

Abstract
Activation of O–H bonds by inorganic metal-oxo complexes has been documented, but no cognate enzymatic process is known. Our mechanistic analysis of 2-hydroxyethylphosphonate dioxygenase (HEPD), which cleaves the C1–C2 bond of its substrate to afford hydroxymethylphosphonate on the biosynthetic pathway to the commercial herbicide phosphinothricin, uncovered an example of such an O–H-bond-cleavage event. Stopped-flow UV–visible absorption and freeze-quench Mössbauer experiments identified a transient iron(IV)-oxo (ferryl) complex. Maximal accumulation of the intermediate required both the presence of deuterium in the substrate and, importantly, the use of 2H2O as solvent. The ferryl complex forms and decays rapidly enough to be on the catalytic pathway. To account for these unanticipated results, a new mechanism that involves activation of an O–H bond by the ferryl complex is proposed. This mechanism accommodates all available data on the HEPD reaction.
Introduction
By mimicking phosphate esters, carboxylic acids, or tetrahedral intermediates in diverse biochemical pathways, phosphonates and phosphinates display a variety of biological activities (Figure 1a).1,2 The phosphinate phosphinothricin (PT) is the active ingredient in the commercial herbicides Liberty, Basta, and Ignite. Investigation of its biosynthetic pathway has revealed several biochemically unusual steps.3 One of them, catalyzed by the enzyme 2-hydroxyethylphosphonate dioxygenase (HEPD), cleaves the unactivated sp3–sp3 carbon–carbon bond of 2-hydroxyethylphosphonate (HEP) to afford hydroxymethylphosphonate (HMP) and formate (Figure 1b).4 HEPD employs iron as its sole cofactor to activate O2. An X-ray crystal structure4 showed that HEPD has the 2-histidine-1-carboxylate “facial triad” ligand set characteristic of a large family of O2-activating mononuclear non-heme-iron enzymes.5 Unlike many members of this family, HEPD requires no reducing co-substrate: all four electrons needed to reduce O2 are extracted from HEP.4 The reaction incorporates one atom of O2 into the formate product without discernible exchange with solvent and a second oxygen atom into HMP with partial (∼40%) solvent exchange.
Figure 1.

(a) Structures of three representative phosphonate natural products. (b) HEPD utilizes Fe(II) and molecular oxygen to convert HEP to HMP and formate during phosphinothricin biosynthesis. (c) A previously postulated chemical mechanism for HEPD.
Current insight into the mechanism of HEPD has arisen through indirect means such as isotope tracking, product analysis with substrate analogues, determination of kinetic isotope effects, and computational modeling.4,6−12 On the basis of these studies, a mechanism featuring abstraction of the pro-S hydrogen from C2 of HEP by a ferric-superoxo species I has been proposed (Figure 1c).6,7,12 Electron transfer from the resulting ketyl radical, II, would reduce the metal center to a ferrous-hydroperoxo intermediate, III.8 Attack on the aldehyde would yield bridged peroxo species, IV, which could undergo homolytic oxygen–oxygen bond cleavage followed by beta scission to afford phosphonomethyl radical, V. Attack of V upon the hydroxo ligand of the Fe(III)–OH cofactor form would yield the HMP product, with solvent exchange of this oxygen ligand leading to the previously observed fractional incorporation of 18O from 18O2 into HMP.8V would likely have a low energy barrier for rotation around the P–C bond,9 thus explaining the racemic HMP product obtained upon turnover of either (R)- or (S)-2-[1-2H1]-HEP.6 Further support for radical V was recently obtained by diversion of the reaction to a different outcome by mutagenesis.13
Although the current mechanistic hypothesis is consistent with all available experimental data, no direct evidence has been reported for any of the postulated intermediates. Ferric-superoxo species analogous to I(14) have been characterized in three other non-heme-iron enzymes, myo-inositol oxygenase (MIOX),15 2,3-homoprotocatechuate dioxygenase (HPCD),16 and isopenicillin N-synthase (IPNS).17 In the case of HPCD, accumulation of the superoxo-Fe(III) species was achieved by employing a less reactive substrate analogue. For MIOX and IPNS, use of deuterium-labeled substrate allowed for both accumulation of the ferric-superoxo complexes for spectroscopic characterization and demonstration that they abstract hydrogen from the substrate. We reasoned on the basis of Figure 1c and the MIOX and IPNS precedent that employing HEP substrate containing deuterium at C2 (D2-HEP) might, by virtue of a similarly large deuterium kinetic isotope effect (2H-KIE) on decay of the intermediate, enable its accumulation. However, as described herein, this approach surprisingly resulted in the observation of an iron(IV)-oxo species, requiring revision of the previously proposed mechanism. The results of additional experiments with substrate isotopologues and in deuterium oxide reveal that the ferryl intermediate abstracts a solvent-exchangeable hydrogen from an O–H bond of an intermediate to initiate the C1–C2 bond cleavage.
Results
Steady-State Kinetic Parameters and Substrate 2H-KIEs for the Reactions of HEPD and HEPD-E176A
By use of a Clark-type electrode to monitor consumption of O2 during oxidation of either HEP or D2-HEP, we demonstrated that the HEPD reaction exhibits a minimal substrate 2H-KIE on kcat/Km,HEP and kcat (Table 1). This result implies that the isotope-sensitive C–H-cleavage step is not primarily rate-limiting for the overall catalytic cycle. However, the reaction does exhibit a significant 2H-KIE on kcat/Km for O2, which implies reversible formation of the C–H-cleaving intermediate. This deduction is consistent with the proposal that a superoxo-Fe(III) complex, rather than an intermediate with a more reduced and cleaved form of O2 (e.g., a ferryl complex), abstracts the hydrogen at C2.12
Table 1. Steady-State Kinetic Parameters for Wild-Type HEPD and HEPD-E176A.
