

Converter HCM mutations cause only small changes in the biophysics of myosin, suggesting an alternative mechanism for hypercontractility.
Keywords: myosin, cardiac myosin, converter domain, hypertrophic cardiomyopathy, cardiomyopathy, contractility
Abstract
Hypertrophic cardiomyopathy (HCM) affects 1 in 500 individuals and is an important cause of arrhythmias and heart failure. Clinically, HCM is characterized as causing hypercontractility, and therapies are aimed toward controlling the hyperactive physiology. Mutations in the β-cardiac myosin comprise ~40% of genetic mutations associated with HCM, and the converter domain of myosin is a hotspot for HCM-causing mutations; however, the underlying primary effects of these mutations on myosin’s biomechanical function remain elusive. We hypothesize that these mutations affect the biomechanical properties of myosin, such as increasing its intrinsic force and/or its duty ratio and therefore the ensemble force of the sarcomere. Using recombinant human β-cardiac myosin, we characterize the molecular effects of three severe HCM-causing converter domain mutations: R719W, R723G, and G741R. Contrary to our hypothesis, the intrinsic forces of R719W and R723G mutant myosins are decreased compared to wild type and unchanged for G741R. Actin and regulated thin filament gliding velocities are ~15% faster for R719W and R723G myosins, whereas there is no change in velocity for G741R. Adenosine triphosphatase activities and the load-dependent velocity change profiles of all three mutant proteins are very similar to those of wild type. These results indicate that the net biomechanical properties of human β-cardiac myosin carrying these converter domain mutations are very similar to those of wild type or are even slightly hypocontractile, leading us to consider an alternative mechanism for the clinically observed hypercontractility. Future work includes how these mutations affect protein interactions within the sarcomere that increase the availability of myosin heads participating in force production.
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is the most frequently occurring inherited cardiac disease, affecting more than 1 in 500 individuals (1), and 10% of HCM patients develop fatal arrhythmia and/or heart failure (2). It is characterized by left ventricular hypertrophy in the absence of predisposing conditions (for example, aortic stenosis, hypertension), ultimately causing decreased left ventricular chamber volume and obstructive physiology during systole. In 1990, Geisterfer-Lowrance et al. (3) reported a missense mutation, R403Q, in the β-cardiac myosin heavy chain gene (MYH7) in a cohort of HCM patients, and since then, hundreds of different mutations in not only myosin but also other sarcomeric proteins [for example, myosin binding protein-C (MyBP-C), troponin I, and cardiac actin] have been identified. It is estimated that missense mutations in the β-cardiac myosin heavy chain (MyHC) are responsible for ~40% of cases of genotype-positive HCM (4, 5). The principle pathology is manifested at the level of the ventricle in HCM (6), and the conventional view is that HCM mutations result in hyperdynamic cardiovascular physiology that is often seen as a supranormal ejection fraction (EF) on echocardiograms (7, 8). However, the molecular mechanisms that determine how missense mutations in the human β-cardiac MyHC cause the hypercontractile phenotype remain unclear. In particular, although there has been more understanding of secondary cellular events that occur during the hypertrophic process, the primary effect of the mutations on the function of the sarcomeric contractile proteins is largely unknown. Hence, there is a paucity of the knowledge that is essential for the development of new therapeutic treatments for this deadly disease, as well as for predicting the phenotypic outcomes in this heterogeneous patient population.
To understand the primary effect of HCM mutations, we focused on the molecular parameters of the contractile properties at the sarcomere level and measured the effect of HCM-causing mutations using in vitro reconstituted systems. These parameters include the following: (i) the intrinsic force (Fintrinsic) of myosin, which can be increased or decreased by mutations that change the spring constant of the elastic element of the motor; (ii) the duty ratio of the myosin, which is the fraction of myosin bound to actin in a force-producing state in the sarcomere at any moment during systole, estimated from the time myosin spends strongly bound to actin (termed ts) divided by the total actin-activated myosin adenosine triphosphatase (ATPase) cycle time (tc) [the duty ratio can be changed in more than one way, such as a change in the adenosine 5′-diphosphate (ADP) release rate (which determines ts) or the weak-to-strong transition time (which determines tc).]; and (iii) the velocity of actin displacement by an ensemble of myosin molecules, which is related to the stroke size (d) divided by the time myosin spends on actin (ts). Finally, the force produced by a given sarcomere can be expressed as the ensemble force (Fensemble), which is the effective force produced by a given sarcomere, expressed as a function of Fintrinsic, the duty ratio of the myosin, and the total number of functionally available myosin heads overlapping with the actin in the sarcomere (Na): Fensemble = Fintrinsic(ts/tc)(Na). The experimental setups for measuring these parameters have been rigorously explored previously (9–11). Our overall hypothesis has been that HCM-causing mutations increase one or more of these parameters and thereby increase the power output of the cardiac muscle, which is the Fensemble times the velocity of contraction. We (10–13) and others (14, 15) have proposed that such changes serve as the initial trigger for the pathologic hypertrophic process.
