

Abstract
The application of circulating tumor DNA(ctDNA) represents a non-invasive method for tumor detection. Its prognostic significance in patients with colorectal cancer is controversial. We performed a systematic review of data from published studies to assess the prognostic values of ctDNA in patients with colorectal cancer. We searched Medline, Embase, Web of Science, the Cochrane Library, and Scopus databases to identify eligible studies reporting disease-free survival (DFS) and overall survival (OS) stratified by ctDNA prior to December 6, 2016. We evaluated the quality and design of these studies. A total of 22 studies were eligible for systematic review. Among them, 11 studies investigated the prognostic value of ctDNA on disease-free survival (DFS). Seven of 11 studies showed that ctDNA was an independent variable to estimate the probability of DFS by multivariate analyses. Thirteen studies assessed the relationship between ctDNA and overall survival (OS). Eight of 13 studies showed that ctDNA was an independent predictor of worse OS through the use of multivariate analyses. This analysis provides evidence that ctDNA may be a prognostic biomarker, negatively correlated with the survival of patients with colorectal cancer.
Introduction
Circulating free DNA with tumor-specific alterations (ctDNA) is found in serum or plasma and represents a small fraction of the total circulating free DNA. It is believed that ctDNA is shed into the bloodstream from tumor cells through apoptosis, necrosis, autophagy, necroptosis, and other physiological processes [1]. CtDNA strands are small fragments (approximately 180–200 base pairs in length), containing tumor-specific alterations in tumor suppressor genes or oncogenes, microsatellite instability, and DNA hypermethylation [2,3]. Some specific genetic alterations detected in ctDNA are driver alterations that are responsible for the initiation and progression of human cancers. Those alterations play broad roles in vivo, such as affecting genomic surveillance mechanisms and reducing cells’ ability to detect and/or repair DNA damage, which increases susceptibility to DNA damage by exogenous and endogenous carcinogens [4]. Epigenetic alterations, such as methylation of CpG islands in promoter regions, are responsible for the silencing of multiple tumor suppressor genes [4]. In some instances, hypermethylation can lead to microsatellite instability.
Virtually every type of cancer harbors genetic/epigenetic alterations. Some studies illustrated that alterations in ctDNA were in concordance with the genomic spectrum of the tumor, providing evidence that ctDNA may be a potential surrogate for the entire tumor genome. Recently, ctDNA has emerged as a non-invasive blood biomarker in tumor precision medicine. CtDNA correlates with tumor stage, tumor burden, and therapy in patients with colorectal cancer (CRC). Patients with early-stage or minimal residual disease usually have lower levels of ctDNA, making it difficult to precisely detect specific alterations. The results of intensive efforts are now evident with the development of new highly sensitive technological methods that can overcome this problem.
Comprehensive analysis of ctDNA is becoming increasingly popular. Potential applications include, but are not limited to, early detection, observation of dynamic tumor changes, assessment of tumor heterogeneity, identification of genetic/epigenetic alterations for targeted therapy, and assessment of drug resistance development [3,5].
Among the numerous possible applications, the prognostic and predictive values of ctDNA in CRC have generated the most intense interest. Studies uncovered that ctDNA could be a reliable prognostic factor correlated with poorer outcome [6]. Positive detection of ctDNA implies a high risk of recurrence or short overall survival (OS) in patients with CRC treated with surgery, chemotherapy, radiotherapy, or targeted therapy [7–9]. However, other studies found no difference in survival between ctDNA-positive and ctDNA-negative CRC [10].
To clarify the prognostic role of ctDNA in CRC, we initiated a systematic literature review to gain a better understanding of its prognostic value in patients with CRC.
Methods
Criteria for inclusion
Eligible studies met the following criteria: inclusion of only patients with CRC; analysis of the correlation between patient survival and ctDNA status; and inclusion of follow-up data for OS, disease-free survival (DFS), and/or cancer-specific mortality. Both prospective and retrospective cohort studies were included. Reviews, comments, and case reports were excluded.
Search methods for identification of studies
We adhered to the Meta-analysis of Observational Studies in Epidemiology guidelines to identify eligible studies. We conducted systematic electronic searches of the Medline, Embase, Web of Science, the Cochrane Library, and Scopus databases to identify eligible studies performed prior to December 6, 2016 (no start date limit was applied). We used combinations of the following search terms: “Colonic Neoplasm,” “Neoplasm, Colonic,” “Neoplasms, Colonic,” “Colon Neoplasms,” “Colon Neoplasm,” “Neoplasm, Colon,” “Neoplasms, Colon,” “Cancer of Colon,” “Colon Cancers,” “Cancer of the Colon,” “Colonic Cancer,” “Cancer, Colonic,” “Cancers, Colonic,” “Colonic Cancers,” “Colon Cancer,” “Cancer, Colon,” “Cancers, Colon,” “colonic,” “colorectal disease,” “rectal neoplasms,” “colorectal polyps,” “sigmoid neoplasms,” “colorectal adenoma,” “circulating tumor DNA,” “ctDNA,” “cell free DNA,” “serum DNA,” “plasma DNA,” “circulating DNA,” “free DNA,” “prognosis,” “survival,” “prognostic,” and “predictive.” No restrictions were placed on the search, and relevant MeSH (Medline) or Emtree (Embase) terms were used where possible. The reference lists of relevant studies were manually searched to identify new studies. In additional to full publications, conference posters and letters that fulfilled the inclusion criteria were documented to capture grey literature. Publications written in languages other than English were also included if sufficient information was available in the abstract.
Each study was independently assessed for inclusion at least by two investigators (Gaowei Fan, Xin Yang), and discrepancies were resolved by discussion. Whenever multiple versions of reports were presented (e.g., same authors, overlapping period of study, same protocol ID), we retained the report with the largest patient population. Duplicates were removed.
Data extraction
The following data were retrieved from the included studies: author, publication year, country in which the study was conducted, publication type, number of patients included in the analysis, percentage of male patients, tumor stage, median patient age, ctDNA panel, detection method, number of patients with ctDNA positivity, treatment, follow-up, sampling time, and outcome (DFS, OS, cancer-specific mortality). Individual investigators of the included studies were also contracted by email if essential information relevant to this systematic review was absent. Data extraction was performed by three independent investigators with a predefined information sheet (Gaowei Fan, Kuo Zhang, Xin Yang, Jiansheng Ding). Any discrepancies were resolved by discussion.
During the entire selection process, none of the authors was blinded to the source of the publications, the authors, or any other details.