| system | kcat (s–1) | Km,HEP (μM) | kcat/Km,HEP (M–1 s–1) | Km,O2 (μM) | kcat/Km,O2 (M–1 s–1) | KIE on kcat | KIE on kcat/Km,HEP | KIE on kcat/Km,O2 |
|---|---|---|---|---|---|---|---|---|
| wt HEPD, HEP | 0.30 ± 0.01 | 7.5 ± 1.5 | (4.1 ± 0.8) × 104 | 19 ± 3 | (1.6 ± 0.3) × 104 | 1.0 ± 0.1 | 1.3 ± 0.3 | 5.5 ± 1.3 |
| wt HEPD, D2-HEP | 0.31 ± 0.02 | 10 ± 2 | (3.0 ± 0.5) × 104 | 110 ± 20 | (2.9 ± 0.5) × 103 | |||
| HEPD-E176A, HEP | 0.75 ± 0.06 | 17 ± 2 | (4.5 ± 0.7) × 104 | 7.1 ± 1.4 | (9.9 ± 1.9) × 104 | 1.2 ± 0.1 | 1.5 ± 0.3 | 4.3 ± 1.0 |
| HEPD-E176A, D2-HEP | 0.65 ± 0.02 | 21 ± 3 | (3.1 ± 0.4) × 104 | 28 ± 3 | (2.3 ± 0.3) × 104 | |||
Because observation of a substrate 2H-KIE on kcat would bode well for accumulation of oxidized intermediates to levels sufficient for spectroscopic characterization,18 we sought a variant of HEPD exhibiting a larger such effect while retaining catalytic efficiency similar to that of the wild-type (wt) enzyme. Substitution of iron ligand E176 with alanine afforded such a variant. Like the wt enzyme, HEPD-E176A produces HMP and formate as its only detectable products upon incubation with Fe(II), HEP, and O2 (Figures S1–S3). Values of kcat/Km,HEP are nearly identical to those of the wt enzyme for both protium- and deuterium-containing substrates (Table 1). Surprisingly, the value of kcat/Km,O2, a measure of the efficiency of O2 activation, is 6–8-fold greater for HEPD-E176A than for wt HEPD with either HEP or D2-HEP as the substrate. As with the wt enzyme, the KIE on kcat/Km,HEP is relatively small, whereas the KIE on kcat/Km,O2 is much larger. In four independent sets of experiments, the HEPD-E176A reaction exhibited a small but reproducible 2H-KIE on kcat (Figure S3), suggesting that a C–H-cleaving intermediate might accumulate to a greater extent in the variant enzyme.
Detection of an Intermediate by Stopped-Flow Absorption (SF-abs) Spectroscopy
To test for accumulation of the postulated superoxo-Fe(III) intermediate (I) in the first turnover, an anoxic solution of the wt HEPD·Fe(II)·D2-HEP reactant complex was rapidly mixed with O2-saturated buffer in a stopped-flow absorption (SF-abs) experiment. No transient absorption in the 300–700 nm regime was detected. By contrast, traces from the identical experiment with the HEPD-E176A variant do exhibit transient UV absorption (Figure 2a) indicative of an accumulating intermediate. The transient absorption is not seen in the same time regime in the reaction of the variant HEPD with unlabeled (all-protium) substrate. The corresponding experiment with the (R) and (S) enantiomers of HEP having a single deuterium on C2 [hereafter (R)-D-HEP and (S)-D-HEP, respectively] showed that the (S) enantiomer, but not the (R) enantiomer, elicits transient absorption similar to that for the reaction with D2-HEP. This result suggests that the detected intermediate decays by transfer of the hydrogen that originates from the C2 pro-S position in HEP. SF-abs traces from the reaction initiated by rapid mixing of the (substrate-free) HEPD-E176A·Fe(II) complex with O2-saturated buffer containing D2-HEP also revealed accumulation of the intermediate. The somewhat delayed and lesser accumulation is consistent with a relatively fast but still kinetically significant HEP-binding step preceding formation of the intermediate (Figure 2).
Figure 2.
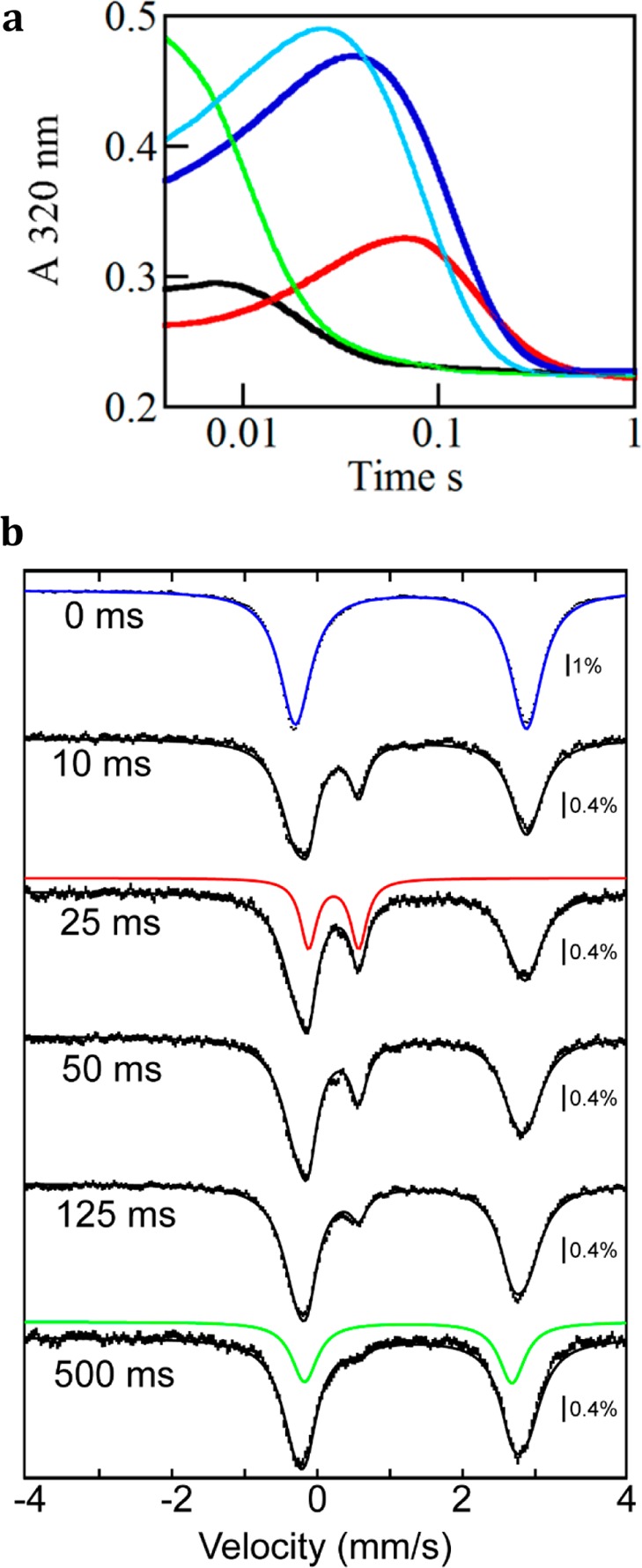