Early studies using reconstituted systems used mouse α-cardiac myosin as a model system. Those studies showed considerable increases in either ATPase activity, velocity of actin filament gliding in in vitro motility assays, or Fintrinsic (14, 15), and hypercontractility was easily explained by the significant increases in these fundamental parameters. However, more than 80 residues are different between mouse α-cardiac and human β-cardiac myosin; it has been reported that the same HCM mutation (R403Q) caused different mechanochemical effects on cardiac myosin depending on the myosin isoform in mouse (16), and there is a significant functional difference between human α-cardiac and β-cardiac myosin (11). Thus, it is important to examine the effects of the HCM mutations in the proper backbone, by using human β-cardiac myosin. Recent studies using human β-cardiac myosin carrying the R453C mutation (10) or the R403Q mutation (12) failed to show these large changes in the same parameters, and whether the net result of the changes observed was contributing to a hypercontractile or hypocontractile state was difficult to assess. Meanwhile, two mutations in human β-cardiac myosin that are associated with early-onset (pediatric) HCM (H251N and D239N) showed a marked increase in mechanochemical function, including ATPase activity, actin gliding velocity, and intrinsic force (17), making a case that hypercontractility manifests at the molecular level, leading to severe HCM phenotype.
Because early-onset HCM mutations are relatively rare, further investigation of more commonly occurring mutations that typically result in adult HCM is necessary to strengthen the argument that HCM mutations result in hypercontractility at the fundamental molecular level. We therefore sought to examine these fundamental parameters for human β-cardiac myosins carrying mutations in the converter region of the molecule, which is a hotspot for HCM mutagenesis (18). To date, every variant known in the converter region in the human population results in HCM pathogenesis (18), which is not true of any other region on the myosin molecule. Generally, HCM-causing mutations in the converter have been reported to result in worse clinical outcomes, whereas a wide range of phenotypes have been observed (19, 20). Prior studies using mammalian and Drosophila muscle myosin showed that the converter is involved in determining the stiffness of the cross-bridge bound to the actin filament (21) and, therefore, the intrinsic force–generating ability of the head. Converter movement is coupled to adenosine 5′-triphosphate (ATP) hydrolysis, phosphate (Pi) release, and the force-generating event (22, 23). Furthermore, converter movement is coupled to load-dependent ADP release, and thus ADP affinity could be changed, which would affect the maximum velocity of the motor, the duty ratio, and, hence, the net Fensemble (22–25). Thus, the converter is pivotal in transducing the energy of ATP hydrolysis to the stroke of the lever arm and force production, and one might suspect that Fintrinsic or one of the other fundamental parameters would show significant increases compared to wild type (WT). Interestingly, this is not what we have found, as shown in this report, and our results suggest that we need to rethink the molecular basis for the clinically observed hypercontractility caused by myosin-based HCM mutations.