Quality assessment
We evaluated the quality of the included studies using the Cochrane Collaboration’s tool for assessing the risk of bias [11]. Specifically, studies were judged on the following criteria: (1) selection bias, defined as a clear description of the inclusion and exclusion criteria; (2) accuracy of measurements, also called measurement bias, defined as an explicit description of the ctDNA detection method; (3) exposure bias, defined as an explicit description of genetic/epigenetic alterations; (4) bias caused by incomplete follow-up, defined as a satisfactory report of the median follow-up length, follow-up range, and loss to follow-up rates, and (5) confounding bias, which included known or commonly discussed confounders in the relationship between ctDNA and survival, such as age, disease status, or other factors for which adjustment was performed.
Measurement of treatment effect
The primary outcome was DFS. The secondary outcome was OS. For the purpose of these analyses, DFS was defined as the time from the initial treatment to the first documentation of relapse/recurrence. OS was defined as the time from the initial treatment to death.
Ethics statement
This study was a literature-based study, and no ethics approval was needed.
Results
Included studies
A total of 2479 potential studies were initially searched. Following eligibility screening by title and abstract, 2371 studies were removed. The main reasons for exclusion were duplicative studies, reviews, non-human studies, no relevance to ctDNA, and incorrect tumor type. Of the remaining 108 studies, the full text was screened, and 86 studies were excluded because of an absence of prognosis information, not ctDNA, overlapping studies, comments, improperly grouped mutations, not DFS/OS, or the inclusion of patients with diseases other than CRC. The reasons for exclusion were listed in S1 Table. Finally, 22 studies met the inclusion criteria, and were included for descriptive summarization (Fig 1).
Fig 1. PRISMA 2009 flow diagram.

PRISMA flow diagram for study selection. From: Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group (2009). Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med 6(6): e1000097. doi:10.1371/journal.pmed1000097. For more information, visit www.prisma-statement.org.
Study characteristics
The included studies, published between 2002 and 2016, analyzed the relationship between ctDNA status and survival outcomes in a total of 2541 patients. The number of patients in each study ranged from 15 to 353. Eighteen studies were prospective studies, and 4 studies were retrospective studies. Of these, 21 studies were published as full publications, and the other one was conference posters.
Most studies were published in the English language. One study was published in a language other than English (with an English abstract). Patients with primary or metastatic CRC with a TNM stage of I-IV or Dukes’ stage of A, B, C, D who received surgery, chemotherapy, radiotherapy, or targeted therapy were included. The study characteristics of the patients enrolled in these studies are summarized in Table 1 and S2 Table.
Table 1. General characteristics of the study populations.
| First author name (year) | Country | Publication type | Study design | Patients included, n | ctDNA-positive patients, n | Male (%) | Tumor stage | Median age | ctDNA panel | Detection methods | Study treatment | Sampling time | Median follow-up | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lecomte (2002) | France | FP | PRO | 37 | 26 | NR | TNM I-IV | NR | KRAS2, p16 | MASA, MSP | Sur, chem, rad | Pre-tr | 22 months | OS |
| Spindler (2013) | Denmark | FP | PRO | 97 | 30KRAS,8BRAF | 65.6% | metastasis | 66 | KRAS2, Braf | ARMS-qPCR | Irinotecan, mono | Pre-tr, | NR | OS |
| Trevisiol (2006) | Italy | FP | PRO | 15 | 7 | NR | Dukes’ A, B, C, D | NR | KRAS2 | ME-PCR | Sur | Pre-tr | 41 months | OS |
| Ryan (2003) | The Netherlands | FP | PRO | 85 | 16 | NR | Dukes’ A, B, C | NR | KRAS2 | SN-PCR, DS | Sur | Post-tr | 3 years | DFS |
| Bai (2013) | China | FP | Retro | 106 | 17low mutation, 16high mutation | 56.6% | metastasis | 55.5 | KRAS | PNA-PCR, nested PCR | Sur, chem, cetuximab | NR | 21.3 months | OS |
| Messaoudi (2016) | France | FP | PRO | 97 | 38KRAS, 5BRAF | 59.8% | metastasis | 66.6 | KRAS, BRAF | Intplex | Chemo, rad | Pre-tr | 36 months | OS |
| Sefrioui (2015) | France | FP | PRO | 16 | 11 | 37.5% | metastasis | NR | KRAS2 | chip-based digital PCR | Chem | NR | NR | OS |
