means analysis. The vertical lines mark the boundaries between cells exhibiting only basal NFAT activation, both basal and enhanced activation, and only enhanced NFAT activation. The red line illustrates a sigmoidal fit to the RS cell data [f(x) = 2.07 + 1/(0.1 + exp(−(x − 16.15))), R2 = 0.865]. Data in (E) and (F) were collected for 3 hours after application of VEGF (n = 76 cells from n = 4 independent experiments).
If the NFAT-dependent Ca2+ signal decoding mechanism functions by maintaining the memory of Ca2+ spikes and pushing the response to RS signals above a threshold, Ca2+ should trigger high NFAT activation through repeated, brief, high-amplitude inputs, rather than through nonrepetitive inputs, irrespective of the average Ca2+ concentration. We tested this prediction by analyzing NFAT responses in cells with LP and RS waveforms, which have wide, overlapping ranges of average Ca2+ concentrations. NFAT activation was the same in cells with LP and NR waveforms, irrespective of average Ca2+ concentration (Fig. 2E). NFAT activation in many RS cells, however, exceeded that of NR cells up to 20-fold across the range of average Ca2+ concentrations observed (Fig. 2E). Thus, although the NFAT activation in cells with RS waveforms depended on average Ca2+ concentration (Fig. 2E), it did not in cells with LP waveforms, suggesting that discrimination of waveforms was not accomplished on the basis of average Ca2+ concentration alone, but also required information about the number of peaks over the duration of the input (Fig. 2F). K-means cluster analysis was used to separate RS cells into two groups based on the amount of NFAT activation. The first group had lower NFAT activation near that observed for NR and LP cells (Fig. 2F), whereas the second group had enhanced NFAT activation, which exceeded that observed for the other two groups. The amount of NFAT activation in cells with RS waveforms roughly correlated with spike frequency in a stepwise fashion. RS cells with less than three spikes per hour had basal amounts of NFAT activation, those with between three and nine spikes per hour had variable amounts of NFAT activation, and those with greater than nine spikes per hour had only enhanced NFAT activation.
Increased cell motility requires sustained and uninterrupted Ca2+ signaling
To determine the impact of VEGF-dependent Ca2+ signaling on cell motility, we simultaneously measured Ca2+-dependent FRET and the path length of cell migration in response to VEGF stimulation (fig. S2A). We found that cells with LP waveforms were more motile than those with NR or RS waveforms (Fig. 3A). In addition, cells with LP waveforms, but not RS waveforms, also maintained Ca2+ concentrations above a minimal amount required to stimulate enhanced cell migration (Fig. 3A). Cells with LP waveforms that did not maintain Ca2+ concentrations at least 5% above baseline clustered (k-means analysis) with NR and RS cells (Fig. 3A) and displayed reduced migration distances indistinguishable from that of cells with NR waveforms (Fig. 3B). Although we used VEGF (5 ng/ml) to obtain nearly equal samples of LP and RS cells, this clustering was also evident using VEGF (50 ng/ml) (fig. S2B). These results confirmed that the initial transient Ca2+ peak of the LP response was not sufficient to sustain the enhanced motility phenotype. To further test whether this low Ca2+ signaling was required to maintain enhanced motility, we inhibited Ca2+ signaling with EGTA after 30 min of VEGF stimulation, which resulted in acute reduction of cell migration distances (fig. S2C), suggesting that instantaneous rather than integrated Ca2+ concentrations were a better predictor of the extent of cell migration. Indeed, we found that, although the ranges of time-integrated values of Ca2+ were similar among LP and RS cells, none of the RS cells displayed increased motility (Fig. 3C and fig. S2D) above that of NR cells. These results suggested that a signaling pathway coding enhanced motility would have a relatively short “memory” of Ca2+ spikes, but would be responsive to persistent, low Ca2+ inputs. This feature would allow LP signals to persistently stimulate migration by remaining above a low threshold value in contrast to RS waveforms, which continually fluctuate below the same low threshold value.
Fig. 3. Signaling that regulates enhanced cell motility requires Ca2+ concentrations to be continuously sustained above a threshold.
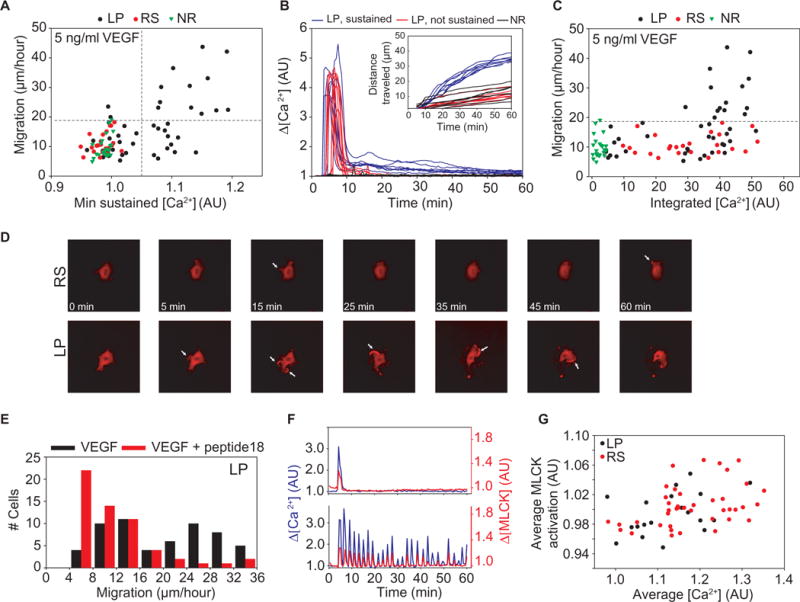
(A) Migration as a function of the minimum Ca2+ concentrations reached during the later portion of Ca2+ signaling (20 to 60 min). Cells that returned to baseline were separated from those with sustained Ca2+ concentrations (vertical gray line). The horizontal gray line indicates the boundary between basal and enhanced motility groups as determined by k-means analysis (n = 92 cells from n = 3 independent experiments). (B) Ca2+ traces for cells displaying LP waveforms that either fully adapt back to baseline (red) or maintained low amounts of signaling (blue) are compared to NR cells, used as a control (black). Corresponding migration is shown in the inset. Data were collected for 1 hour after simulation with VEGF. (C) Migration as a function of integrated Ca2+ concentrations. (D) Representative images showing that cells with RS waveforms (movie S5) did not change shape and displayed limited actin recruitment to small lamellipodia and filopodia (white arrows). Cells exhibiting LP waveforms (movie S4) markedly changed shape and formed large lamellipodia with greater actin recruitment (white arrows). (E) Distribution of migration values for cells exhibiting LP waveforms after VEGF stimulation alone (black, n = 58 cells from n = 3 independent experiments) and with 30-min preincubation with the MLCK-specific inhibitor peptide 18 (red, n = 57 cells from n = 3 independent experiments). (F) MLCK activation corresponding to Ca2+ waveforms with LP (top) and RS (bottom) was synchronized in time. Traces are representative of n = 64 cells from three independent experiments. (G) Average MLCK activation, taken after 1 hour of VEGF stimulation, shown as a function of the average amount of Ca2+ for both LP and RS (n = 64 cells from n =3 independent experiments).