SF-abs and freeze-quench Mössbauer experiments monitoring the reaction of HEPD-E176A·Fe(II)·HEP with O2. (a) ΔA320 kinetic traces obtained after mixing of an anoxic solution of the HEPD-E176A·Fe(II)·HEP complex with an equal volume of O2-saturated buffer at 4 °C. Final concentrations after mixing: 0.50 mM HEPD-E176A, 0.40 mM Fe(II), 3.0 mM HEP isotopologue, 50 mM HEPES pH 7.5, and ∼0.85 mM O2. The traces shown are for unlabeled HEP (black), D2-HEP (dark blue), (R)-D-HEP (green), and (S)-D-HEP (light blue). The red line shows the trace obtained after mixing at 4 °C of an anoxic solution of the HEPD-E176A·Fe(II) complex with an equal volume of O2-saturated buffer containing HEP. The enantiomeric purity of the labeled substrates was 93:7, and thus some (S)-D-HEP is present in the experiment with (R)-D-HEP and vice versa. (b) Mössbauer spectra (4.2-K/53-mT) of samples prepared by mixing at 4 °C a solution of the HEPD-E176A·57Fe(II)·D2-HEP complex with two equivalent volumes of O2-saturated buffer and freeze-quenching after the indicated reaction times. Final concentrations: 1.1 mM HEPD-E176A, 1.1 mM 57Fe(II), 2.3 mM D2-HEP, 50 mM HEPES pH 7.5, and ∼1.3 mM O2. The blue, red, and green lines are simulations of the spectra of the HEPD-E176A·Fe(II)·D2-HEP reactant complex, the Fe(IV) intermediate, and the HEPD-E176A·Fe(II)·HMP product complex, respectively, with parameters provided in the main text. The summed contributions of the three components are shown as black lines overlaid with the experimental spectra.
Identification of the Intermediate as an Fe(IV) Complex by Freeze-Quench Mössbauer Spectroscopy
Freeze-quench (FQ) Mössbauer experiments were employed to determine the nature of the intermediate detected by SF-abs. The 4.2-K/53-mT Mössbauer spectra of samples freeze-quenched at varying reaction times (Figure 2b, vertical bars) can all be satisfactorily analyzed as superposition of three quadrupole-doublet components (Figure 2b; see also Figure S4). The first component has an isomer shift (δ) of 1.29 mm/s and a quadrupole splitting (ΔEQ) of 3.18 mm/s (blue line in Figure 2b), parameters characteristic of high-spin Fe(II) complexes with oxygen and nitrogen ligands. Comparison to the spectrum of an O2-free control sample shows that this first component arises from the reactant complex. The third component also has parameters (δ = 1.25 mm/s, ΔEQ = 2.86 mm/s, green line in Figure 2b) typical of high-spin Fe(II). This component is thus assigned to the Fe(II)-containing enzyme·product(s) complex. The second and most mechanistically informative component is transient and exhibits a prominent peak at ∼0.55 mm/s. This peak is the high-energy line of a quadrupole doublet with parameters (δ = 0.22 mm/s, ΔEQ = 0.69 mm/s) typical of Fe(IV) (Figure 2b, red line). Analysis of spectra recorded in variable (1, 4, and 8 T) magnetic fields shows that the species has electronic-structural parameters similar to those of high-spin Fe(IV)-oxo intermediates in other mononuclear non-heme-iron enzymes19−22 (Figure S5, Table S1). Analysis of the Mössbauer spectra recorded in zero-field and 53-mT magnetic field (Figure S6) reveals that the features expected for an Fe(III)-superoxo complex17 are undetectable. The time-dependence of formation and decay of the detected Fe(IV) complex, defined by quantitative analysis of the Mössbauer spectra, agrees well with the ΔA320 kinetic trace observed in the SF-abs experiment (Figure 3), suggesting that both spectroscopic features are associated with the same complex.
Figure 3.
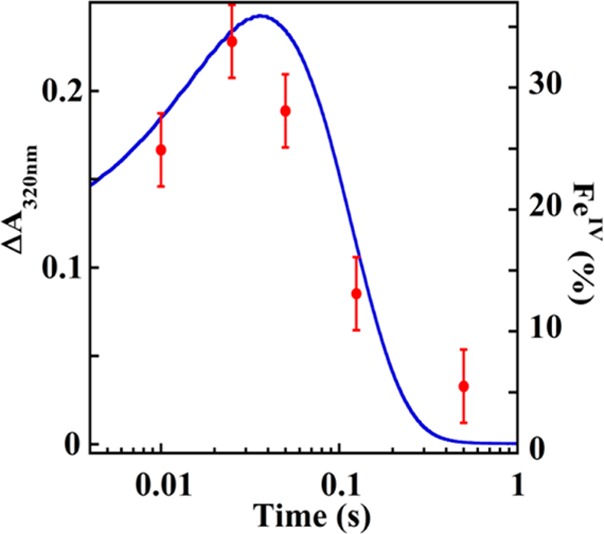
Kinetics of the accumulation and decay of the Fe(IV) intermediate. Overlay of the quantities of Fe(IV) determined by freeze-quench Mössbauer spectroscopy (red dots, right axis) with the SF-abs trace (blue trace, left axis). Both sets of data were from the reaction of a solution of the HEPD-E176A·Fe(II)·D2-HEP complex with O2 as described in the legend of Figure 2. The estimated uncertainty of the total quantity of the Fe(IV) complex determined by analysis of the Mössbauer spectra is ±3% of total intensity, as determined by iterative simulations evaluated by visual inspection.