| Tie (2014) | America | Meeting | PRO | 78 | 6 | NR | TNM II | 66 | TP53, APC, KRAS, NRAS, BRAF, PIK3CA, CTNNB1, SMAD4, FBXW7 | Safe-SeqS | Sur, chem | NR | 2 years | RFS |
| Tie (2016) | Australia | FP | PRO | 230 | 20 | 57% | TNM II | 65 | TP53, APC, KRAS | Safe-SeqS | Sur with chem or not | Post-tr | 27 months | RFS |
| Bazan (2006) | Italy | FP | PRO | 50 | 8KRAS, 8TP 53 | NR | Primary | NR | TP53 KRAS p16INK4A | SSCP-PCR, MSP | Sur | NR | 26 months | DFS, OS |
| Lin (2014) | Taiwan | FP | PRO | 133 | 41 | 41.3% | TNM I-IV | NR | 74 genes | MassArray | Sur | Pre-tr | 62 months | OS |
| Lindforss (2005) | Sweden | FP | PRO | 25 | 9 | 36% | TNM I-III | 72 | KRAS | TGGE | Sur | Pre-tr &Pro-tr | 38 months | DFS |
| Wang (2004) | Taiwan | FP | PRO | 104 | 36 | 48.7% | Dukes’ A, B, C, D | DD62.1; DU65.9 | 36 | PCR-SSCP | Sur | Pre-tr | 20 months | DFS |
| Herbs t(2009) | Germany | FP | Retro | 106 | 13 | NR | UICC I-III | 66 | HLTF, HPP1, TPEF | MethyLight | Sur | Pre-tr | 5 years | RFS |
| Lee (2013) | Korea | FP | PRO | 101 | 37 | 32.7% | TNM I-IV | NR | SEPT9 | Real-time PCR | Sur, chem, rad | Pre-tr | 518 days | DFS |
| Leung (2005) | Hong Kong | FP | PRO | 49 | 28 | 36.7% | TNM I-IV | 57 | APC, hMLH1, HLTF | MethyLight | Any treat | Pre-tr | 13.6 months | OS |
| Philipp (2012) | Germany | FP | PRO | 311 | 48HLTF; 64HPP1 | 55% | TNM IV | NR | HLTF, HPP1 | MSP | NR | Pre-tr, | 8 years | OS |
| Tham (2014) | Singapore | FP | Retro | 150 | NR | 56.7% | TNM I-II | NR | TAC1, SEPT9, NELL1 | MSP | Sur | Pre-tr, | 59 months | DFS |
| Wallner (2006) | Germany | FP | Retro | 77 | 20 | NR | TNM I-III | NR | HPP1, HLTF | MethyLight | NR | Pre-tr | 5 years | DFS |
| Matthaios (2016) | Greece | FP | PRO | 155 | 22RASSF1A, 29APC | 57.4% | Dukes’ A,B,C,D | 70 | RASSF1A, APC | MSP | Sur | Pre-tr | NR | OS |
| Liu (2016) | Singapore | FP | PRO | 165 | 82 | 55.2% | TNM I-IV | 67 | SST | MSP | Sur | Pre-tr | 56 months | DFS, OS |
| Lin (2016) | Taipei | FP | PRO | 353 | 129 | 60.06% | TNM I-IV | 67 | AGBL4, FLI1, TWIST1 | Sequenom MassCLEAVE and MALDI-TOF | Sur | Pre-tr | 56 months | DFS |
PRO, prospective study; retro, retrospective study; FP, full publication article; pt, patient; NR, no report; chem, chemotherapy; FU, follow-up; pre-tr, pretreatment; sur, surgery; rad, radiotherapy; mono, monotherapy; meeting, ASCO meeting abstract; post-tr, after treatment; DS, direct sequencing; PCR, polymerase chain reaction; MASA, mutant allele-specific amplification; MSP, methylation-specific PCR; ARMS-qPCR, allele refractory mutation systems-based quantitative PCR; ME-PCR, mutant-enriched PCR; SN-PCR, semi-nested PCR; SSCP-PCR, single-strand conformation polymorphism-PCR; RFS, recurrence-free survival; OS, overall survival; DFS, disease-free survival; SSCP-PCR, single-strand conformation polymorphism-PCR; MALDI-TOF, matrix-assisted laser desorption ionization—time of flight mass spectrometry; TGGE, temperature gradient gel electrophoresis; DD, circulating DNA-detectable; DU, circulating DNA-undetectable.
We assessed risk of bias using the Cochrane Collaboration’s tool for assessing the risk of bias in randomized trials. We categorized bias according to five domains: selection bias, ctDNA detection bias, follow-up bias, selective reporting bias, and confounding bias. We expressed risk of bias as “low risk,” “high risk,” or “unclear risk.” In most studies, inclusion and exclusion criteria for patient selection were clearly defined. These studies were rated as having a “low risk” of selection bias. Most full publication studies described the ctDNA detection method and the time to blood sample explicitly, and we rated these studies as having a “low risk” of ctDNA detection bias. Studies that reported the median follow-up length and range and the loss-to-follow-up rate clearly were rated as “low risk.” Other known factors such as tumor stage, patient age, and lymph node involvement have an effect on patient prognosis. If these factors were considered and adjusted, we rated these studies as “low risk.” The risk of bias in each included study is summarized in Fig 2, and each risk of bias feature, presented as a percentage across all included studies, can be found in S1 Fig.
Fig 2. The risk of bias evaluation of the included studies.
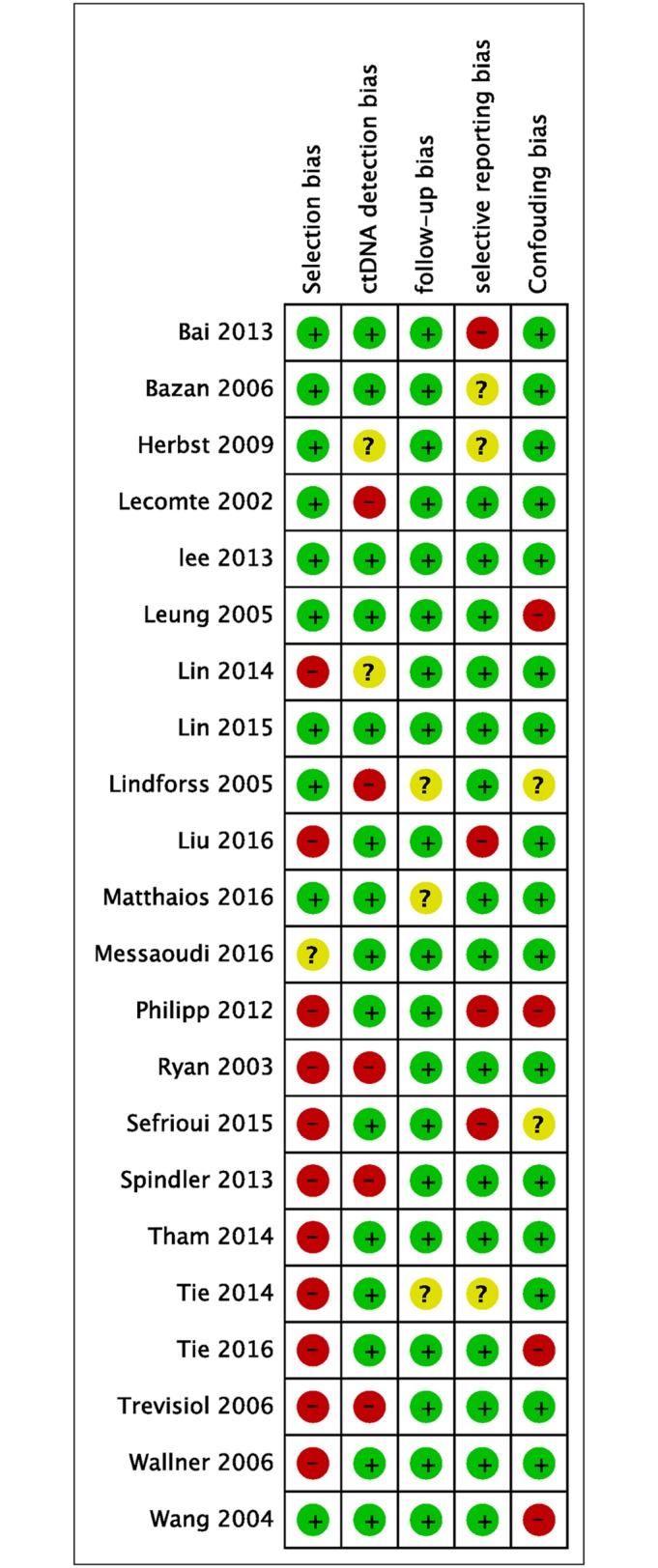
Red circles represent studies with a high risk of bias, green circles represent studies with a low risk of bias, and yellow circles represent studies with an uncertain risk of bias.