Myosin light chain kinase (MLCK) is a Ca2+-sensitive kinase that activates myosin light chain through phosphorylation, thereby facilitating the stress fiber formation and cell contraction required for movement. We investigated MLCK as a possible effector of VEGF-induced cell motility. We found that cells exhibiting LP waveforms formed a greater number of lamellipodia than those with RS waveforms (fig. S2E). Such lamellipodia, which were larger and more persistent compared to those observed in cells with RS waveforms (fig. S2E), have been previously implicated in control of cell migration in an MLCK-dependent fashion (33). We indeed found that cells with the LP Ca2+ waveforms drastically changed shape in response to VEGF (Fig. 3D, LP, and movie S4). In contrast, cells with RS waveforms generally had small filopodia (Fig. 3D, RS, and movie S5) and showed little change in shape. Inhibition of MLCK with the MLCK-specific peptide 18 decreased VEGF-induced cell motility, suggesting that the observed cell changes were indeed MLCK-dependent (Fig. 3E).
Our data suggested that cells become motile in response to acute changes in Ca2+ concentration and that this response was MLCK-dependent. Thus, we predicted that the dynamics of MLCK activation should closely reflect the dynamics of the Ca2+ waveforms, with sufficiently rapid activation and inactivation kinetics. Using an MLCK FRET reporter, we found that MLCK activation correlated with both LP (Fig. 3F, top) and RS (Fig. 3F, bottom) waveforms with transient MLCK peaks coinciding with Ca2+ spikes. In contrast to NFAT activation, average MLCK activation was similar for LP and RS waveforms across a range of time-averaged Ca2+ concentrations (Fig. 3G), indicating that MLCK was not capable of integrating repeated Ca2+ spikes. Thus, neither waveform was capable of sustaining MLCK activation in the absence of sustained Ca2+ concentrations, confirming the role of MLCK in enabling the migration pathway to behave in an acute and instantaneous fashion.
Decoding of dynamic Ca2+ signals results in deterministic phenotype selection coupled with noisy response amplitudes
The results above suggested that a VEGF-triggered dynamic signal, intracellular Ca2+, was decoded differently by two signaling systems to obtain distinct and mutually exclusive cell responses—that is, to achieve signal “multiplexing” through dynamic signal encoding (Fig. 4A, top). We showed that the selection of either of the two signaling pathways, NFAT and MLCK, is achieved by their respective memory of the Ca2+ signals—that is, retention of information about the duration of Ca2+ signaling activity after a transient Ca2+ increase. This type of memory by signaling pathways is analogous to a physical ratcheting mechanism, which permits movement in a single direction while preventing travel in the opposite direction. If, as is the case with the NFAT activation, the memory of the transient Ca2+ increase is sufficiently long compared to the characteristic time scale of the Ca2+ waveform dynamics—for example, the interspike intervals in the RS waveforms—then the signaling pathway could act as an effective ratcheting mechanism, integrating sequential transient inputs into a high and sustained output (Fig. 4A, bottom, orange). Several Ca2+ spikes, operating within a wide range of stochastic spike distributions, can thus sustain the signaling output above a response threshold to ensure robust phenotypic selection (Fig. 4B, orange box). When presented with an LP waveform, lack of repeated Ca2+ transients would prevent the pathway from sustaining signaling above the response threshold (Fig. 4B, orange box). Alternatively, if the memory of a signaling pathway is comparatively short, as is the case with the MLCK activation, making it an ineffective ratchet, the response to RS waveforms would be negligible because each individual spike would be insufficient to sustain an appreciable signaling output (Fig. 4A, bottom, blue). To achieve a sustained output, a signaling pathway behaving as an ineffective ratchet therefore requires a continuous input. The required amount of input may be high enough to prevent activation through the baseline Ca2+ concentrations found in-between RS peaks, but low enough such that the later phase of the LP waveforms are effective in sustaining downstream signaling (Fig. 4B, blue box). In this fashion, signaling pathways responding to Ca2+ transients as effective and ineffective ratchets could selectively and robustly respond to RS and LP waveforms, respectively.
Fig. 4. Waveform discrimination through signal “ratcheting.”.
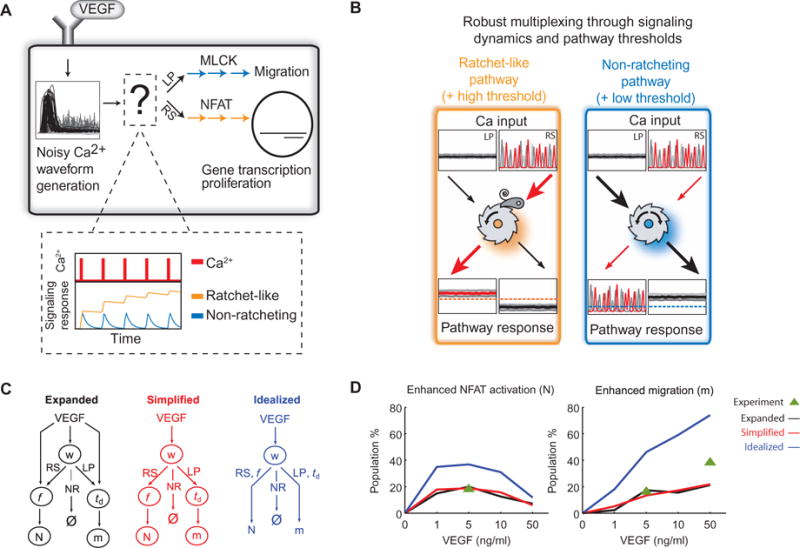
(A) Schematic description of Ca2+ waveform discrimination that translates VEGF input into specific phenotypes. The inset depicts the theoretically possible activities of Ca2+ effectors responding to Ca2+ spikes (red) in analogy to a ratchet. An effective ratchet (orange) resists deactivation, and an ineffective ratchet (blue) readily deactivates. (B) Conceptual model of information processing by a ratchet-like pathway (left, orange) and a non-ratcheting pathway (right, blue). The red lines and arrows denote how the sustained Ca2+ signaling after an RS waveform is translated into a response, and the black lines show the same for LP. The threshold for triggering Ca2+ effector activation differs for NFAT and cell motility phenotypes (dashed lines). (C) Illustration of the probabilistic model structure when considering the distributions of f and td either dependent on VEGF (“expanded model,” black) or independent of VEGF (“simplified model,” red). Also shown is the model structure without stochasticity downstream of waveform (w) selection (“idealized,” blue). (D) Predicted percentage of each enhanced phenotype, either NFAT activation (N, left) or migration (m, right). Lines are colored corresponding to the model illustrations in (C). Experimental measurements are denoted by the green triangle.
Whereas the model above supports an experimentally observed deterministic selection of responses, in which cells with LP waveforms did not show enhanced NFAT signaling (Fig. 1E) and cells with RS waveforms did not display enhanced cell motility (Fig. 1D), it leaves open the question of the magnitude of each response. Indeed, we found that the degree of NFAT activation by RS cells and the extent of cellular motility displayed by LP cells showed a high degree of variability. To better understand this response variability, we constructed a Bayesian decision model, accounting for the three stochastic elements: (i) the selection of Ca2+ waveforms, (ii) the magnitude of their characteristics—that is, Ca2+ spike frequency (f) for RS and late-phase Ca2+ signal duration (td) for LP, and (iii) the phenotype responses NFAT activation or cell motility (text S1).