Three HCM mutations were selected for this study: R719W is a mutation found in the early days of HCM genetics and has been characterized to be one of the most lethal mutations in MYH7, the gene encoding the β-cardiac MyHC (26). R723G is a mutation first reported in families in Barcelona who manifested progressive heart failure as well as sudden death (27). G741R is also one of the longest recognized mutations found in multiple families (28). Prior studies have shown that slow skeletal muscle fibers from soleus, which use the same human β-cardiac MyHC as the heart, albeit different light chains, containing the R719W mutation from HCM patients showed increased isometric force, increased stiffness in rigor, and increased ATPase activity, but no change in kinetics of active cross-bridge cycling (21, 29). A mouse model harboring the R719W mutation in α-cardiac myosin, generated by the Seidman group (30), was shown to have activation of proliferative and profibrotic signals in nonmyocyte cells that promotes pathological remodeling leading to HCM (30). The R723G mutation has been studied using human skeletal muscle biopsy samples and showed similar changes, whereas there was no change in ATPase activity (29). These mutations are also known to cause variable calcium sensitivity (31). A recent study by Kraft et al. (32) showed that in mechanically isolated human cardiomyocytes from biopsy samples with the R723G mutation, maximum force was significantly lower and Ca2+ sensitivity was unchanged. Conversely, R723G mutant soleus fibers showed significantly higher maximum force and reduced Ca2+ sensitivity than controls. These discrepancies were speculated to be due to the changes in protein phosphorylation in different muscle types (32). G741R myosin was also studied from human soleus fibers and showed decreased velocity and force production (33), whereas a study using a mouse myoblast cell line showed increased tetanic fusion frequency without significant impact on the contractile kinetics (34). These previous studies suggest that there is a significant impact on myosin function when converter domain residues are altered (22–25, 35), although conclusions are variable and complicated by differences in light chain composition, the isoform that is being studied, posttranslational modifications, disease state of the tissue used, and so forth. To date, there has been no report studying the effect of converter domain mutations on biomechanical properties of myosin function using purified human β-cardiac myosin. Here, we report the primary effects of these mutations in the converter domain of human β-cardiac myosin on fundamental parameters of power output using homogeneous, expressed, and purified human β-cardiac short subfragment 1 (sS1), the motor domain of myosin. Our approach enables the direct comparison of WT and HCM mutant forms of human β-cardiac myosin and eliminates the background variability inherent in using patient-derived samples that may contain nonsynonymous variants (36).
RESULTS
The intrinsic forces of R719W and R723G human β-cardiac sS1s are slightly decreased, whereas G741R shows no change compared to WT myosin
For all of our experiments, we used the purified sS1 construct of human β-cardiac myosin from residues 1 to 808 (Fig. 1A and fig. S1A), which contains the catalytic domain and a short lever arm with only the human β-cardiac myosin essential light chain (ELC) and is known to be the motor domain of the molecule (37–39). The same construct was used previously in other mutational studies of HCM (10–12).
Fig. 1. Structure of a homology-modeled human β-cardiac sS1 domain and intrinsic force measurements for the three converter domain HCM mutant forms of the protein.
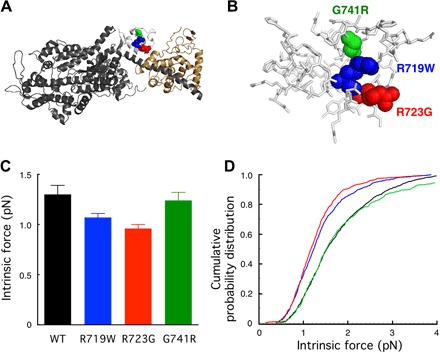
(A) Structure of the homology-modeled (see Materials and Methods) human β-cardiac sS1 containing residues 1 to 808 of the MyHC (dark gray) and the ELC (beige) based on the structure of Winkelmann et al. (56). Positions of the HCM mutations R719W (blue), R723G (red), and G741R (green) in the converter domain (white). (B) Blowup of the converter domain shown in (A), with all residues shown in stick form except for the three mutated residues. (C) Intrinsic force measurements using a dual-beam laser trap. More than three independent protein preparations were made for each mutant and for WT human β-cardiac sS1; 800 to 900 binding events of six to seven individual molecules were analyzed. P = 0.0067, one-way analysis of variance (ANOVA). The error bars show the SEM. (D) Cumulative probability distributions from the same intrinsic force measurements shown in (C).
For reasons outlined in the introduction, we expected that the converter mutations R719W, R723G, and G741R (Fig. 1, A and B) might cause an increase in Fintrinsic of the human β-cardiac sS1 and thereby explain the clinical hypercontractility caused by HCM mutations. We used a single-molecule laser trap (9, 10, 12) to measure the intrinsic forces of the three converter HCM mutations compared to the WT human β-cardiac sS1 (see Materials and Methods).
The force data were analyzed by multiple methods to confirm that the comparison of force-producing ability between WT and mutants is unbiased (see Materials and Methods). Figure 1C shows the summary of average Fintrinsic measurements for the three converter domain mutant proteins and WT. Compared to WT, R719W and R723G showed ~15 to 30% reduction in Fintrinsic, whereas there was no significant difference for G741R human β-cardiac sS1. An alternative way to approach the data is to look at the cumulative probability distribution of all individual intrinsic forces measured (Fig. 1D). This measures the probability of occurrence of any particular force event; for example, at a cumulative probability distribution of 0.5, the intrinsic force value was lower for R719W and R723G than for WT, suggesting that single molecules of R719W and R723G are more probable to generate a lower intrinsic force than WT. Thus, these changes would contribute to either no change (G741R) or a small hypocontractility contribution (R719W and R723G) toward power output.