Heterogeneity in ctDNA definition
In our included studies, ctDNA status was classified in a dichotomized manner (ctDNA-positive vs. ctDNA-negative or low-level vs. high-level), with the exception of one study that classified ctDNA status in a trichotomized manner (wild-type group, low-mutation group, and high-mutation group) [12]. Some studies used both tissue and serum/plasma to detect alterations, and patients with the same detectable alterations in both tissue and serum were defined as ctDNA-positive, whereas patients with detectable tissue alterations but undetectable serum alterations were defined as ctDNA-negative [8,13–15]. Other studies defined ctDNA-positive and ctDNA-negative as detectable and undetectable serum alterations, respectively. In one study, two consecutive serum samples from the same patient had to test positive to assign a ctDNA-positive status [7].
The ctDNA panel, detection method, and time of blood sample collection also varied significantly across the included studies.
CtDNA detection panel
The detection panels contained both genetic and epigenetic alterations. Some studies contained only genetic mutations in their detection panel, some devised a detection panel with epigenetic alterations only, and a few contained both genetic and epigenetic alterations in their detection panels.
The most commonly detected genetic mutations were KRAS mutations. In total, 6 of 22 studies analyzed KRAS only. Other mutations such as BRAF, RAS, TP53 and APC mutations were often observed, usually in combination with KRAS mutations. For epigenetic alterations, the most commonly investigated genes were HLTF and HPP1. Other panels, such as those used by Wallner et al. (HPP1, HLTF, and hMLH1), Leung et al. (APC, hMLH1, and HLTF), Tham et al. (TAC1, SEPT9, or NELL1), Lin et al. (AGBL4, FLI1, TWIST1), Liu et al. (SST) and Mattaios et al. (APC, RASSF1A)were also included [16–21].
Detection methods
The following methods were used to detect genetic mutations: single-strand conformation polymorphism-PCR, mutant allele-specific amplification, in-house assay based on allele refractory mutation systems-based quantitative PCR, mutant-enriched PCR, semi-nested enrichment technology, direct sequencing, peptide nucleic acid clamp PCR, nested primer PCR, Intplex, personalized Safe-SeqS assays and chip-based digital PCR. To detect epigenetic alterations, the most commonly used method was methylation-specific PCR and MethyLight. Besides, Sequenom MassCLEAVE base-specific cleavage method and matrix-assisted laser desorption ionization—time of flight (MALDI-TOF) mass spectrometry were also used. In some studies, ctDNA could only be detected in patients who harbored tissue alterations. Using highly sensitive technology, some studies concluded that the serum ctDNA status was in high concordance with that of the tissue.
Sample type
Plasma or serum was used to detect ctDNA. Ten studies preferred plasma [8,9,12–14,19,22–25], whereas seven studies used serum [15,17,18,20,21,26,27,28]. The other studies did not report the sample type [6,16,29–31].
Sample timing
Most studies obtained blood samples for ctDNA detection prior to treatment [6,8,19–21,24,26,27]. Three study collected blood samples from untreated patients before treatment or from previously treated patients at least 1 month after treatment [14,15,28]. Four studies continuously obtained samples both prior to treatment and at each visit during the follow-up period [9,16,18,25]. Three studies chose to obtain postoperative blood samples and follow-up samples [7,13,29]. The rest of the studies did not report the time at which the blood samples were collected.
Treatment
Treatment varied across studies. The different treatments included surgery, chemotherapy, targeted therapy, and radiotherapy as well as a number of other therapeutic intervention options.
Relationship between ctDNA and DFS
Eleven studies provided information regarding the association between ctDNA status and DFS [7,13,16,19,20,23,25,27,28,32,33]. Six of them calculated DFS according to the Kaplan-Meier method. Eight of 11 studies performed multivariable analyses to evaluate the independent prognostic effects of ctDNA on prognosis. However, adjustments for potential risk factors varied.
CtDNA mutations and DFS
Ryan et al. invested serum KRAS2 mutations in patients ranging from early to advanced CRC, and found that postoperative serum mutant KRAS2 was an independent factor of disease recurrence [7]. In fact, serum KRAS2 was stronger than the influence of Dukes’ stage or of treatment with adjuvant chemotherapy in the predict of CRC recurrence by a Cox regression multivariate analysis [7].
Tie et al. analyzed hotspot mutations in TP53, APC, KRAS, NRAS, BRAF, PIK3CA, CTNNB1, SMAD4, and FBXW7 in tumor tissue in CRC patients with stage II [32]. The identified mutations were then detected in plasma. An exploratory analysis of the correlation between ctDNA and clinicopathologic features showed that ctDNA was an independent factor, associated with a shorter recurrence-free survival (RFS) [32].
Lindforss et al. assessed the prognostic values of circulating KRAS in CRC patients with stagesI-III and found that no significant correlation between relapse of disease and KRAS mutation status in circulating DNA postoperatively on day three [25].
Wang et al. aimed to determined the presence of APC, KRAS, and P53 mutations in serum from CRC patients with Dukes’ stage A-D [28]. The correlation between the detection of ctDNA and the development of postoperative recurrence was significant (p<0.001) [28].
Tie et al. focused on TP53, APC, KRAS mutations in CRC patients with stage II [13]. They found that ctDNA detected postoperatively had a significantly reduced RFS (P<0.01). A multivariate analysis showed that ctDNA status was an independent variable to estimate the probability of RFS after adjustment for T stage, lymph node yield, and lymphovascular invasion [13].
CtDNA methylation and DFS
After controlling for the classic risk factors such as tumor size, lymph node status, and age at diagnosis, Herbst et al. showed that serum methylation of HLTF was associated with a high risk of disease recurrence, and serum methylation of HLTF proved to be an independent prognostic factor for patients with stages I-III in a multivariate analysis [27].
Lee et al. focused SEPT9 methylation (mSEPT9) in ctDNA among patients with stages I-II, and found that mSEP9 in ctDNA was significantly associated with lower DFS by univariate analysis. However, mSEP9 in ctDNA was not an independent prognostic factor in a multivariate analysis [24].
Tham et al. investigated the prognostic values of serum methylation of TAC1, SEPT9 and NELL1 among patients with stages I-II [16]. Their study showed that high serum methylation of TAC1 and SEPT9 but not NELL1 were independent predictors of unfavorable DFS after adjustment for vascular embolism, perineural invasion and serum CEA [16].