To determine whether phenotypic selection depended on the direct modulation of waveform characteristics by VEGF or solely through its effect on waveform selection, we trained the Bayesian decision model using either expanded or simplified model topologies (Fig. 4C). In the expanded model, we considered the distributions of f and td for each concentration of VEGF (1, 5, 10, and 50 ng/ml). This expanded model predicted waveform distributions and characteristics that generally agreed with the experimental data (fig. S3, A to C), and simulations using the expanded model (see fig. S3, D to F, for model parameters) predicted distributions of phenotypes across VEGF concentrations consistent with the distribution of waveforms. The percentage of cells with enhanced NFAT activation was biphasic (Fig. 4D, left), and the percentage of cells with enhanced motility increased monotonically as a function of increasing VEGF concentration (Fig. 4D, right).
In the simplified Bayesian decision model, we made the distributions off and td independent of VEGF concentration by pooling f and td values across all concentrations of VEGF. The predictions of the simplified model were different (P < 0.01, Student’s t test) in the percentage of cells with enhanced NFAT or enhanced motility as compared to the expanded model (Fig. 4D). However, the magnitude of the difference was less than 5%, demonstrating that the modulation of waveform characteristics by VEGF in the expanded model (fig. S3, B and C) minimally affected the selection of enhanced NFAT activation or enhanced cell motility phenotypes. We also evaluated the effect of the stochasticity downstream of waveform selection by training the model, assuming that every RS waveform fully activated NFAT and every LP waveform fully enhanced motility (Fig. 4C, idealized topology). We found that this idealized model overestimated the percent of cells showing both enhanced motility and NFAT activation (Fig. 4D, blue lines).
Overall, our results suggest that in cells with LP and RS waveforms, the stochasticity in the quantitative phenotypic features f and td introduces variability into the number of cells with enhanced motility or NFAT activation, largely independently of the VEGF dose. The VEGF dose thus mostly serves to control the relative fractions of the waveforms rather than define the quantitative waveform characteristics.
Probabilistic modeling can predict cell phenotypes in a spatial gradient of VEGF
To investigate whether the simplified Bayesian decision model could predict cell phenotype selection under more complex and dynamic VEGF signaling regimes, we analyzed cells migrating in vitro in a gradient of VEGF concentrations. Cells migrating in a gradient are exposed to changing VEGF concentrations and could respond differently than those exposed to a static concentration of VEGF. Thus, within this experimental setup, a single cell over the course of the experiment can experience different amounts of VEGF signaling, which might be reflected in the dynamically changing selection of Ca2+ signaling waveforms. We seeded PAECs in a microfluidic device engineered to create a stable linear gradient of VEGF concentrations from 1 to 50 ng/ml over a distance of 800 μm (fig. S4A). We monitored individual cell migration over a period of 4 to 5 hours and found that the net migration of most of the cells was toward increasing concentrations of VEGF (fig. S4B), consistent with previous reports (34). Moreover, cells that migrated greater overall distances were more likely to show net migration toward increasing concentrations of VEGF than less migratory cells (fig. S4B, correlation coefficient of 0.55). Therefore, we hypothesized that a cell’s ability to migrate toward a gradient of VEGF in this experimental setup is modulated by the enhanced motility phenotype. In addition, we hypothesized that over the duration of several hours, cells could switch their Ca2+ waveform in response to changes in the magnitude of VEGF signaling.
To address these hypotheses and predict cell phenotypes in a gradient of VEGF, we implemented the simplified Bayesian decision model (text S1), allowing cells to periodically reevaluate responses to VEGF (fig. S5A) and display higher directional bias when selecting the enhanced motility phenotype. This migration model predicted stochastic migration trajectories (Fig. 5A) that were evaluated as a function of td or local VEGF concentration. The model predicted that the migration velocity (vmig), defined as positive in the direction of increasing VEGF, correlated with td (Fig. 5B) and, to a lesser degree, with the local VEGF concentration (fig. S5B). Cells that started at higher local VEGF concentrations had a greater probability of sustained Ca2+ signaling (Fig. 5B, top), consistent with increased vmig. The predictions of the migration model were consistent with experimental observations in that the measured vmig correlated with td (Fig. 5C) but not with local VEGF concentration. The migration model also predicted that, over the duration of 4 hours, the sensitivity of results to the frequency of reevaluation of the local VEGF input was relatively low in terms of the dependence of vmig on td (fig. S5C). However, the range of the migration data was most consistent with a reevaluation period of about 15 min (fig. S5C, insert). In the model implementation, cells selecting enhanced migration, those with moderate and high values of td, were considered to have enhanced directional guidance. This consideration was essential because simulations conducted without the assumption of directional sensing, only enhanced motility, did not predict increased directed migration with larger values of td (fig. S5D). Application of the migration model also predicted that cells at lower VEGF inputs tend to have a higher probability of responding with repeated spikes (Fig. 5D). Cells that remained at lower VEGF concentrations were able to continue uninterrupted spiking for longer durations and thus exhibited more extensive NFAT signaling (Fig. 5D).
Fig. 5. A combination of stochastic and deterministic decoding governs phenotype selection in a gradient of VEGF.
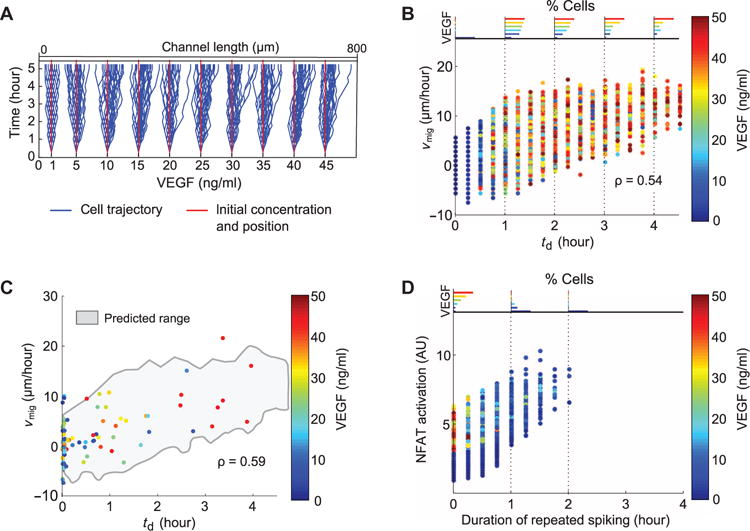
(A) Migration trajectories from cells simulated in a 0.0625 ng ml−1 μm−1 gradient of VEGF. Cells were assumed to reevaluate decisions every 15 min for these simulation results and those described below. (B) Predicted migration velocity vmig shown as a function of td for cells simulated in a gradient of VEGF (correlation coefficient of 0.54, P < 0.01). Each point shows data from an individual cell and is colored by starting VEGF concentration. Histograms showing the distribution of VEGF concentrations at discrete values of td are shown at the top of the plot and are also colored by VEGF concentration. (C) Experimentally measured vmig as a function of td for cells placed in a microfluidic gradient of VEGF (0.0625 ng ml−1 μm−1) (correlation coefficient of 0.59, P < 0.01) (n = 91 cells from n = 3 independent experiments). Range of simulation results is superimposed (gray envelope) for comparison. (D) Predicted NFAT activation for cells as a function of the duration of RS waveforms in a gradient of VEGF.