ATPase activity was not affected by the converter domain mutations
Next, we compared the actin-activated ATPase activity of recombinant sS1 carrying the converter domain mutations with that of WT human β-cardiac sS1 (for example, see Fig. 2A and fig. S1, B and C). The actin-activated myosin ATPase activity of each mutant myosin was normalized to the WT myosin from protein preparations made simultaneously to minimize the effect of variability between protein batches (Fig. 2B). All three mutations showed no significant change in the maximal rate of actin-activated myosin ATP hydrolysis (kcat) (Fig. 2B and fig. S2), and there were no significant differences in Km for actin for any of the mutants (20 to 30 μM actin). The inverse of kcat defines the overall cycle time tc. Together, the tc of the ATPase cycle was unchanged with the converter domain mutations. Thus, both the Fintrinsic and ATPase measurements for the three converter HCM mutations failed to reveal changes in parameters that could account for the hypercontractility seen clinically for myosin-based HCM mutations.
Fig. 2. Actin-activated ATPase activity of recombinant human β-cardiac sS1 with mutations in the converter domain compared with WT human β-cardiac sS1.
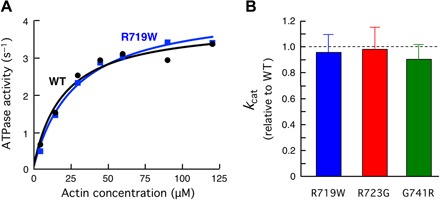
(A) Representative actin-activated sS1 ATPase curves. (B) Mutant human β-cardiac sS1 kcat values relative to WT (error bars: 95% confidence interval).
Actin and regulated thin filament gliding velocities were faster in R719W and R723G human β-cardiac sS1 and unchanged in the G741R myosin
Because power output is the product of Fensemble and velocity, we next assessed the mechanical property of the myosins with HCM mutations using the in vitro motility assay, where sS1 was anchored to surface-attached PDZ domain peptides that bind tightly to an eight-residue affinity clamp sequence on the C terminus of the sS1 (see Materials and Methods). A significant increase in velocity by the mutant proteins could offset the apparent small decrease in Fensemble, resulting in a net increase in power output. We therefore measured the velocities of actin filaments and regulated thin filaments (RTFs; consisting of actin saturated with the tropomyosin-troponin complex) gliding along surfaces coated with the various myosin preparations.
The actin/RTF filaments were tracked using the previously reported FAST (Fast Automated Spud Trekker) program (11). FAST enables tracking and analysis of hundreds of actin filaments and provides a plot of filament length-velocity relationships as well as histograms (for example, see Fig. 3A). Here, we focus on three parameters: the top 5% of velocities for each myosin preparation (TOP5%), the mean velocity (MVEL), and the percent of the filaments that are mobile (% mobile) (see Materials and Methods).
Fig. 3. In vitro motility data for the three mutant human β-cardiac sS1 proteins compared to WT.
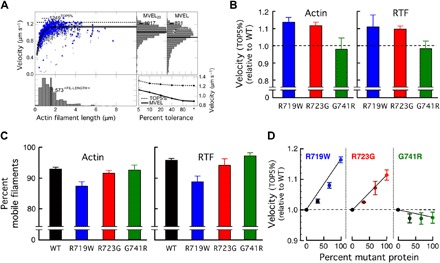
(A) An example of automatic analysis of an in vitro motility movie with FAST (11) (available for download at http://spudlab.stanford.edu). Scatterplot of actin filament velocities as a function of filament length (gray, all velocity points for each filament; blue, maximum velocity of each filament). The dashed line indicates the TOP5% of filament velocities. At the top right, two histograms of velocities are shown: a histogram of all filament velocities with its MVEL marked as a black line and a histogram of velocities with tolerance filtering to eliminate intermittently moving filaments with a velocity dispersion higher than 20% of their mean within a five-frame window (MVEL20). At the bottom right, the effects of various levels of tolerance filtering on the TOP5% and the MVELs are shown. (B) The TOP5% velocities relative to WT for gliding actin filaments (left) or RTFs (in 10−5 M Ca2+) (right) driven by the three mutant human β-cardiac sS1 proteins. Each mutant protein was normalized against its matching WT protein prepared and assayed on the same days. Each bar graph is a mean of relative velocity (error bars represent ±95% confidence interval). (C) The percent mobile filaments for the velocity measurements using WT and mutant human β-cardiac sS1 proteins, for actin (left) and RTFs (right). Data are presented as means ± SEM. (D) In vitro motility assays with mixtures of WT and mutant protein at varying ratios.