The detecting panel of Liu et al contained seven serum methylation markers: SST, MAL, TAC1, SEPT9, EYA4 and CRABP1 as well as NELL1 [20]. Only methylation of SST was found to be an independent predictor of unfavorable DFS in a multivariable Cox analysis, with adjustment for the effects of lymphovascular invasion, perineura invasion and serum CEA in combined group of stages II and III. When studying in individual subsets of stage II or III along, the researchers found that the statistical significance of serum methylation of SST was no longer retained [20].
Lin et al. explored the prognostic values of plasma AGBL4, FL11 and TWIST1 methylation in patients with stages I-IV. In univariate analysis, these three hypermathylated markers or serum GL11 hypermethylation were associated with poorer DFS (P<0.05). However, after stepwise elimination of tumor stage, lymphovascular invasion, preoperative CEA, mucinous histology and differentiation, none of these three markers were associated with patient outcome [19].
CtDNA mutation/methylation and DFS
Bazan et al. prospectively evaluated KRAS and TP53 mutations as well as p16INK4A methylation status in primary CRC patients [23]. The univariate analysis showed that only KRAS mutations were associated with quicker relapse (P<0.01) [23]. No multivariate analysis was performed in their study.
Relationship between ctDNA and OS
Thirteen studies assessing the relationship between ctDNA status and OS were eligible for the systematic review [6,8,9,12,14,17,18,20,21,23,26,29,34]. Eleven of them used the Kaplan-Meier method to estimate survival distribution curves [8,9,12,14,17,18,20,21,23,26,29]. Nine of 13 studies performed multivariate Cox regression analyses to explore the association between ctDNA status and OS [6,8,9,12,14,17,18,21,26].
CtDNA mutation and OS
Messaoudi et al. detected cfDNA KRAS and BRAF mutations in mCRC patients [34]. They found no statistic differences in OS between the KRAS-WT and KRAS-mutant mCRC patients. However, there was a statistically high significant difference between the median OS of BRAF-mutant patients and BRAF-WT patients. Compared to patients WT for BRAF V600E, BRAF-mutant mCRC patients had a shorter OS. A multivariate COX proportional hazards model showed that after controlling for CEA, tumor localization and age, BRAF-mutant status is a independent prognostic value (p = 0.002, HR = 7.33, 95% CI [1.04–2.89]) [34].
Spindler et al. came up with a quite different conclusion. They investigated the clinical implications of KRAS and BRAF mutations in ctDNA in patients with mCRC [9]. They found that KRAS mutations in ctDNA were independent prognostic factors and were associated with decreased OS by multivariate analysis, controlling for effects of age and performance status (PS). However, the effect of BRAF mutations in ctDNA on OS did not reach significance [9].
Sefrioui et al. analyzed KRAS mutations in ctDNA in patients with mCRC, and found that KRAS mutations in ctDNA were predictors of worse OS [29]. However, no multivariate analysis was performed.
Trevisiol et al. investigated the prognostic significance of circulating KRAS2 mutations in patients with Dukes’ A, B, C, D [6]. Serum KRAS2 mutations were significantly associated with a worse OS in multivariate analysis when adjusting for effects of CEA and stage [6].
Bai et al assessed the prognostic value of KRAS mutations in ctDNA in mCRC [12]. They classified ctDNA in a trichotomized manner (wild-type group, low-mutation group, and high-mutation group) and found that KRAS mutations were significantly associated with poorer prognosis by a multivariate analysis after adjusting for performance status and times of surgery as well as metastatic sites [12].
A panel of 74 genes were detected in circulating DNA in patients with stages I-III by Lin et al. [14]. They conducted a univariate analysis and found that the 5-year OS among patients who harbored a mutation in circulating DNA was significantly poorer than those without a ctDNA mutation. However, a multivariate analysis showed that the mutation status of ctDNA was no longer associated with patients’ survival when controlling for TNM stage, status of lymphovascular invasion and preoperative CEA level [14].
CtDNA methylation and OS
Leung et al. tested the role of serum methylation of APC, hMHL1 and HLTF in CRC with stages I-IV [17], and found that none of these three markers individually were associated with OS. When combining these three markers together, there is a trend to a poor chance of OS in patients harboring any one of these three markers (p = 0.08) [17].
Liu et al. assessed the prognostic potentials of methylation SST, NMV, MAL, TAC1, SEPT9 and EYA4 in the serum of CRC patients with stages I-IV [20]. Univariate analyses showed that serum methylation levels of MAL and SST were significantly predictive of cancer-specific death. Multivariate Cox regression model revealed the independent prognostic effect of serum methylation of SST on OS, adjusting for stage, perineural invasion, lymphovascular invasion [20]. The effects of serum methylation of SST were also investigated based on tumor stages. In the combined group of stages II and III, serum mSST remained as a significant independent predictor of worse OS (HR = 2.797, 95% CI, 1.34–5.84; P = 0.006) [20]. When analyzed in patients with each stage individually, independent predictive effect of serum mSST on OS remained significance only in stage III subgroup (HR = 2.52, 95% CI, 1.02–6.25; P = 0.045) [20].
Philipp et al. focused on serum methylation of HLTF and HPP1 in CRC patients with stages I-IV [26]. Their results showed that the presence of HLTF or HPP1 was significantly associated with poorer prognosis [26]. The researchers then examined the prognostic values of serum HLTF and HPP1 methylation in subgroup analyses according to tumor stage. Serum HLTF but not HPP1 methylation was associated with shorter survival in patients with stage I. Neither serum HLTF or HPP1 methylation was associated with OS in patients with stages II and III or in patients with the combined stages I-III. In patients with stage IV, serum HLTF and HPP1 methylation conferred a significantly worse OS. A multivariate analysis showed that the methylation of HLTF and the methylation of HPP1 were independent prognostic factors in patients with stage IV, whereas controlling for effects of CEA.
Wallner et al. studied the prognostic potential of ctDNA methylation in CRC patients with stage I-III [18]. Three genes HPP1/TPEF, HLTF and hMLH1 were studied to assess their prognostic effects on OS. The multivariate analysis showed that HPP1/TPEP and HLTF along or in combination were independent prognostic factor of worse OS after adjusting for lymph node metastases, distant metastases, age, tumor size [18].