Random Ca2+ spikes impede tip cell migration in vivo
We found that increased cell motility requires sustained and uninterrupted Ca2+ signaling (Fig. 3, A and B). Moreover, the migration velocity of endothelial cells moving in a gradient of VEGF was dependent on the length of time Ca2+ signaling was sustained (Fig. 5, B and C). Interruption of this signaling by Ca2+ spikes resulted in reduced migration velocities. To confirm whether this finding also applies to endothelial cells in vivo, we examined angiogenesis in developing zebrafish (Danio rerio) embryos. These animals expressed a bicistronic transgenic reporter containing mCherry and the Ca2+-sensitive protein GCaMP6m under an endothelial cell-specific promoter. Tip cell migration was imaged as new blood vessels sprouted from the dorsal aorta, migrated dorsally, and eventually formed the intersegmental vessels. After this migration, the tip cells eventually branched and fused to form the dorsal longitudinal anastomotic vessel (Fig. 6A and movie S6).
Fig. 6. Repeated Ca2+ spikes reduce tip cell migration during angiogenesis in developing zebrafish.
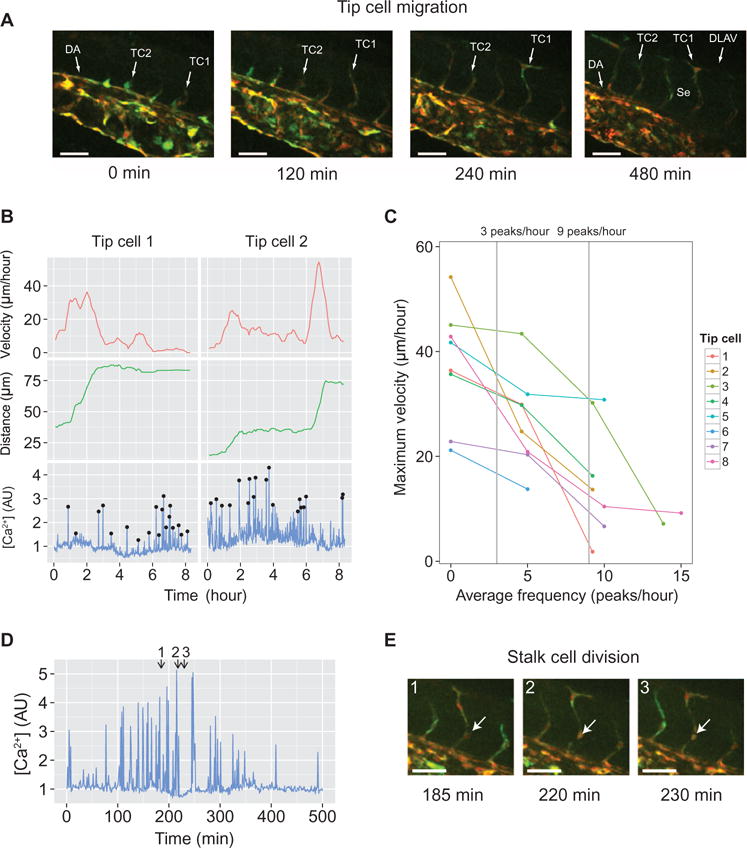
(A) Time-lapse images showing angiogenic sprout formation originating from the dorsal aorta (DA). GCaMP6m was used to monitor cytoplasmic Ca2+ regulation and is shown in green. mCherry was used to determine the location of vascular cells and is shown in red. Sprouts eventually split and fuse to form the dorsal longitudinal anastomotic vessel (DLAV) and remain as intersegmental vessels (Se). Examples of migrating tip cells are labeled TC1 and TC2. Scale bars, 50 μm. (B) Detailed Ca2+ signaling and migration profiles for tip cells TC1 and TC2 are also indicated in (A). Black circles indicate Ca2+ peaks that were distinguished from noise. The tip cell migration distance (middle panel) is measured from the tip of the sprouting vessel to the branch point along the DA. The migration velocity (top panel) is calculated from the tip cell migration distance over time (determined as a rolling average, see Materials and Methods). (C) Maximum tip cell migration velocity at each observed Ca2+ spike frequency (n = 8 angiogenic sprouts from n = 2 zebrafish embryos). (D and E) Ca2+ signaling during cell division. Individual frames from the image tile in (E) are indicated at the top of the plot. (E) Image tile showing stalk cell division (white arrow) during sprout formation. Scale bars, 50 μm.
The velocity of an individual tip cell was highly variable (Fig. 6B, top), with each cell exhibiting random Ca2+ spiking during angiogenesis (Fig. 6B, bottom). Generally, time intervals during which Ca2+ spiking became less frequent or stopped completely led to tip cells acquiring higher migration velocities (Fig. 6B, top) and, accordingly, traveling greater distances toward their target location (Fig. 6B, middle). Analysis of eight growing vessels revealed that the maximum achievable migration velocity was inversely related to the spike frequencies (Fig. 6C). These results were consistent with those obtained from single human cells, including the maximal velocity achieved (20 to 50 μm/hour) and the range of spike frequencies over which the maximal velocity dropped and tip extension arrested (between three and nine peaks/hour). This same frequency range defined the switch to enhanced NFAT signaling in single cells (Fig. 2F and fig. S3D; also note that three peaks/hour corresponded to td = 20 min, at which the transition between low to enhanced migration speed occurred). Cells closely trailing the tip cells (namely, stalk cells) also exhibited random Ca2+ spiking (Fig. 6D) with higher frequencies, and cell divisions were observed during periods of continuous spiking, again consistent with the expectations based on the single cell behavior (Fig. 6E). Overall, these results were consistent with the decoding mechanism suggested by the analysis of single endothelial cells, suggesting the proposed signal decoding could be relevant for angiogenic vasculature development.
DISCUSSION
Cellular decision-making is based on complex signal processing, which can display a high degree of cell-to-cell variability. This variability can enhance the repertoire of cell decision possibilities but also can limit the precision of cell responses to extracellular cues. Thus, signaling and cell processes display complex mixtures of mechanisms for both preventing and enhancing stochastic responses. Our results suggest that VEGF signaling through Ca2+-dependent pathways is an example of a mixed signal decoding strategy combining both deterministic and stochastic signal processing. The magnitude of VEGF signaling stochastically determines the distributions of three distinct Ca2+ waveforms, two of which are interpreted by at least two distinct signaling processes. As a result, these two waveforms deterministically specify a distinct cellular phenotype, either NFAT activation coupled to cell proliferation or MLCK activation coupled to enhanced cell migration. However, the absolute degree of NFAT activation or cell migration above a defined threshold is stochastic and largely independent of the magnitude of VEGF signaling. Robust phenotype selection is achieved because the repeated spikes and low persistent Ca2+ waveforms each allow the selective activation of only one corresponding signaling pathway. This selective activation arises because of different dynamic characteristics of each pathway’s activation, which can be seen as shorter or longer memory of the Ca2+ input dynamics. Furthermore, a diverse set of these repeated spikes or low persistent Ca2+ waveforms can exceed the corresponding thresholds for phenotype activation without restricting the magnitude of phenotype response to a single value.