In the corresponding reactions of either the HEPD-E176A variant with unlabeled HEP or the wt enzyme with D2-HEP, the prominent high-energy line of the Fe(IV) complex in the Mössbauer spectra is either much less intense (10% of total intensity) or below the detection limit of the method (∼3% of total intensity), respectively (Figure S7). The intermediate therefore either does not accumulate in these reactions or completely decays within the minimum reaction time accessible by the conventional FQ method (≥10 ms). Samples prepared by reaction of the wt-HEPD·Fe(II)·D2-HEP complex with the much higher levels of O2 (∼10 mM) that can be accessed using the chlorite/chlorite-dismutase method23do exhibit the high-energy line of the Fe(IV) intermediate in the Mössbauer spectrum (Figure S7). Thus, the Fe(IV) intermediate can accumulate also in the reaction of the wt enzyme, but to a lesser extent and in a less readily accessible time regime than in the reaction of the HEPD-E176A variant. All subsequent experiments were, therefore, performed with the variant.
Demonstration of Enhanced Accumulation of the Intermediate in 2H2O
Accumulation of an iron(IV)-oxo intermediate, while not anticipated, can be rationalized by invoking hydrogen abstraction by the ferryl from the hydroxyl group of a gem-diolate intermediate generated by initial hydroxylation of C2 of HEP (see Discussion). Because this hydrogen is expected to exchange rapidly with solvent, the impact of carrying out the reaction in 2H2O (D2O) was examined. In matched experiments, the HEPD-E176A·Fe(II)·D2-HEP complex prepared in buffered H2O or D2O was rapidly mixed with O2-saturated buffer in the same solvent. The amplitude of the feature observed in the SF-abs trace is significantly greater for the reaction in D2O solvent, consistent with enhanced accumulation of the intermediate (Figure 4a). FQ Mössbauer experiments were then carried out to verify accumulation of the same complex. The 4.2-K/zero-field Mössbauer spectra (Figure 4b) confirm that the same Fe(IV) complex accumulates and to a greater extent in the reaction carried out in D2O (27% of the total absorption area in D2O, compared to 15% in H2O). The diminished accumulation of the Fe(IV) complex relative to that in Figure 2b is the result of partial loss of enzyme activity (in both the H2O and D2O experiments) associated with the prolonged buffer-exchange procedure used for both experiments.
Figure 4.
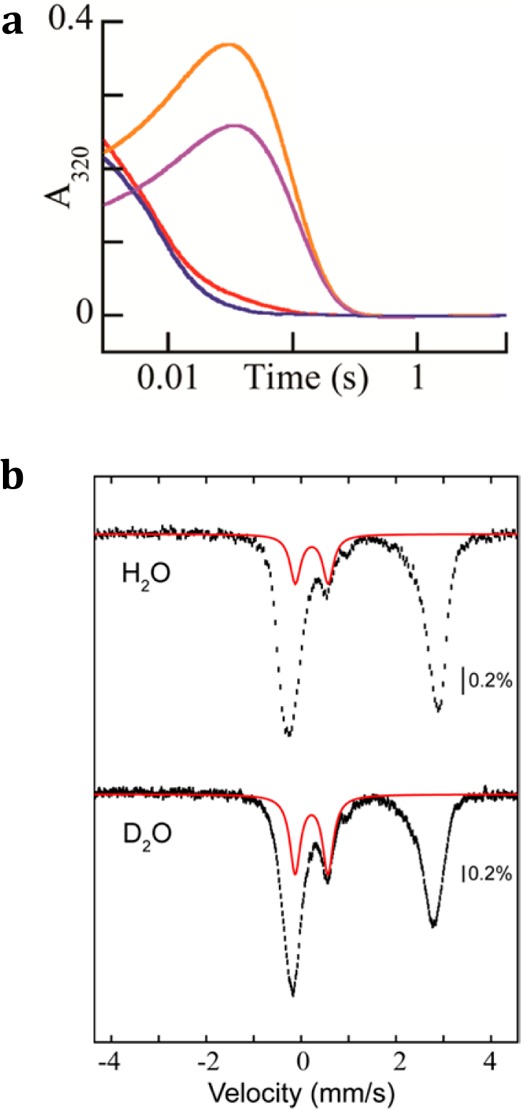
Results of SF-abs and freeze-quench Mössbauer experiments in H2O and D2O. (a) A solution of HEPD-E176A was exchanged into buffer made in H2O or D2O, concentrated, rendered anoxic, and treated with Fe(II) and D2-HEP. This solution was mixed at 4 °C with an equal volume of an O2-saturated solution in buffer of the same isotopic composition to give final concentrations of 0.5 mM HEPD-E176A, 0.4 mM Fe(II), 3.0 mM D2-HEP, and ∼0.85 mM O2, and 50 mM HEPES, pH/pD 7.5. The purple and brown lines are ΔA320 kinetic traces from the reactions carried out in buffer of natural isotopic abundance and buffer enriched in D2O, respectively. The red and blue lines are the corresponding traces with unlabeled HEP substrate in D2O-enriched buffer and natural-abundance buffer, respectively. Control experiments demonstrated that the activity of the enzyme was diminished by the buffer-exchange procedure to approximately the same extent as the amplitudes of the ΔA320 kinetic traces (compare panel a and Figure 2a). (b) 4.2-K/zero-field Mössbauer spectra of samples prepared by mixing at 4 °C of an anoxic solution of HEPD-E176A, Fe(II), and D2-HEP with an equal volume of an O2-saturated buffer. Final concentrations: 0.87 mM HEPD-E176A, 0.85 mM Fe(II), 5.0 mM D2-HEP, and 50 mM sodium HEPES, pH/pD 7.5. The reaction was quenched after 25 ms. The solid red lines are simulations of the spectrum of the Fe(IV) intermediate with the parameters quoted in the text. These theoretical spectra correspond to 15% and 27% of the total spectral intensity for the samples prepared in H2O and D2O, respectively.