R719W and R723G human β-cardiac sS1 showed ~12 to 15% increase in TOP5% gliding velocity, whereas no significant difference was observed for the G741R sS1 (Fig. 3B). However, when MVEL was calculated, R719W human β-cardiac sS1 no longer showed an increase compared to WT, whereas R723G still retained a small ~5% increase in velocity for actin and for RTF (fig. S3).
The difference between the TOP5% and MVEL levels are likely explained by the difference in the amount of stuck filaments for each mutant, which is plotted as percent mobile filaments in Fig. 3C. The motility surface of the R719W mutant showed significantly more stuck filaments than WT (and hence, less percent mobile filaments), adding more drag force to moving filaments. Whereas the R719W showed faster velocity when we only scored the fastest moving filaments (that is, TOP5%), which are least affected by the drag force from stuck filaments, the average velocity (that is, MVEL) taken from the entire histogram is brought down by the presence of this drag force to a level similar to WT. The important conclusions from the abovementioned data are that all the abovementioned changes are small and vary from slightly hypocontractile to slightly hypercontractile contributions and that it is not possible to explain the clinical hypercontractility seen in HCM patients by the changes in these parameters.
Note that all of the abovementioned studies were carried out with pure populations of mutant or WT proteins, whereas patients carrying these mutations are heterozygous. It was previously reported that HCM patients with R719W and R723G mutations express ~70% of mutant protein in their cardiomyocytes (40). Thus, it is possible that one would observe significant alterations in, for example, the velocity of actin filament movement if mixtures of mutant and WT proteins were examined. Although we observed a dose-dependent increase in the velocity of gliding filaments as the percent of R719W and R723G mutant proteins was increased, there were no marked cooperative effects seen (Fig. 3D). Rather, the mixtures showed velocities that were near those expected by weight-averaging the velocities of the mutant and WT proteins, with perhaps a slightly higher influence by the WT human β-cardiac sS1 for the R719W/WT mixtures and a slightly higher influence by the G741R human β-cardiac sS1 for the G741R/WT mixtures (Fig. 3D).
The three converter HCM human β-cardiac sS1 proteins showed load-dependent velocity change profiles that are very similar to WT
The abovementioned velocity experiments were carried out under “unloaded” conditions, meaning that there was no additional load applied to the myosin in the in vitro motility assays. However, in cardiovascular physiology, the heart is always operating under varying amounts of preload and afterload, and we therefore compared the behaviors of the three converter HCM mutant human β-cardiac sS1 proteins to WT human β-cardiac sS1 using a loaded in vitro motility assay.
Thus, we added to the myosin-coated surface varying amounts of a recombinant protein that binds to actin and places a load on the myosin (11). The loaded in vitro motility assay shows a relationship between filament velocity and the concentration of load molecule that relates to the force-velocity relationship of contracting muscle. As a loading molecule, we used a utrophin construct with the same eight-residue C-terminal affinity clamp tag that is at the C terminus of the sS1 construct (11, 12). At zero utrophin concentration, the actin filaments glide at their maximal velocity, because there is no additional load on the myosin. As the concentration of the utrophin increases, the velocity of filaments decreases. The differences in the degree change in velocity intuitively correspond to the differences in ensemble force produced by the myosin molecules on the surface; the higher the ensemble force, the less effect is seen by the utrophin load molecule. The concept of a loaded in vitro motility assay has been used in characterizing the ensemble force of motor proteins for many years (11, 12, 41, 42).
Figure 4 shows the loaded motility assay applied to the three converter domain mutant proteins and WT, using either actin or RTF as gliding filaments. Each curve represents the compilation of 2 to 11 experiments from each of two to five fresh protein preparations, where a fresh preparation of the WT human β-cardiac sS1 was made simultaneously with every mutant protein preparation so they could be compared under identical preparation and experimental conditions. The marked result is that, perhaps with the exception of the R723G human β-cardiac sS1 driving RTF gliding, the curves are nearly identical between the mutant proteins and WT, in keeping with the very small changes in Fintrinsic, kcat, and unloaded velocities described above. Thus, unexpectedly, the fundamental parameters for power output that we report here for the converter mutant human β-cardiac sS1 proteins are very similar to the WT human β-cardiac sS1, and we are driven to consider a different molecular basis for hypercontractility for myosin-based HCM mutations (see Discussion).
Fig. 4. Loaded in vitro motility assays for the three mutant proteins compared to WT controls.