Matthaios et al. investigated the prognostic value of the methylation status of APC and RASSF1A in ctDNA in patients with early operative CRC and metastatic CRC [21]. Multivariate Cox proportional hazards regression analysis was performed to investigate the independent effects of methylated APC and RASSF1A promoter status on OS, adjusting for patients’ gender, age, clinical stage, tumor differentiation, lymph node status, CEA and CA199 levels. The results showed that methylated APC promoter status have a significant negative impact on the survival of patients with (adjusted HR = 3.47, 95% CI 1.35–8.92, P = 0.017) or without metastases (adjusted HR = 7.88, 95% CI = 2.73–22.73, P<0.001). Methylated RASSF1A promoter status was also associated with a worse survival in patients with (adjusted HR = 5.76, 95% CI 2.44–14.82, P = 0.001) or without metastases (adjusted HR = 3.06, 95% CI 1.25–7.50, P = 0.038)(21).
CtDNA mutation/methylation and OS
Lecome et al. focused on KRAS2 mutations and p16 hypermethylation in CRC patients with stages I-III [8]. For OS, patients with dateable ctDNA had a significantly worse survival than those without detectable ctDNA by the univariate analysis. When adjusting for TNM’s stage, the multivariate analysis revealed that this significance disappeared [8].
Bazan et al. investigated whether the detection of TP53, KRAS mutations and p16INK4A methylation in ctDNA was associated with OS [23]. Their results showed that no significant association had been found between these alterations in ctDNA and OS [23].
Discussion
To the best of our knowledge, this is the first systematic review to explore the relationship between ctDNA status and prognosis in patients with CRC.
Most papers included in our review found that ctDNA was associated with a worse DFS/OS in patients with CRC. Some of them declared that ctDNA had an independent significant effect on patients’ prognosis. Some showed that not all of genetic/epigenetic alterations detected in ctDNA in their detection panels were correlated with patients’ prognosis. A few studies showed that the prognostic effects of ctDNA could only be found in certain patients, such as patients with advanced stages. The divergent conclusions of the included papers may result from detection panels, patients, detection methods, matrixes (plasma or serum), treatment and sampling time.
In our systematic review, one of the major confounding factors was the absence of a standardized definition of ctDNA. Additionally, there was no consensus regarding the detecting panel, marker threshold, detection method, time of sample collection, or sample type. The included studies revealed that both genetic and epigenetic alterations could be detected in ctDNA in patients with CRC. It was reported that certain genetic alterations such as KRAS mutations in ctDNA were highly specific for colorectal neoplasia. Compared with genetic alterations, epigenetic alterations appeared to be less specific as tumor markers. Epigenetic alterations could also be detected in non-tumor cells, and their frequency may increase in older patients [35]. Additionally, the frequency of methylation varies in different suppressor tumor genes and changes with tumor stage. In most studies, aberrant hypermethylation of ctDNA was found to be associated with a worse prognosis. However, contradicting results were also reported by other studies because of the use of different detecting panels. Because of a lack of a recommended standard marker panel, most studies chose markers that had been reported to occur more frequently in patients with CRC than in healthy patients. In our included studies, the number and type of alterations varied. Hence, in future studies, a standard marker panel with both satisfactory sensitivity and specificity will be needed.
The low level of ctDNA is another challenge for successful detection. In early studies, ctDNA alterations were in low agreement with those in tumor tissue because of the low levels of ctDNA. For example, a study by Lecomte et al. found that serum alterations could be detected in less than half of the patients harboring tissue alterations [8]. The discordance in the alteration detection rate between tumor tissue and plasma or serum could be resolved by high-sensitivity detection methods. In our included studies, the detection sensitivity varied according to the detection method. Regarding KRAS mutation detection, for example, the sensitivity and specificity of next-generation sequencing-based SafeSeq technology were 87.2 and 99.2%, respectively, whereas for the optimized clamp-PCR Intplex test, the sensitivity and specificity were 92 and 98%, respectively [5,36]. Droplet-based PCR had a sensitivity of 80%. In chip-based digital PCR, only 69% of KRAS-positive patients were detected [29], whereas ARMS-based PCR methodology could detect a mutant gene with a frequency as low as 0.1% [37]. Inevitably, a low-sensitivity method may miss low amounts of tumor-associated alterations in circulating. Data should be interpreted with caution because low sensitivity methods can only detect ctDNA in patients if present at a high level.
Sampling time also has an important effect on the successful detection of ctDNA. CtDNA dynamically changes during therapy [38]. It has been reported that previously detected mutations are undetectable one week after surgery. In chemotherapy-responsive patients, ctDNA levels can decrease or increase to detectable levels [39,40]. Tumor-associated alterations in circulating DNA may emerge during the follow-up period [7]. In vivo, ctDNA has a short half-life. Its appearance indicates that an occult tumor exists after therapy. Another aspect that needs to be addressed is clonal selection. Clonal selection is normally induced by treatment and causes the ratio between wild-type and mutated cells to change over time [41]. Taking these variables into consideration, the timing of the blood sample collection is critical for successful detection. Hence, it is necessary to continuously collect blood samples from patients both before and at least 1 month after therapy.
In the eligible studies, either serum or plasma was used for ctDNA detection. Strictly speaking, the DNA content of serum and plasma can be drastically different. Serum samples contain higher total free DNA yields, perhaps because of the release of DNA caused by cell lysis during coagulation. This large amount of non-tumor DNA released by normal cells in serum means that the fraction of tumor DNA in serum is lower than that in plasma. This difference may affect ctDNA detection.
Our comprehensive review revealed that a ctDNA-positive status is associated with a worse prognosis. However, some limitations should be considered when interpreting our findings. The major limitations included clinical and methodological heterogeneity. Although some studies excluded patients with familial adenomatous polyposis or hereditary non-polyposis colorectal cancer [6,15], the detection panels varied across the studies. Additionally, the different follow-up times, detection methods, and intrinsic differences in treatment regimens and blood sample collection times also contributed to the heterogeneity. Some included studies only analyzed ctDNA status in patients with tissue alterations [8,15]. Some studies included in our review investigated the prognosis effects of ctDNA by a multivariate analysis. However, the confounders adjusted for also differed between studies. To include the maximal number of relevant studies, we treated RFS and DFS synonymously, knowing that a small percentage of patients with secondary primary cancers could be included.
The current gold standards in determining prognosis in CRC are Dukes’ staging and TNM staging [42]. However, for patients in intermediate groups (stage II-III tumors), these staging systems are less informative. Until now, only the serum biomarker carcinoembryonic antigen has been confirmed to provide prognostic information in Dukes’ B, or an equivalent, stage of CRC. One included study showed that after adjustment for other significant factors, the correlation between ctDNA and prognosis was still strong, stronger than Dukes’ stage of disease [7].