This decoding process, which we explain by drawing an analogy with a mechanical ratchet, accounts for signal “multiplexing” in a manner analogous to a simple Fourier transform (35). This signaling architecture is composed of two pathways with different thresholds of activation and dynamic characteristics that are activated by the same dynamically changing molecular input. It allows for one of the pathways, and the corresponding functional response, to be sensitive to the input frequency, whereas the other pathway and the alternative functional response are only sensitive to the current input amplitude. In addition, the distinct memory properties of the signaling pathways used for phenotype selection can convey further functional benefits. For instance, integration of multiple Ca2+ spikes over time, required for a higher than threshold NFAT activation, is an effective way to ensure robustness to interspike fluctuations. In contrast, the persistent and instantaneous evaluation of the environment—that is, decoding the Ca2+ signal as an imperfect ratchet through MLCK signaling—allows cells to rapidly assess and react to environment changes by altering the migration response. It can also provide cells with added sensitivity to complex in vivo VEGF signals. Ca2+ can therefore serve as an effective signaling “hub” with the potential to integrate several different input signals and elicit distinct downstream responses, but without prohibiting downstream cell responses from using the decoding schemes that best suit their functionality.
These considerations are particularly relevant for endothelial cell responses in various complex physiological settings, including angiogenesis. Indeed, multiple soluble ligands, elements of extracellular matrix, and neighboring cells connected to a particular cell through gap junctions can all contribute to the definition of the Ca2+ signaling profile. Thus, here, Ca2+ can be a powerful integrator of diverse inputs. The sequential, hybrid stochastic-deterministic signaling scheme described here allows for considerable flexibility in the context of multiple inputs that can impinge on Ca2+ concentration regulation. If any of the particular cues, such as VEGF, was fully deterministic in the control of the cell responses, extending its effects beyond the control of Ca2+ dynamics, it could limit the capacity of Ca2+ as a signal integrator. This would restrict the possible conditioning of dynamic Ca2+ signaling characteristics, such as the effective spike frequency or signal duration, that potentially arise in response to a rich palette of environmental inputs. For instance, in microvessels stimulated with ATP (adenosine 5′-triphosphate), short-term Ca2+ regulation in the neighboring cells of intact blood vessels was heterogeneous in timing, duration, and amplitude (36). This heterogeneity in Ca2+ regulation correlated with differing amounts of cytoskeleton restructuring and cell contraction, resulting in differences in local vascular permeability (37). We thus suggest that Ca2+ responsiveness is a dynamic and stochastic property of endothelial cells, which can be filtered through sophisticated decoding schemes, including those unraveled by our analysis, contributing to complex regulation of dynamic reorganization of the endothelial tissue by VEGF and other cues.
Our analysis further suggests that the stochastic properties of cellular responses we observed under uniform and static VEGF simulation can be predicted with high fidelity under conditions with more complex and dynamic VEGF signaling, such as the migration of cells in our microfluidic experiments and in vivo angiogenesis during zebrafish development. Although the presence and functional significance of VEGF gradients in vivo are still subject to debate, VEGF gradient sensing by endothelial cells has been observed in vitro (34, 38). Our results provide interesting insights into this process. In particular, they suggest that during VEGF gradient sensing, the duration of the sustained increase in Ca2+ signaling can be used as the predictor not only of enhanced cell motility but also of higher persistence of cell migration in the direction of the gradient. This observation suggests that coupling of gradient sensing and the motility apparatus allows more effective tracking of the VEGF source. Gradual cell migration up the gradient of VEGF can further bias the cells toward enhanced cell migration, whereas cells further away from the source are being more strongly biased to undergo NFAT-dependent gene transcription and cell proliferation.
Although there is little evidence that VEGF is present in a gradient in vivo, the underlying Ca2+ signal decoding scheme we describe in regard to VEGF signaling is consistent with our observations of Ca2+ signaling in the developing vasculature in the tail portions of zebrafish embryos. Indeed, we observed cells with switching patterns of Ca2+ fluctuations, with higher frequencies associated with stalling of the tip cells and division of stalk cells, and the absence of spiking during rapid tip cell migration. However, because angiogenesis takes place in a complex physiological environment, we cannot ascribe these patterns unambiguously to VEGF alone as we could in the case of the in vitro experiments. Still, our single cell analysis suggests that VEGF stimulation, even on its own, might translate into diverse phenotypic outputs, such that the resulting cell population behavior can be quite diverse. This result confirms that VEGF can be a potent regulator of the resulting complex patterns of novel tissue formation and de novo forming vascular beds complete with anastomosis and capillary loops (39).
In summary, we suggest that cellular decisions are made by decoding complex temporal stochastic signaling profiles. This signal decoding allows deterministic qualitative decisions that are robust to signal variability, such as phenotype determination, and stochastic quantitative refinement of these decisions. We hypothesize that this form of decision-making can both ensure that signaling output is not completely dominated by multistep noisy signal-transduction processes (1) and convey the advantages of response diversification and integration.
MATERIALS AND METHODS
Cell culture
Primary HUVECs were acquired from Lonza Inc. and cultured in EGM2 media. PAECs exogenously expressing VEGFR2 were gifts from S. Soker (Wake Forest School of Medicine, North Carolina) and were cultured in Ham’s F-12 media supplemented with 5% calf bovine serum, gentamycin, and G418 (400 μg/ml). Note that VEGFRs are not present in PAECs, so only the VEGFR2 was present on the cells used in this study. Cells were cultured at 37°C with 5% CO2 and serum-starved in EBM media with 2 mM Ca2+ for 18 to 24 hours before conducting experiments.
In vitro live cell imaging
Imaging data were collected using an Axiovert 200M epifluorescence microscope (Zeiss) equipped with a motorized stage, filter wheels, and shutter system. Experiments were conducted at 37°C under 5% CO2 using a Zeiss microscope incubation system. All measurements were taken under ×40 magnification at 30-s intervals. Fluorescent light was generated using an X-Cite Exacte (Lumen Dynamics). Images were acquired with a Photometrics Cascade 512B CCD (charge-coupled device) camera. Microscope automation and image acquisition were controlled using SlideBook software (Intelligent Imaging Innovations). CFP (cyan fluorescent protein)/YFP (yellow fluorescent protein) FRET measurements were obtained using Chroma excitation/emission filter sets to collect CFP donor (430 nm/470 nm) and YFP acceptor (430 nm/535 nm) intensities. Images were also collected for YFP alone (500 nm/535 nm) to assay any photobleaching. Indo-1 measurements were conducted to assay the amount of Ca2+-bound (365 nm/405 nm) and Ca2+-unbound (380 nm/485 nm) dye. Measurements made with mCherry fusion proteins (NFAT and actin probes) were imaged using Texas Red filters (572 nm/628 nm). Semiautomatic cell segmentation, and background and photobleaching correction were made after acquisition using a custom MATLAB (MathWorks) program.