A different kind of solvent exchange was documented previously for the HEPD reaction.4 These studies showed that the oxygen atom incorporated into the HMP product derives in part from molecular oxygen (60%) and in part from solvent water (40%). Because use of D2O and D2-HEP enhances accumulation of the ferryl complex, presumably by increasing its lifetime, substrate and solvent deuteration could also potentially allow for an increased extent of “washout” of the oxygen in the ferryl complex (originally derived from O2) and thus an increase in the extent of solvent oxygen incorporation into HMP. To test this possibility, we first established that the HEPD-E176A variant also incorporates ∼60% of 18O from 18O2 into the HMP product formed from unlabeled HEP. We then challenged the variant protein with D2-HEP and found, unexpectedly, little effect of substrate labeling on the extent of 18O incorporation (from either 18O2 or H218O) into HMP. Moreover, use of D2O solvent along with D2-HEP, which would be expected to maximize the lifetime of the ferryl complex, actually resulted in a slight decrease in the “washout” of the label from 18O2 (Figure S8). Thus, enhanced accumulation of the ferryl complex is not associated with increased incorporation of oxygen derived from solvent into the hydroxyl group of HMP. This result implies that the partial (∼60%) gas-oxygen incorporation into HMP must result from a mechanism distinct from solvent-mediated proton–deuterium exchange in and/or leading up to the ferryl state.
Discussion
Previous studies invoked an Fe(III)-superoxo species as the oxidant that cleaves the pro-S C2–H bond of HEP (Figure 1c). Attempts to trap this species in this study resulted in identification of an intermediate with UV–vis absorption and Mössbauer characteristics strongly resembling those of Fe(IV)-oxo species trapped in other enzymes.19−22 Each of the latter enzymes requires two electrons from the Fe(II) center and two electrons from a co-substrate to form the Fe(IV)-oxo species. By contrast, HEPD requires no reducing co-substrate for catalysis. Whereas two tyrosine residues in the active site could, in principle, each donate a single electron to cleave O2 prior to substrate activation, substitution of these residues with phenylalanine was shown not to abolish activity.7 Therefore, formation of the ferryl intermediate almost certainly involves extraction of two electrons from the substrate. Attempts to use chemical quenched-flow methods to define the chemical state of the substrate during accumulation and decay of the intermediate under the same reaction conditions used for the SF and FQ Mössbauer studies were unsuccessful because of technical difficulties (see Supporting Information). However, comparison of the steady-state kinetic parameters with the kinetics of formation and decay of the Fe(IV)-oxo complex clearly shows that the transient species is kinetically competent to be on-pathway. We obtained most of our results with HEPD-E176A, a variant with a first shell substitution that could have perturbed the chemistry compared to wt HEPD. However, because the variant generates the same products as the wt enzyme and with very similar steady-state kinetics, and because at high O2 concentrations the wt enzyme also produces the same ferryl species, we presume that the chemical mechanisms of wt and variant enzymes are not greatly different.
As discussed previously, all available data indicate that the reaction is initiated by abstraction of the pro-S-hydrogen by a Fe(III)-superoxo intermediate. However, the finding that the ferryl complex is observed only in the reaction with (S)-D-HEP requires that this unexpected intermediate also decay by abstraction of the hydrogen atom initially on C2. In other words, this hydrogen is abstracted at least twice during the course of the catalytic cycle, once by the Fe(III)-superoxo species and once by the ferryl intermediate. A reformulated mechanism that can account for the data reported here and the findings of previous studies is presented in Figure 5. The ferric-superoxo complex, I, abstracts the pro-S hydrogen from C2 of the substrate. After formation of the ferric-hydroperoxo intermediate, II, at least two possible pathways could explain the formation of the observed Fe(IV)-oxo species. In one route, homolytic peroxide-bond cleavage would directly yield an iron(IV) oxo species, VI, and a gem-diolate. Alternatively, density functional theory (DFT) calculations on HEPD8 and observations made with the related enzyme, 2-hydroxypropylphosphonate epoxidase, suggest rapid electron transfer from the ketyl radical in intermediate II to the Fe(III).24,25 The resulting ferrous-hydroperoxo intermediate, III, could then undergo heterolytic O–O bond scission with hydration of the nearby aldehyde to form intermediate VI.
Figure 5.

Reformulated mechanism for HEPD that explains both the accumulation of a ferryl intermediate (VI) and the observed effects of the substrate and solvent hydrogen isotope on the extent of its accumulation. Exchange of the oxygenic ligand derived from O2 with solvent in intermediate VII or V, or partitioning of the attack of the phosphonomethyl radical in V on two oxygenic ligands derived from O2 or solvent (blue and red arrows), can account for the observed fractional incorporation of label from O2 into HMP.
In either of these routes, several intermediates could undergo exchange of the original pro-S hydrogen at C2 of HEP (blue in Figure 5) with deuterium derived from solvent. The observation that the ferryl does not accumulate with HEP but does with D2-HEP in H2O is consistent with incomplete wash-out of the deuterium from the substrate in the intermediates leading up to and including ferryl intermediate, VI. Indeed, as predicted, when the reaction occurs in D2O buffer, a significantly greater quantity of the ferryl complex accumulates, consistent with the abstraction of a partially solvent-exchangeable hydrogen. We note that a previous DFT study also suggested the retention of the initially abstracted hydrogen on the gem-diolate oxygen.8
The mechanism in Figure 5 can also explain several other observations. The inability to observe any intermediate in the reaction with unlabeled HEP in H2O suggests that conversion of unlabeled intermediate VI to VII is rapid. If so, it may be too fast for solvent exchange, explaining why the reaction with unlabeled HEP in D2O does not accumulate the ferryl species in the same time regime. By contrast, when the substrate is deuterium labeled, the hydroxyl group in VI is predicted to contain deuterium, at least initially (Figure 5). Apparently the KIE on the conversion of VI to VII is sufficiently large to allow detection of VI. This enhanced lifetime might, in turn, allow for exchange of the hydroxyl hydrogen with solvent, resulting in greater accumulation of the intermediate when the reaction is conducted in D2O, which maintains complete deuteration of the otherwise partially exchanged C2 hydroxyl group.