Reports illustrated that tissue genotyping could contribute to prognostic and predictive biomarker evaluation in patients with CRC [43–45]. Because of inherent tumor heterogeneity and invasive sampling, tumor tissue genotyping has limitations. Other studies suggested that the primary tumor has a different genomic landscape than metastases. Additionally, the invasive nature of the procedure means continuous sampling cannot be performed. Mutated DNA in circulating is of tumor origin. It was reported that plasma mutations have better prognostic value than tumor tissue mutations [9]. Lecomte et al. found that among patients with tissue alterations, ctDNA alteration-positive patients had a worse prognosis [8]. Furthermore, a study by Philipp et al. uncovered that the presence of ctDNA was correlated with carcinoembryonic antigen expression and that ctDNA status was a stronger prognostic biomarker than tumor stage in CRC. These studies shed some light on the prognostic value of ctDNA in CRC.
In summary, this is the first comprehensive analysis to assess the prognostic value of ctDNA in patients with CRC using the currently available literature. The findings of this systematic review strongly suggest that patients with ctDNA-positive CRC have an unfavorable prognosis. In view of some of the limitations of this study, we conclude that a large cooperative study is needed to address the prognostic and predictive value of ctDNA in patients with tumors.
Supporting information
(TIFF)
(DOCX)
(DOCX)
(DOC)
Acknowledgments
There was no funding source associated with the study design, collection analysis, interpretation of data, or writing of the report. We thank Dr. Sun Min for his kind help with the data analysis.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
There was no funding source associated with the study design, collection analysis, interpretation of data, or writing of the report.
References
- 1.Warton K, Samimi G. Methylation of cell-free circulating DNA in the diagnosis of cancer. Front Mol Biosci. 2015;2: 13 10.3389/fmolb.2015.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol Off J Am Soc Clin Oncol. 2014;32: 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heitzer E, Auer M, Ulz P, Geigl JB, Speicher MR. Circulating tumor cells and DNA as liquid biopsies. Genome Med. 2013;5: 73 10.1186/gm477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13: 795–806. 10.1038/nrg3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6: 224ra24 10.1126/scitranslmed.3007094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trevisiol C, Di Fabio F, Nascimbeni R, Peloso L, Salbe C, Ferruzzi E, et al. Prognostic value of circulating KRAS2 gene mutations in colorectal cancer with distant metastases. Int J Biol Markers. 2006;21: 223–228. [DOI] [PubMed] [Google Scholar]
- 7.Ryan BM, Lefort F, McManus R, Daly J, Keeling PWN, Weir DG, et al. A prospective study of circulating mutant KRAS2 in the serum of patients with colorectal neoplasia: strong prognostic indicator in postoperative follow up. J Clin Pathol-Mol Pathol. 2003;56: 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lecomte T, Berger A, Zinzindohoue F, Micard S, Landi B, Blons H, et al. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int J Cancer. 2002;100: 542–548. 10.1002/ijc.10526 [DOI] [PubMed] [Google Scholar]
- 9.Spindler KG, Appelt AL, Pallisgaard N, Andersen RF, Jakobsen A. KRAS-mutated plasma DNA as predictor of outcome from irinotecan monotherapy in metastatic colorectal cancer. Br J Cancer. 2013;109: 3067–3072. 10.1038/bjc.2013.633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jo P, Jung K, Grade M, Conradi LC, Wolff HA, Kitz J, et al. CpG island methylator phenotype infers a poor disease-free survival in locally advanced rectal cancer. Surgery. 2012;151: 564–570. 10.1016/j.surg.2011.08.013 [DOI] [PubMed] [Google Scholar]
- 11.Higgins J, Green S, Editors. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]. Cochrane Libr. 2005; (3).
- 12.Bai YQ, Liu XJ, Wang Y, Ge FJ, Zhao CH, Fu YL, et al. [Correlation analysis between abundance of K-ras mutation in plasma free DNA and its correlation with clinical outcome and prognosis in patients with metastatic colorectal cancer]. Zhonghua Zhong Liu Za Zhi. 2013;35: 666–671. [PubMed] [Google Scholar]
- 13.Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8: 346ra92 10.1126/scitranslmed.aaf6219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin J-K, Lin P-C, Lin C-H, Jiang J-K, Yang S-H, Liang W-Y, et al. Clinical relevance of alterations in quantity and quality of plasma DNA in colorectal cancer patients: based on the mutation spectra detected in primary tumors. Ann Surg Oncol. 2014;Suppl 4: S680–686. [DOI] [PubMed] [Google Scholar]
- 15.Lefebure B, Charbonnier F, Di Fiore F, Tuech JJ, Le Pessot F, Michot F, et al. Prognostic value of circulating mutant DNA in unresectable metastatic colorectal cancer. Ann Surg. 2010;251: 275–280. 10.1097/SLA.0b013e3181c35c87 [DOI] [PubMed] [Google Scholar]
- 16.Tham C, Chew M, Soong R, Lim J, Ang M, Tang C, et al. Postoperative serum methylation levels of TAC1 and SEPT9 are independent predictors of recurrence and survival of patients with colorectal cancer. Cancer. 2014;120: 3131–3141. 10.1002/cncr.28802 [DOI] [PubMed] [Google Scholar]
- 17.Leung WK, To KF, Man EPS, Chan MWY, Bai AHC, Hui AJ, et al. Quantitative detection of promoter hypermethylation in multiple genes in the serum of patients with colorectal cancer. Am J Gastroenterol. 2005;100: 2274–2279. 10.1111/j.1572-0241.2005.50412.x [DOI] [PubMed] [Google Scholar]
- 18.Wallner M, Herbst A, Behrens A, Crispin A, Stieber P, Göke B, et al. Methylation of Serum DNA Is an Independent Prognostic Marker in Colorectal Cancer. Clin Cancer Res. 2006;12: 7347–7352. 10.1158/1078-0432.CCR-06-1264 [DOI] [PubMed] [Google Scholar]
- 19.Lin P-C, Lin J-K, Lin C-H, Lin H-H, Yang S-H, Jiang J-K, et al. Clinical Relevance of Plasma DNA Methylation in Colorectal Cancer Patients Identified by Using a Genome-Wide High-Resolution Array. Ann Surg Oncol. 2015;Suppl 3: S1419–S1427. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Chew MH, Tham CK, Tang CL, Ong SY, Zhao Y. Methylation of serum SST gene is an independent prognostic marker in colorectal cancer. Am J Cancer Res. 2016;6: 2098–2108. [PMC free article] [PubMed] [Google Scholar]