In vitro Ca2+ measurement
The Ca2+-sensitive dye Indo-1 was used to measure Ca2+ regulation in HUVECs. Cells were incubated with 4 μM Indo-1 in imaging solution (Hanks’ balanced salt solution with 2 mM Ca2+) for 1 hour and then washed three times. Imaging was conducted 40 min after incubation. Changes in Ca2+ concentration were assayed by examining the intensity ratio of bound (405-nm emission) and unbound (485-nm emission) Indo-1 dye. Ca2+ measurements in PAECs were carried out using the FRET reporter YC3.6, provided by R. Tsien (University of California, San Diego, California). PAECs were transfected using FuGENE 6 according to the manufacturer’s protocol, and stable transfectants were obtained through clonal selection. FRET was assayed as the intensity ratio between the YFP acceptor and the CFP donor.
NFAT measurement
To assay NFAT translocation, we created an NFAT-mCherry probe by cloning NFATC1 into pcDNA3.1/Hygro(−) through restriction digest (Nhe I/Bam HI). mCherry was placed downstream of the construct (Bam HI/Hind III). Cells stably expressing YC3.6 were transfected with NFAT-mCherry for 24 hours using FuGENE HD, serum-starved for 12 hours, and then imaged using FRET and Texas Red filters as described above. NFAT activation was reported as the ratio of nuclear intensity to cytoplasmic intensity.
Simultaneous Ca2+ and MLCK measurement
We assayed MLCK activation using a CFP/YFP-based FRET reporter, a gift of J. Stull (University of Texas Southwestern Medical Center, Texas). The FRET activity was calculated by taking the ratio of the CFP donor intensity to the YFP acceptor intensity (40). Cells were transfected using FuGENE 6 as described above. Simultaneous measurement of Ca2+ in PAECs was accomplished using Indo-1 as previously described (41).
Cell motility assay
We tracked PAE-YC cells for 1 hour while simultaneously recording Ca2+ regulation as described above. Individual cell shapes were segmented from each image semiautomatically using MATLAB. Cell positions were determined at 2.5-min intervals by calculating the center of mass of segmented cells. The difference in position measured between each time interval was tallied, and the total distance traveled was used as a measure of cell motility (fig. S2A).
Lamellipodia measurement
PAE-YC cells were tracked for 1 hour, and the movement of the cell periphery was analyzed using the Quimp11a plugin (42) for ImageJ (43). Quimp11a was used to generate motility maps, which allowed easy determination of lamellipodia characteristics like width and duration. These characteristics were then confirmed by manually inspecting the video recording of cell movement.
Cell proliferation assay
HUVECs were cultured in endothelial cell growth medium (Lonza, CC-3162) and seeded onto glass-bottom dishes previously coated with fibronectin (10 μg/ml) for 2 hours at room temperature. HUVECs were serum-starved overnight in endothelial cell growth medium containing only 1% fetal bovine serum and then exposed to VEGF (Gibco, PHC9394) and VIVIT (Tocris Bioscience, 3930) for 24 hours. Cells were incubated with BrdU reagent (Life Technologies, 00-0103) for 16 hours before fixation. BrdU-stained nuclei were then counted for quantification.
Microfluidic chemotaxis assay
We designed a microfluidic device, similar to that shown in (44), capable of generating a stable linear gradient without subjecting cells to fluid shear forces (fig. S4A). The device consists of an array of shallow test chambers (9 μm in height) that are connected on each side to a perpendicular flow chamber (350 μm in height). The device was coated with gelatin for 16 hours at 4°C before experimentation. After perfusing the microfluidic chip with full media, PAE-YC cells were seeded into the gradient test chambers, allowed to attach, and placed in a 37°C incubator for 12 hours. The media were then exchanged with serum-free EBM with 0.1% bovine serum albumin, and cells were serum-starved overnight. During experimentation, a linear VEGF gradient was created along each test chamber by molecular diffusion between a flow channel with VEGF (50 ng/ml) and an adjacent flow channel which held only serum-free media. Ca2+ regulation was assayed using CFP/YFP FRET as described above. Cell movement was determined as described for the cell motility assay. Migration distance, vmig, was normalized by the time each cell was observed (minimum of 4.5 to 5 hours).
Device microfabrication
The device layout was designed in AutoCAD, exported in EPS (Encapsulated PostScript) format, and printed at high resolution on a Mylar transparency (In Tandem Design). Fabrication was accomplished using standard soft lithography (45). The master mold used to create the fluidic channels was fabricated using SU-8 photoresist (Microchem) according to the manufacturer’s protocol. Here, we used two sequential applications of SU-8 to achieve dual-height channels (46). The first SU-8 layer, which was used to form the inlet, outlet, and observation channels, was 9 μm in height, and a taller layer of 380 μm was used to form the side flow channels (fig. S4A). Sylgard PDMS (polydimethylsiloxane) (Dow Corning) at 10:1 (monomer/curing agent) was applied to the mold and cured at 85°C for 1 hour. After peeling off the PDMS and partitioning it into separate chips, a syringe tip was used to bore the inlet, outlet, and seeding holes. Microfluidic chips were cleaned with ethanol and then reversibly bonded to glass coverslips by baking at 85°C overnight.
In vivo zebrafish angiogenesis assay
The zebrafish line Tg (Tol2 fliEbasP::mCherry V2A GCaMP6m)ric100 was created to examine Ca2+ regulation during angiogenesis. A plasmid construct containing Tol2 transposon arms and the endothelial-specific Ca2+ sensor was created. The fliEbasP endothelial cell-specific promoter (gift from N. Lawson, University of Massachusetts Medical School) was used to drive expression of a bicistronic cassette consisting of an mCherry open reading frame followed by the GCaMP6m (47) open reading frame. The protein coding sequences were separated by a viral peptide 2A sequence, allowing production of separate proteins (48). Expression of the mCherry from the same transcript allowed for standardization of the GCaMP6m signal. The plasmid was injected with mRNA encoding the Tol2 transposase (49) into one-cell zebrafish embryos, and a line expressing the mCherry and GCaMP6m specifically in vascular endothelial cells was identified (RIC-100). Embryos expressing the reporter at 24 hours post-fertilization (hpf) were mounted in 1.2% low-melt agarose with 0.04% tricaine to prevent movement and 0.003% 1-phenyl 2-thiourea (PTU) to suppress pigment formation.
An Andor spinning-disk confocal microscope with Olympus IX83 base, Yokogawa CSU-W1 scan head, and Andor iXon 888 electron multiplying (EM) CCD camera was used to obtain images in the trunk and tail beginning at about 28 hpf. Angiogenic sprouts were imaged for 8 hours at a frequency of one image per min at 27°C. Tip cell positions were determined at 5-min intervals using a custom MATLAB program (MathWorks). Because the zebrafish embryos were alive, growing, and expanding during the experiment, the displacement of the tip cell was determined relative to the displacement of the sprout branching point. This displacement was used to calculate the tip cell velocity, which was then filtered using a moving average (eight points) to reduce noise. We used the distance of the tip cell from the branch point as the migration distance. Because the Ca2+ spikes did not occur at a fixed frequency during the experiment, a moving average (13 points) was used to approximate the frequency at any given time.