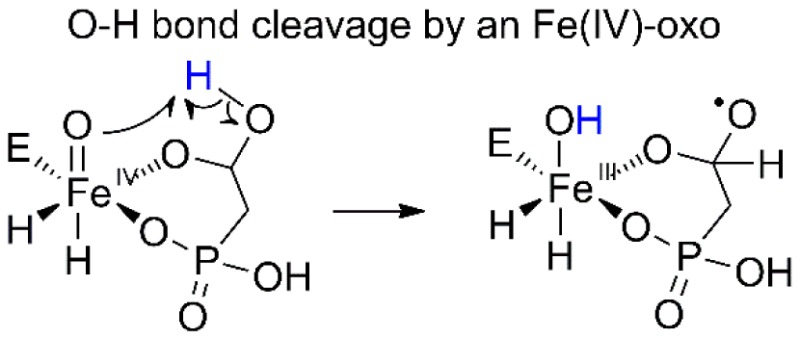
One unusual facet of this mechanism is that it involves homolytic O–H bond activation. The bond dissociation energy for an O–H bond is at least 7–10 kcal/mol greater than for the C–H bonds typically activated by iron(IV)-oxo species.26,27 However, the greater kinetic reactivity of O–H bonds for hydrogen atom transfer (HAT) is well documented.26 In addition, the process may involve a proton-coupled electron transfer (PCET) mechanism rather than HAT. Indeed, previous computational studies on HEPD suggested that O–H activation by an iron(IV)-oxo intermediate is energetically feasible.8−10 Two separate theoretical studies postulated that π attack28−30 by a ferryl in HEPD could cleave the O–H bond depicted in Figure 5.8,10 The detection herein of a kinetically competent ferryl intermediate provides experimental support for its intermediacy during HEPD catalysis, and our observed substrate and solvent KIEs provide evidence for its removal of a hydrogen atom in an O–H bond.
After hydrogen abstraction by ferryl VI, the proposed catalytic mechanism is unchanged from previous proposals. Cleavage of the O–H bond of VI would yield a gem-diolyl radical that could undergo beta scission to generate a phosphonomethyl radical and ferric hydroxide, V, which would then form HMP by radical coupling. Subsequent product release is likely rate-determining for multiple turnovers, as evidenced by the small substrate KIEs on kcat with wt HEPD under steady-state conditions7 and the accumulation of an Fe(II)-product complex in the Mössbauer experiment even in the presence of excess substrate.
As mentioned, previous studies showed that the hydroxyl oxygen in the HMP product derives 60% from O2 and 40% from solvent.4 Precedent suggests that this incorporation of solvent oxygen could arise most simply by exchange of the ferryl oxygen, initially derived from the O2, with the H2O solvent.31−39 Because it retards decay of the ferryl complex in the hydrogen atom abstraction step, the use of either D2-HEP or D2O as solvent promotes accumulation of the intermediate. This increased lifetime of the complex would be expected to also increase solvent exchange of the ferryl oxygen by affording more time for the exchange to proceed. In contrast to these expectations, ratiometric quantification of H16OCH2PO32– and H18OCH2PO32– by mass spectrometry in experiments carried out either with D2-HEP substrate or in D2O showed either no change (with D2-HEP) or a slight diminution (with D2O) in the fraction of solvent-derived oxygen in HMP. This unexpected trend argues against solvent exchange in the ferryl state, but rationalizes how the 2H removed from C2 by the Fe(III)-superoxo complex can fail to exchange completely with solvent and thereby exert a downstream retarding effect on the decay of the ferryl complex: apparently, the enzyme shields states leading up to the C1–C2-bond-cleavage step from solvent.
Because the extent of exchange of the 18O derived from O2 with 16O derived from solvent did not change upon slowing down ferryl decay, exchange must take place after this step, by one of two mechanisms. It is possible that the state containing the Fe(III) and the product radical V has two coordinated water/hydroxide molecules, one derived from O2 in formation of the ferryl complex and one derived from solvent (Figure 5). A partition between attack of the radical on the two oxygenic ligands (red and blue arrows) could yield the observed fractional incorporation. Alternatively, the Fe(III)/·CH2PO32– state could persist for sufficient time for exchange of the single (initially O2-derived) oxygenic ligand with solvent. Either possibility could rationalize the results, including the apparent discrepancy in the solvent-exchange behavior of the C2 hydron and the incorporated oxygen. The recombination of a substrate-derived radical with a hydroxyl ligand is typically thought to occur rapidly when this “oxygen-rebound” step occurs to the atom from which the hydrogen atom was abstracted.8,40 However, in certain P450 systems, this rebound is slow enough to permit racemization at the targeted carbon atom.41,42 Furthermore, in HEPD, the “rebound” occurs with a radical that is (i) located on an atom that is two bonds removed from the atom from which the hydrogen was abstracted, and (ii) generated by an intervening β-scission step. As such, the active site of HEPD may require a rearrangement that impedes very rapid recombination, thus explaining the previously observed racemization at C1 and oxygen exchange with solvent in this state. Alternatively, we cannot rule out that a larger distance of the phosphonomethyl radical to the ferric-hydroxide could result in preferential electron transfer from the radical to the cofactor to form the corresponding cation. Subsequent trapping of the cation by water/hydroxide in the active site could then explain the observed exchange of the oxygen in HMP.