- 21.Matthaios D, Balgkouranidou I, Karayiannakis A, Bolanaki H, Xenidis N, Amarantidis K, et al. Methylation status of the APC and RASSF1A promoter in cell-free circulating DNA and its prognostic role in patients with colorectal cancer. Oncol Lett. 2016;12: 748–756. 10.3892/ol.2016.4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong ALA, Lim JSJ, Sinha A, Gopinathan A, Lim R, Tan CS, et al. Tumour pharmacodynamics and circulating cell free DNA in patients with refractory colorectal carcinoma treated with regorafenib. J Transl Med [Internet]. 2015;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bazan V, Bruno L, Augello C, Agnese V, Calo V, Corsale S, et al. Molecular detection of TP53, Ki-Ras and p16INK4A promoter methylation in plasma of patients with colorectal cancer and its association with prognosis. Results of a 3-year GOIM (Gruppo Oncologico dell’Italia Meridionale) prospective study. Ann Oncol. 2006;17 Suppl 7: vii84–90. [DOI] [PubMed] [Google Scholar]
- 24.Lee HS, Hwang SM, Kim TS, Kim DW, Park do J, Kang SB, et al. Circulating methylated septin 9 nucleic Acid in the plasma of patients with gastrointestinal cancer in the stomach and colon. Transl Oncol. 2013;6: 290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindforss U, Zetterquist H, Papadogiannakis N, Olivecrona H. Persistence of K-ras mutations in plasma after colorectal tumor resection. Anticancer Res. 2005;25: 657–661. [PubMed] [Google Scholar]
- 26.Philipp AB, Stieber P, Nagel D, Neumann J, Spelsberg F, Jung A, et al. Prognostic role of methylated free circulating DNA in colorectal cancer. Int J Cancer. 2012;131: 2308–2319. 10.1002/ijc.27505 [DOI] [PubMed] [Google Scholar]
- 27.Herbst A, Wallner M, Rahmig K, Stieber P, Crispin A, Lamerz R, et al. Methylation of helicase-like transcription factor in serum of patients with colorectal cancer is an independent predictor of disease recurrence. Eur J Gastroenterol Hepatol. 2009;2: 565–569. [DOI] [PubMed] [Google Scholar]
- 28.Wang J-Y, Hsieh J-S, Chang M-Y, Huang T-J, Chen F-M, Cheng T-L, et al. Molecular detection of APC, K- ras, and p53 mutations in the serum of colorectal cancer patients as circulating biomarkers. World J Surg. 2004;28: 721–726. 10.1007/s00268-004-7366-8 [DOI] [PubMed] [Google Scholar]
- 29.Sefrioui D, Sarafan-Vasseur N, Beaussire L, Baretti M, Gangloff A, Blanchard F, et al. Clinical value of chip-based digital-PCR platform for the detection of circulating DNA in metastatic colorectal cancer. Dig Liver Dis [Internet]. 2015;47: 884–890 [DOI] [PubMed] [Google Scholar]
- 30.Tie J, Wang Y, Kinde I, Steel M, Elsaleh H, Singh MS, et al. Circulating tumor DNA (ctDNA) in nonmetastatic colorectal cancer (CRC): Potential role as a screening tool. J Clin Oncol [Internet]. 2015;33: 518 [Google Scholar]
- 31.El Messaoudi S, Mouliere F, Mollevi C, Gillet B, Nouaille M, Loriot V, et al. Circulating DNA as a strong multimarker prognostic tool in metastatic colorectal cancer patients. J Clin Oncol [Internet]. 2014;32. [DOI] [PubMed] [Google Scholar]
- 32.Tie J, Kinde I, Wang Y, Wong HL, Skinner I, Wong R, et al. Circulating tumor DNA (ctDNA) as a marker of recurrence risk in stage II colon cancer (CC). J Clin Oncol [Internet]. 2014;32: 11015. [Google Scholar]
- 33.Lee CK, Brown C, Gralla RJ, Hirsh V, Thongprasert S, Tsai C-M, et al. Impact of EGFR Inhibitor in Non-Small Cell Lung Cancer on Progression-Free and Overall Survival: A Meta-Analysis. JNCI J Natl Cancer Inst. 2013;105: 595–605. 10.1093/jnci/djt072 [DOI] [PubMed] [Google Scholar]
- 34.El Messaoudi S, Mouliere F, Du Manoir S, Bascoul-Mollevi C, Gillet B, Nouaille M, et al. Circulating DNA as a Strong Multimarker Prognostic Tool for Metastatic Colorectal Cancer Patient Management Care. Clin Cancer Res Off J Am Assoc Cancer Res. 2016;22: 3067–3077. [DOI] [PubMed] [Google Scholar]
- 35.Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32: 579–586. 10.1200/JCO.2012.45.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20: 430–435. 10.1038/nm.3511 [DOI] [PubMed] [Google Scholar]
- 37.Fox J. C. E J. The detection of K-ras mutations in colorectal cancer using the amplification-refractory mutation system. Br J Cancer. 1998; 77: 1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mok T, Wu Y-L, Lee JS, Yu C-J, Sriuranpong V, Sandoval-Tan J, et al. Detection and Dynamic Changes of EGFR Mutations from Circulating Tumor DNA as a Predictor of Survival Outcomes in NSCLC Patients Treated with First-line Intercalated Erlotinib and Chemotherapy. Clin Cancer Res. 2015;21: 3196–3203. 10.1158/1078-0432.CCR-14-2594 [DOI] [PubMed] [Google Scholar]
- 39.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368: 1199–1209. 10.1056/NEJMoa1213261 [DOI] [PubMed] [Google Scholar]
- 40.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14: 985–990. 10.1038/nm.1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486: 537–540. 10.1038/nature11219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duffy MJ, van Dalen A, Haglund C, Hansson L, Klapdor R, Lamerz R, et al. Clinical utility of biochemical markers in colorectal cancer: European Group on Tumour Markers (EGTM) guidelines. Eur J Cancer. 2003;39: 718–727. [DOI] [PubMed] [Google Scholar]
- 43.Juo YY, Johnston FM, Zhang DY, Juo HH, Wang H, Pappou EP, et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol. 2014;25: 2314–2327. 10.1093/annonc/mdu149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9: 962–972. 10.1016/S1470-2045(08)70206-7 [DOI] [PubMed] [Google Scholar]
- 45.Qiu LX, Mao C, Zhang J, Zhu XD, Liao RY, Xue K, et al. Predictive and prognostic value of KRAS mutations in metastatic colorectal cancer patients treated with cetuximab: a meta-analysis of 22 studies. Eur J Cancer. 2010;46: 2781–2787. 10.1016/j.ejca.2010.05.022 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIFF)
(DOCX)
(DOCX)
(DOC)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.