Probabilistic model
Details of the model formulation, training, and generation of simulated data are given in the Supplementary Materials. All model calculations were performed using MATLAB (MathWorks).
Statistical analyses
Statistical analysis was performed using the MATLAB Statistics Toolbox (MathWorks).
Supplementary Material
Movie S1. Time-lapse video showing NR, RS, and LP waveforms in PAECs.
Movie S2. Time-lapse video showing NR, RS, and LP waveforms in HUVECs.
Movie S3. Time-lapse video showing simultaneous Ca2+ and NFAT signaling.
Movie S4. Cells exhibiting LP waveforms formed large lamellipodia with pronounced actin translocation.
Movie S5. Cells exhibiting RS waveforms displayed only limited actin recruitment and small filopodia.
Movie S6. Angiogenic sprout formation in zebrafish.
Text S1. Detailed description of the probabilistic model.
Fig. S1. Distribution of averaged Ca2+ concentrations.
Fig. S2. Additional analysis of cell motility.
Fig. S3. Probabilistic model simulation of responses to spatially uniform VEGF.
Fig. S4. Analysis of cell migration in a microfluidic gradient.
Fig. S5. Probabilistic model simulation of cells in a gradient of VEGF.
Acknowledgments
We would like to thank R. Cheong and K. Hess for reviewing drafts of the manuscript. We thank C. Rivera and J. Zhang for insightful discussions. We are grateful to S. Soker for providing PAE/VEGFR2 cells, R. Tsien for the YC3.6 reporter, J. Stull for the MLCK FRET reporter, and N. Lawson for the fliEbasP promoter.
Funding: This work was supported by NIH R01 HL101200, R01 CA138264 (D.P.N. and A.S.P.), GM072024, GM084332, CA65145, RR140073 (A.W.), and RR020839 (D.P.N. and A.L.).
Footnotes
Author contributions: Study concept and design: D.P.N., A.S.P., and A.L.; acquisition and analysis of data: D.P.N., W.H.C., S.H.L., A.A.Q., A.W., and D.S.W.; probabilistic model: D.P.N.; drafting of the manuscript: D.P.N., A.S.P., and A.L.; obtainedfunding: A.L., A.S.P., A.A.Q., and D.S.W.
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Cheong R, Rhee A, Wang CJ, Nemenman I, Levchenko A. Information transduction capacity of noisy biochemical signaling networks. Science. 2011;334:354–358. doi: 10.1126/science.1204553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459:428–432. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samadani A, Mettetal J, van Oudenaarden A. Cellular asymmetry and individuality in directional sensing. Proc Natl Acad Sci USA. 2006;103:11549–11554. doi: 10.1073/pnas.0601909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rimchala T, Kamm RD, Lauffenburger DA. Endothelial cell phenotypic behaviors cluster into dynamic state transition programs modulated by angiogenic and angiostatic cytokines. Integr Biol. 2013;5:510–522. doi: 10.1039/c2ib20090b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamalice L, Houle F, Huot J. Phosphorylation of Tyr1214 within VEGFR-2 triggers the recruitment of Nck and activation of Fyn leading to SAPK2/p38 activation and endothelial cell migration in response to VEGF. J Biol Chem. 2006;281:34009–34020. doi: 10.1074/jbc.M603928200. [DOI] [PubMed] [Google Scholar]
- 7.Rousseau S, Houle F, Kotanides H, Witte L, Waltenberger J, Landry J, Huot J. Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J Biol Chem. 2000;275:10661–10672. doi: 10.1074/jbc.275.14.10661. [DOI] [PubMed] [Google Scholar]
- 8.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 9.Kohn EC, Alessandro R, Spoonster J, Wersto RP, Liotta LA. Angiogenesis: Role of calcium-mediated signal transduction. Proc Natl Acad Sci USA. 1995;92:1307–1311. doi: 10.1073/pnas.92.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- 11.Antoniotti S, Pla AF, Pregnolato S, Mottola A, Lovisolo D, Munaron L. Control of endothelial cell proliferation by calcium influx and arachidonic acid metabolism: A pharmacological approach. J Cell Physiol. 2003;197:370–378. doi: 10.1002/jcp.10359. [DOI] [PubMed] [Google Scholar]
- 12.Faehling M, Kroll J, Föhr KJ, Fellbrich G, Mayr U, Trischler G, Waltenberger J. Essential role of calcium in vascular endothelial growth factor A-induced signaling: Mechanism of the antiangiogenic effect of carboxyamidotriazole. FASEB J. 2002;16:1805–1807. doi: 10.1096/fj.01-0938fje. [DOI] [PubMed] [Google Scholar]
- 13.Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–1061. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- 14.Marhl M, Gosak M, Perc M, Roux E. Importance of cell variability for calcium signaling in rat airway myocytes. Biophys Chem. 2010;148:42–50. doi: 10.1016/j.bpc.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura N, Yamazawa T, Okubo Y, lino M. Temporal switching and cell-to-cell variability in Ca2+ release activity in mammalian cells. Mol Syst Biol. 2009;5:247. doi: 10.1038/msb.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maurya MR, Subramaniam S. A kinetic model for calcium dynamics in RAW 264.7 cells: 1. Mechanisms, parameters, and subpopulational variability. Biophys J. 2007;93:709–728. doi: 10.1529/biophysj.106.097469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dragoni S, Laforenza U, Bonetti E, Lodola F, Bottino C, Berra-Romani R, Cario Bongio G, Cinelli MP, Guerra G, Pedrazzoli P, Rosti V, Tanzi F, Moccia F. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells. 2011;29:1898–1907. doi: 10.1002/stem.734. [DOI] [PubMed] [Google Scholar]
- 18.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 19.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 20.Purvis JE, Lahav G. Encoding and decoding cellular information through signaling dynamics. Cell. 2013;152:945–956. doi: 10.1016/j.cell.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernández GL, Volpert OV, Íñiguez MA, Lorenzo E, Martínez-Martínez S, Grau R, Fresno M, Redondo JM. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin a: Roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med. 2001;193:607–620. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dixelius J, Olsson A-K, Thulin Å, Lee C, Johansson I, Claesson-Welsh L. Minimal active domain and mechanism of action of the angiogenesis inhibitor histidine-rich glycoprotein. Cancer Res. 2006;66:2089–2097. doi: 10.1158/0008-5472.CAN-05-2217. [DOI] [PubMed] [Google Scholar]
- 23.Rafiee P, Heidemann J, Ogawa H, Johnson NA, Fisher PJ, Li MS, Otterson MF, Johnson CP, Binion DG. Cyclosporin A differentially inhibits multiple steps in VEGF induced angiogenesis in human microvascular endothelial cells through altered intracellular signaling. Cell Commun Signal. 2004;2:3. doi: 10.1186/1478-811X-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ewan LC, Jopling HM, Jia H, Mittar S, Bagherzadeh A, Howell GJ, Walker JH, Zachary IC, Ponnambalam S. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic. 2006;7:1270–1282. doi: 10.1111/j.1600-0854.2006.00462.x. [DOI] [PubMed] [Google Scholar]