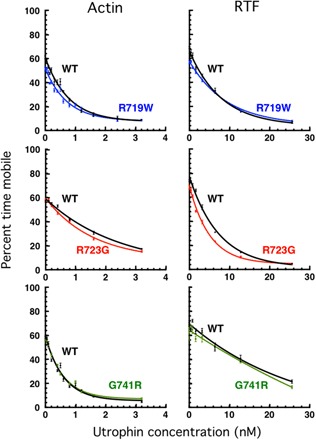
The percent time mobile (11) is a measure of the relative effectiveness of the load molecule utrophin to overcome the Fensemble of the sS1 on the surface (see Materials and Methods) (11). The effect of utrophin concentration on the velocity of actin (left) and RTFs (in 10−5 M Ca2+) (right) is shown. For WT and each mutant protein, ≥5 independent protein preparations and ≥5 separate sets of curves were combined.
DISCUSSION
The conventional view on clinical hypercontractility resulting from HCM mutations is emphasized by the work of Ho and colleagues (7) on patients who carry known HCM mutations but have normal left ventricular thickness and left ventricular size (so-called genotype+/phenotype− individuals). These patients show a combination of diastolic dysfunction and increased ejection fraction, as measured by echocardiography with two-dimensional (2D) tissue Doppler imaging (7), suggesting that the increase in ejection fraction could not be attributed to a decrease in stroke volume, which is commonly seen in more advanced patients who manifest left ventricular hypertrophy and decrease in left ventricular chamber size. Hence, it is generally thought that HCM mutations result in gain of function. This idea is further supported by the recent development of a small molecule that inhibits cardiac myosin’s ATPase activity and that had a clear benefit in preventing the occurrence of hypertrophy in three mouse models of HCM (43). This molecule is currently being investigated in a phase 2 clinical trial, and the initial phase 1 trial data suggest that the drug is well tolerated and results in decreased contractility and a modest reduction in left ventricular outflow tract gradient due to septal hypertrophy (result presented in American Heart Association Scientific Sessions 2016).
The converter domain is the most frequently occurring domain for HCM mutations within the entire myosin molecule (18, 19), and its function has important implications in force generation, load-dependent ADP release, ATP hydrolysis, and Pi release. Our examination of the relevant biochemical and biophysical parameters for three key converter domain HCM mutations, R719W, R723G, and G741R, showed that R719W and R723G both showed significant, but small, reductions in Fintrinsic and increases in velocities in the in vitro motility assay, likely due to a decrease in ts. They also showed small downward shifts in the loaded in vitro motility assay. The similarity in the effect of these two mutations may be explained by the observation that the aliphatic side chains of both arginines pack against one another in the second helix of the converter domain and could disrupt interactions that may be important to the packing and stabilization of the converter domain (fig. S4). On the other hand, the G741R mutation had no obvious change measured with the present experimental assays, and it appears that the mutation has a neutral effect on the biochemical and biophysical functions of the molecule. This is surprising from a structural point of view, because introducing a large side chain like arginine might clash with adjacent residues P731 and I736 and destabilize the converter domain linking to the lever arm, on the basis of the structure of the WT human β-cardiac motor domain (fig. S4). Nonetheless, it is clear functionally at both the molecular level and the clinical level that these mutations must not grossly alter the structure-function of the converter because the effects are small at the molecular level and individuals with this mutation can remain without clinical symptoms for decades. In an attempt to further understand the structural aspects of the effects of these mutations, we turned our attention to intramolecular interactions at the converter domain to see how else these mutations could affect overall biomechanical properties, which is discussed below.
Strikingly, the affected parameters in the present study showed even smaller magnitudes of change than either R403Q (12) or R453C human β-cardiac sS1 (10), and the direction of change was, for the most part, opposite to our hypothesis that HCM mutations cause gain of function. It should be emphasized that the three mutations in the present study have been associated with a severe clinical phenotype for many years (26–28), similar to R403Q and R453C. It can be argued that the hypocontractile changes we describe here could lead to left ventricular pressure and/or volume overload due to mild left ventricular systolic dysfunction, leading to compensatory hypertrophy (44, 45). It is also important to point out from this study that the effects of the mutations were variable between different locations and amino acid changes, even within the same domain of the molecule. Hence, it is possible that the mutations selected for this study do not result in hypercontractile changes at the sarcomere level, and there are alternative mechanisms involved in pathogenesis.