Conclusion
Non-heme-Fe(IV)-oxo intermediates are involved in diverse reactions including hydroxylation,19−21,43 halogenation,22 and epimerization.44 In these processes, the ferryl complexes cleave C–H bonds. High-valent iron model complexes have been reported also to activate O–H bonds,45 and a ferryl intermediate has been hypothesized to cleave N–H and O–H bonds in the non-heme-iron enzymes 1-aminocyclopropane-1-carboxylic acid oxidase (ACCO) and clavaminate synthase, respectively.46,47 For the latter enzyme, a DFT study suggested a number of potential reaction mechanisms, including one in which a ferryl abstracts a hydrogen atom from an alcohol followed by a β-scission reaction, not unlike the pathway suggested in Figure 5. In the case of ACCO, the activated oxygen species has been proposed on the basis of measured solvent and 18O KIEs to be a ferryl complex that abstracts a hydrogen atom from the amine of its substrate.48 The current study is, to our knowledge, the first direct experimental support for cleavage of a hydrogen-heteroatom bond by a mononuclear iron(IV)-oxo enzyme intermediate. This investigation thus adds to the already impressive mechanistic diversity of this versatile enzyme family.49,50
Methods
Steady-State Kinetics of HEPD and HEPD-E176A
The kinetics of oxidation of HEP and D2-HEP by wt HEPD and HEPD-E176A were determined by using a Clark-type Oxytherm Electrode Unit (Hansatech, Inc.). The dependence of activity on Fe(II) concentration was defined by assaying enzyme activated by prior addition (to 40 μM wt HEPD or 70 μM HEPD-E176A in 25 mM sodium HEPES, pH 7.5) of a varying quantity of a solution of Fe(NH4SO4)2·6H2O in the absence of O2 and incubation on ice for 10 min. Turnover was quantified by O2 consumption in air-saturated buffer (25 mM HEPES pH 7.5, 280 μM O2) at 20 °C in the presence of wt HEPD or HEPD-E176A (2 μM). Reactions were initiated by addition of HEP (250 μM for wt HEPD; 400 μM for HEPD-E176A). Assays were carried out in triplicate.
Routine activity assays were carried out with 1 equiv. Fe(II) for wt HEPD and 3 equiv. Fe(II) for HEPD-E176A. Reactions with varying HEP and constant O2 were initiated by dilution of the enzyme to a final concentration of 1 μM with air-saturated 50 mM sodium HEPES (pH 7.5) buffer (giving ∼280 μM O2 at 20 °C) followed by addition of HEP (5–100 μM for wt HEPD, 10–250 μM for HEPD-E176A). For varying the concentration of O2 (10–500 μM), the assay buffer was sparged with O2 or N2, and the concentration of O2 was monitored by the oxygen electrode until the desired concentration was reached. The enzyme and HEP were then added (to 1 μM and 250 μM, respectively), and the rate of O2 consumption was monitored.
The procedures for the 18O tracer and chemical quench experiments are provided in the Supporting Information.
Transient-State Kinetic/Spectroscopic Experiments
Procedures and apparatus for the SF-abs and FQ Mössbauer experiments have been described elsewhere.21,23 Reaction conditions and details of spectral acquisition are provided in the appropriate figure legends. FQ Mössbauer experiments were carried out with slightly substoichiometric amounts of Fe(II) to avoid observation of unbound Fe. The SF-abs experiments were performed under the same conditions to allow comparison.
Acknowledgments
This work was supported by the National Institutes of Health (GM 077596 to W.A.V. and GM 069657 to C.K. and J.M.B.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b12147.
Molecular biology procedures, details of Mössbauer analysis and 18O labeling experiments, Figures S1–S8, and Table S1 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Metcalf W. W.; van der Donk W. A. Annu. Rev. Biochem. 2009, 78, 65. 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck S. C.; van der Donk W. A. Curr. Opin. Chem. Biol. 2013, 17, 580. 10.1016/j.cbpa.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blodgett J. A.; Thomas P. M.; Li G.; Velasquez J. E.; van der Donk W. A.; Kelleher N. L.; Metcalf W. W. Nat. Chem. Biol. 2007, 3, 480. 10.1038/nchembio.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchillo R. M.; Zhang H.; Blodgett J. A. V.; Whitteck J. T.; Li G.; Nair S. K.; van der Donk W. A.; Metcalf W. W. Nature 2009, 459, 871. 10.1038/nature07972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehntop K. D.; Emerson J. P.; Que L. Jr. J. Biol. Inorg. Chem. 2005, 10, 87. 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- Whitteck J. T.; Malova P.; Peck S. C.; Cicchillo R. M.; Hammerschmidt F.; van der Donk W. A. J. Am. Chem. Soc. 2011, 133, 4236. 10.1021/ja1113326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck S. C.; Cooke H. A.; Cicchillo R. M.; Malova P.; Hammerschmidt F.; Nair S. K.; van der Donk W. A. Biochemistry 2011, 50, 6598. 10.1021/bi200804r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao H.; Morokuma K. J. Am. Chem. Soc. 2010, 132, 17901. 10.1021/ja108174d. [DOI] [PubMed] [Google Scholar]
- Hirao H.; Morokuma K. J. Am. Chem. Soc. 2011, 133, 14550. 10.1021/ja206222f. [DOI] [PubMed] [Google Scholar]
- Du L.; Gao J.; Liu Y.; Liu C. J. Phys. Chem. B 2012, 116, 11837. 10.1021/jp305454m. [DOI] [PubMed] [Google Scholar]