- 25.Robbs BK, Cruz ALS, Werneck MBF, Mognol GP, Viola JPB. Dual roles for NFAT transcription factor genes as oncogenes and tumor suppressors. Mol Cell Biol. 2008;28:7168–7181. doi: 10.1128/MCB.00256-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buchholz M, Schatz A, Wagner M, Michl P, Linhart T, Adler G, Gress TM, Ellenrieder V. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBOJ. 2006;25:3714–3724. doi: 10.1038/sj.emboj.7601246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heit JJ, Apelqvist ÅA, Gu X, Winslow MM, Neilson JR, Crabtree GR, Kim SK. Calcineurin/NFAT signalling regulates pancreatic β-cell growth and function. Nature. 2006;443:345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 28.Köenig A, Linhart T, Schlengemann K, Reutlinger K, Wegele J, Adler G, Singh G, Hofmann L, Kunsch S, Büch T, Schäfer E, Gress TM, Femandez-Zapico ME, Ellenrieder V. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology. 2010;138:1189–1199.e2. doi: 10.1053/j.gastro.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh G, Singh SK, König A, Reutlinger K, Nye MD, Adhikary T, Eilers M, Gress TM, Fernandez-Zapico ME, Ellenrieder V. Sequential activation of NFAT and c-Myc transcription factors mediates the TGF-β switch from a suppressor to a promoter of cancer cell proliferation. J Biol Chem. 2010;285:27241–27250. doi: 10.1074/jbc.M110.100438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neal JW, Clipstone NA. A constitutively active NFATc1 mutant induces a transformed phenotype in 3T3-L1 fibroblasts. J Biol Chem. 2003;278:17246–17254. doi: 10.1074/jbc.M300528200. [DOI] [PubMed] [Google Scholar]
- 31.Yissachar N, Sharar Fischler T, Cohen AA, Reich-Zeliger S, Russ D, Shifrut E, Porat Z, Friedman N. Dynamic response diversity of NFAT isoforms in individual living cells. Mol Cell. 2013;49:322–330. doi: 10.1016/j.molcel.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: The calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 33.Chew TL, Wolf WA, Gallagher PJ, Matsumura F, Chisholm RL. A fluorescent resonant energy transfer-based biosensor reveals transient and regional myosin light chain kinase activation in lamella and cleavage furrows. J Cell Biol. 2002;156:543–553. doi: 10.1083/jcb.200110161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barkefors I, Le Jan S, Jakobsson L, Hejll E, Carlson G, Johansson H, Jarvius J, Park JW, Li Jeon N, Kreuger J. Endothelial cell migration in stable gradients of vascular endothelial growth factor A and fibroblast growth factor 2: Effects on chemotaxis and chemokinesis. J Biol Chem. 2008;283:13905–13912. doi: 10.1074/jbc.M704917200. [DOI] [PubMed] [Google Scholar]
- 35.Cheong R, Levchenko A. Oscillatory signaling processes: The how, the why and the where. Curr Opin Genet Dev. 2010;20:665–669. doi: 10.1016/j.gde.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKnight TR, Curry FE. Mechanisms of heterogeneous endothelial cytoplasmic calcium increases in venular microvessels. Microcirculation. 2002;9:537–550. doi: 10.1038/sj.mn.7800166. [DOI] [PubMed] [Google Scholar]
- 37.Pagakis SN, Curry FE. Imaging of Ca2+ transients in endothelial cells of single perfused capillaries: Correlation of peak [Ca2+]i with sites of macromolecule leakage. Microcirculation. 1994;1:213–230. doi: 10.3109/10739689409146749. [DOI] [PubMed] [Google Scholar]
- 38.Shamloo A, Ma N, Poo M-m, Sohn LL, Heilshorn SC. Endothelial cell polarization and chemotaxis in a microfluidic device. Lab Chip. 2008;8:1292–1299. doi: 10.1039/b719788h. [DOI] [PubMed] [Google Scholar]
- 39.Stokes CL, Lauffenburger DA, Williams SK. Migration of individual microvessel endothelial cells: Stochastic model and parameter measurement. J Cell Sci. 1991;99:419–430. doi: 10.1242/jcs.99.2.419. [DOI] [PubMed] [Google Scholar]
- 40.Isotani E, Zhi G, Lau KS, Huang J, Mizuno Y, Persechini A, Geguchadze R, Kamm KE, Stull JT. Real-time evaluation of myosin light chain kinase activation in smooth muscle tissues from a transgenic calmodulin-biosensor mouse. Proc Natl Acad Sci USA. 2004;101:6279–6284. doi: 10.1073/pnas.0308742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tay LH, Griesbeck O, Yue DT. Live-cell transforms between Ca2+ transients and FRET responses for a troponin-C-based Ca2+ sensor. Biophys J. 2007;93:4031–4040. doi: 10.1529/biophysj.107.109629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tyson RA, Epstein DBA, Anderson KI, Bretschneider T. High resolution tracking of cell membrane dynamics in moving cells: An electrifying approach. Math Model Nat Phenom. 2010;5:34–55. [Google Scholar]
- 43.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paliwal S, Iglesias PA, Campbell K, Hilioti Z, Groisman A, Levchenko A. MAPK-mediated bimodal gene expression and adaptive gradient sensing in yeast. Nature. 2007;446:46–51. doi: 10.1038/nature05561. [DOI] [PubMed] [Google Scholar]
- 45.Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 46.Mata A, Fleischman AJ, Roy S. Fabrication of multi-layer SU-8 microstructures. J Micromech Microeng. 2006;16:276–284. [Google Scholar]
- 47.Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Provost E, Rhee J, Leach SD. Viral 2A peptides allow expression of multiple proteins from a single ORF in transgenic zebrafish embryos. Genesis. 2007;45:625–629. doi: 10.1002/dvg.20338. [DOI] [PubMed] [Google Scholar]
- 49.Kawakami K, Shima A, Kawakami N. Identification of a functional transposase of the Tol2 element, an Ac-like element from the Japanese medaka fish, and its transposition in the zebrafish germ lineage. Proc Natl Acad Sci USA. 2000;97:11403–11408. doi: 10.1073/pnas.97.21.11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Time-lapse video showing NR, RS, and LP waveforms in PAECs.
Movie S2. Time-lapse video showing NR, RS, and LP waveforms in HUVECs.
Movie S3. Time-lapse video showing simultaneous Ca2+ and NFAT signaling.
Movie S4. Cells exhibiting LP waveforms formed large lamellipodia with pronounced actin translocation.
Movie S5. Cells exhibiting RS waveforms displayed only limited actin recruitment and small filopodia.
Movie S6. Angiogenic sprout formation in zebrafish.
Text S1. Detailed description of the probabilistic model.
Fig. S1. Distribution of averaged Ca2+ concentrations.
Fig. S2. Additional analysis of cell motility.
Fig. S3. Probabilistic model simulation of responses to spatially uniform VEGF.
Fig. S4. Analysis of cell migration in a microfluidic gradient.
Fig. S5. Probabilistic model simulation of cells in a gradient of VEGF.