Yet, there is one pivotal parameter that has been largely neglected in studies of HCM mutations: the number of functionally accessible heads (Na) for interaction with actin. We have recently proposed that many, if not most, of the known myosin-based HCM mutations cause an increase in Na, which results in an increase in Fensemble, and therefore in hypercontractility seen clinically (46, 47). For reasons described below, we hypothesize that the three converter mutations studied here fall into this category.
There are many studies that demonstrate that striated muscle myosins exist both in an open state, with each head [subfragment 1 of myosin (S1)] available for interaction with actin, and in a closed state, where the heads are folded back onto their own coiled-coiled tail [subfragment 2 of myosin (S2)] (Fig. 5A) (48–53). The first folded structure of this type dates back to 2001 when Wendt et al. (54) showed for smooth muscle myosin that the “blocked head” (so-called because its actin-binding face is not accessible for binding to actin) (Fig. 5B, dark gray head) interacts with the converter domain of the “free head” (Fig. 5B, light gray head with white converter). Subsequently, the same folded structure has been seen in a variety of other myosins, including skeletal (48) and cardiac (55) myosins, and Fig. 5 shows the tarantula skeletal myosin structure of Alamo et al. (49), which we homology-modeled to the human β-cardiac myosin form using the Winkelmann structure (56), as described in Materials and Methods.
Fig. 5. Homology-modeled structures of human β-cardiac short heavy meromyosin (MyHC ending at residue 961) in the open and closed states.
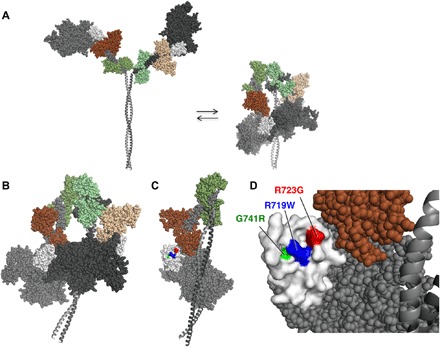
(A) Illustration of the equilibrium between open and closed states. (B) The closed state, shown enlarged and slightly rotated to the left about its vertical axis compared to that in (A). The interface of the blocked head (dark gray) and the converter (white) of the free head (light gray) is seen from the side. (C) Face-on view of the converter domain surface involved in the S1-S1 interaction. This view was obtained by rotating the molecule in (B) 90° to the left about its vertical axis and removing the blocked head from the image. (D) Blowup of the binding interface of the converter in (C), showing the positions of the three converter domain HCM mutations studied here.
What is striking is what one observes when rotating the molecule in Fig. 5B 90° to the left about its vertical axis and removing the blocked head so that, one is looking at the binding interface of the converter (Fig. 5, C and D). R719 and R723 are on the surface of the binding interface, and G741 is slightly below the surface where a change to an arginine is likely to be generally disruptive to the surface architecture and weaken the shown S1-S1 interaction. The binding interface of the blocked head is generally negatively charged (47), such that R719 and R723 seem certainly important for the shown S1-S1 interaction.
As a precedent for this hypothesis, Oshima et al. (57) showed that electrostatic interactions between the converter domain and the 50-kDa domain accounted for the intermolecular and intramolecular head-head interactions observed in intact resting frog skeletal muscle by x-ray fiber diffraction. In addition, Nag et al. (47) have provided biochemical experimental evidence for an interaction between sS1 and the proximal region of the S2 coiled-coil tail, as well as with the C0-C2 domain of MyBP-C, both hypothesized to be occurring on the other side of the folded molecule from that shown in Fig. 5B. Nag et al. (47) further showed that these interactions are weakened as a result of myosin HCM mutations on the myosin mesa, in a manner consistent with the structural model shown in Fig. 5.
Thus, our future work on these and other converter mutations, nearly all of which are near the S1-S1 binding interface (47), will be to experimentally explore our hypothesis that the converter HCM mutations are generally weakening the S1-S1 interaction, causing an increase in Na and therefore causing the hypercontractility observed clinically. It will also be important to extend our studies to more physiological, two-headed motor species that retain regulatory elements, such as the phosphorylatable regulatory light chain, as these species may conceivably behave differently from the sS1 motor domain we have studied to date.