- Whitteck J. T.; Cicchillo R. M.; van der Donk W. A. J. Am. Chem. Soc. 2009, 131, 16225. 10.1021/ja906238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H.; Peck S. C.; Bonnot F.; van der Donk W. A.; Klinman J. P. J. Am. Chem. Soc. 2015, 137, 10448. 10.1021/jacs.5b03907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck S. C.; Chekan J. R.; Ulrich E. C.; Nair S. K.; van der Donk W. A. J. Am. Chem. Soc. 2015, 137, 3217. 10.1021/jacs.5b00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Donk W. A.; Krebs C.; Bollinger J. M. Jr. Curr. Opin. Struct. Biol. 2010, 20, 673. 10.1016/j.sbi.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing G.; Diao Y.; Hoffart L. M.; Barr E. W.; Prabhu K. S.; Arner R. J.; Reddy C. C.; Krebs C.; Bollinger J. M. Jr. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 6130. 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbughuni M. M.; Chakrabarti M.; Hayden J. A.; Bominaar E. L.; Hendrich M. P.; Münck E.; Lipscomb J. D. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16788. 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamanaha E.; Zhang B.; Guo Y.; Chang W. C.; Barr E. W.; Xing G.; St. Clair J.; Ye S.; Neese F.; Bollinger J. M. Jr.; Krebs C. J. Am. Chem. Soc. 2016, 138, 8862. 10.1021/jacs.6b04065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger J. M. Jr.; Krebs C. J. Inorg. Biochem. 2006, 100, 586. 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Eser B. E.; Barr E. W.; Frantom P. A.; Saleh L.; Bollinger J. M. Jr.; Krebs C.; Fitzpatrick P. F. J. Am. Chem. Soc. 2007, 129, 11334. 10.1021/ja074446s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffart L. M.; Barr E. W.; Guyer R. B.; Bollinger J. M. Jr.; Krebs C. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 14738. 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Tirupati B.; Bollinger J. M. Jr.; Krebs C. Biochemistry 2003, 42, 7497. 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- Galonić D. P.; Barr E. W.; Walsh C. T.; Bollinger J. M. Jr.; Krebs C. Nat. Chem. Biol. 2007, 3, 113. 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- Dassama L. M. K.; Yosca T. H.; Conner D. A.; Lee M. H.; Blanc B.; Streit B. R.; Green M. T.; DuBois J. L.; Krebs C.; Bollinger J. M. Jr. Biochemistry 2012, 51, 1607. 10.1021/bi201906x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Chang W.-c.; Lin G.-M.; Romo A.; Pai P.-J.; Russell W. K.; Russell D. H.; Liu H.-w. J. Am. Chem. Soc. 2014, 136, 2944. 10.1021/ja4100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Chang W.-c.; Pai P.-J.; Romo A.; Mansoorabadi S. O.; Russell D. H.; Liu H.-w. J. Am. Chem. Soc. 2012, 134, 16171. 10.1021/ja3078126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer J. M. Acc. Chem. Res. 2011, 44, 36. 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillen D. F.; Golden D. M. Annu. Rev. Phys. Chem. 1982, 33, 493. 10.1146/annurev.pc.33.100182.002425. [DOI] [Google Scholar]
- Ye S.; Neese F. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1228. 10.1073/pnas.1008411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srnec M.; Wong S. D.; England J.; Que L. Jr.; Solomon E. I. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 14326. 10.1073/pnas.1212693109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I.; Wong S. D.; Liu L. V.; Decker A.; Chow M. S. Curr. Opin. Chem. Biol. 2009, 13, 99. 10.1016/j.cbpa.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad B.; Lindstedt G.; Lindstedt S. J. Am. Chem. Soc. 1970, 92, 7446. 10.1021/ja00728a032. [DOI] [PubMed] [Google Scholar]
- Sabourin P. J.; Bieber L. L. J. Biol. Chem. 1982, 257, 7468. [PubMed] [Google Scholar]
- Kikuchi Y.; Suzuki Y.; Tamiya N. Biochem. J. 1983, 213, 507. 10.1042/bj2130507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wackett L. P.; Kwart L. D.; Gibson D. T. Biochemistry 1988, 27, 1360. 10.1021/bi00404a041. [DOI] [PubMed] [Google Scholar]
- Baldwin J. E.; Adlington R. M.; Crouch N. P.; Pereira I. A. C. Tetrahedron 1993, 49, 7499. 10.1016/S0040-4020(01)87226-4. [DOI] [Google Scholar]
- Seo M. S.; In J. H.; Kim S. O.; Oh N. Y.; Hong J.; Kim J.; Que L. Jr.; Nam W. Angew. Chem., Int. Ed. 2004, 43, 2417. 10.1002/anie.200353497. [DOI] [PubMed] [Google Scholar]
- Pestovsky O.; Bakac A. Inorg. Chem. 2006, 45, 814. 10.1021/ic051868z. [DOI] [PubMed] [Google Scholar]
- Chang W.-c.; Li J.; Lee J. L.; Cronican A. A.; Guo Y. J. Am. Chem. Soc. 2016, 138, 10390. 10.1021/jacs.6b05400. [DOI] [PubMed] [Google Scholar]
- Puri M.; Company A.; Sabenya G.; Costas M.; Que L. Jr. Inorg. Chem. 2016, 55, 5818. 10.1021/acs.inorgchem.6b00023. [DOI] [PubMed] [Google Scholar]
- Cooper H. L. R.; Groves J. T. Arch. Biochem. Biophys. 2011, 507, 111. 10.1016/j.abb.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb M. H.; Heimbrook D. C.; Malkonen P.; Sligar S. G. Biochemistry 1982, 21, 370. 10.1021/bi00531a026. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; He X.; Ortiz de Montellano P. R. Biochemistry 2006, 45, 533. 10.1021/bi051840z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Glass T. E.; Krebs C.; Bollinger J. M. Jr. J. Am. Chem. Soc. 2003, 125, 13008. 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- Chang W.-c.; Guo Y.; Wang C.; Butch S. E.; Rosenzweig A. C.; Boal A. K.; Krebs C.; Bollinger J. M. Jr. Science 2014, 343, 1140. 10.1126/science.1248000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Farquhar E. R.; Stubna A.; Münck E.; Que L. Jr. Nat. Chem. 2009, 1, 145. 10.1038/nchem.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowski T.; de Marothy S.; Broclawik E.; Schofield C. J.; Siegbahn P. E. M. Biochemistry 2007, 46, 3682. 10.1021/bi602458m. [DOI] [PubMed] [Google Scholar]
- Thrower J.; Mirica L. M.; McCusker K. P.; Klinman J. P. Biochemistry 2006, 45, 13108. 10.1021/bi061097q. [DOI] [PubMed] [Google Scholar]
- Mirica L. M.; Klinman J. P. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1814. 10.1073/pnas.0711626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleva E. G.; Lipscomb J. D. Nat. Chem. Biol. 2008, 4, 186. 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger J. M. Jr.; Chang W.-c.; Matthews M. L.; Martinie R. J.; Boal A. K.; Krebs C. In 2-Oxoglutarate-Dependent Oxygenases; Hausinger R. P., Schofield C. J., Eds.; Royal Society of Chemistry: London, 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.