MATERIALs AND METHODS
Myosin constructs and protein expression
Human β-cardiac myosin S1s with three converter domain mutations (R719W, R723G, and G741R) were constructed and produced using a modified AdEasy Vector system (Qbiogene Inc.). The cloning, expression, and purification methodologies are described in detail elsewhere (10, 12). Briefly, complementary DNA (cDNA) for MYH7 (human β-cardiac MyHC) and MYL3 (human ventricular ELC) was purchased from Open Biosystems (Thermo). A truncated version of MYH7 (residues 1 to 808), corresponding to sS1, followed by a flexible GSG (Gly-Ser-Gly) linker was made with either a C-terminal enhanced green fluorescent protein (eGFP) linker (for ATPase and single-molecule optical trap measurements) or a C-terminal eight-residue (RGSIDTWV) PDZ binding peptide (for motility and ADP release experiments) and was coexpressed with human ventricular ELC containing an N-terminal FLAG tag (DYKDDDDK) and tobacco etch virus (TEV) protease site in mouse myoblast C2C12 cells (fig. S1A). Purified fractions were stored in column buffer [10 mM imidazole, 4 mM MgCl2, 1 mM ATP, 1 mM dithiothreitol (DTT), and ~200 mM NaCl] containing 10% sucrose and were flash-frozen in liquid nitrogen before storage at −80°C. Frozen proteins exhibited similar ATPase and motility properties as compared to their fresh counterparts. Before any experiment, the myosin constructs were exchanged into the appropriate buffer conditions using Amicon centrifugal filter units (Millipore) followed by centrifugation at 350,000g for at least 10 min to remove any aggregated protein.
Additional protein purification
Chicken and bovine α-cardiac actin were prepared from muscle acetone powders, using slight modifications of previously described protocols (58). In addition, we used purified bovine G-actin provided by MyoKardia Inc. After preparation, actin was stored in its F form in 2 mM tris (pH 8), 50 mM KCl, 0.2 mM CaCl2, 2 mM ATP, 2 mM MgCl2, 1 mM DTT, and 0.02% sodium azide. Actin was freshly cycled from G- to F-actin for each assay and used only for up to a week before being recycled again. For optical trap measurements, biotin-labeled rabbit skeletal G-actin was purchased from Cytoskeleton. F-actin was stored at 4°C, and G-actin was frozen at −80°C for future use. ATPase experiments were done with chicken skeletal actin, and motility assays were done with α-cardiac actin.
Full-length human gelsolin was expressed and purified on the basis of previous methods (10, 59). Final fractions of gelsolin were dialyzed into buffer D (59), flash-frozen in liquid nitrogen, and stored at −80°C.
Tropomyosin was purified from bovine cardiac tissue according to the protocol of Smillie (60) with modifications described by Sommese et al. (61). Purified tropomyosin was dialyzed in 20 mM imidazole (pH 7.5), 300 mM KCl, and 1 mM DTT before flash-freezing and storing at −80°C.
Human adult cardiac troponin subunit (TNNT2, TNNI3, and TNNC2) expression and purification were based on previously published methods (12, 61–63). TnT, TnI, and TnC were purified and stored in storage buffer containing 20 mM imidazole (pH 7.5), 1 M KCl, 1 mM MgCl2, and 1 mM DTT. These proteins were then flash-frozen and stored at −80°C for future use. Troponin complexes were formed according to Szczesna et al. (64) with slight modifications. Components were mixed at a molar ratio of 1.3:1.3:1 (TnI/TnT/TnC) for 1 hour on ice. Complexes were then dialyzed at 4°C in six sequential steps into complex buffer [20 mM imidazole (pH 7.5), 2 mM MgCl2, and 1 mM DTT] containing 0.7, 0.5, 0.3, 0.1, and 0.01 M KCl twice, for 6 to 12 hours each. Complexes were flash-frozen in complex buffer before storing them at −80°C. The cDNAs for human adult cardiac TnI, TnC, and TnT in carbenicillin-selective pET-3d plasmids were obtained from J. Potter (University of Miami).
For all experiments involving the RTFs, RTFs were formed by mixing excess tropomyosin and troponin complex to actin on ice and then incubating for at least 1 hour before use. The final molar ratio was 7:2:2 of actin/tropomyosin/troponin for all experiments.
Mouse utrophin with an eight-residue (RGSIDTWV) C-terminal PDZ binding peptide was expressed in bacterial cells, as previously published (11). The purified protein was concentrated and dialyzed overnight against 150 mM NaCl, 25 mM tris, and 1 mM DTT (pH 8.0) at 4°C before flash-freezing in liquid nitrogen and storing at −80°C.
The SNAP-PDZ18 fusion construct was expressed in bacterial cells as previously described (11). Eluted protein was concentrated, and the buffer was exchanged to 150 mM NaCl and 25 mM tris (pH 8.0). The purified protein was flash-frozen in liquid nitrogen and stored at −80°C.
Actin-activated ATPase assay
For actin-activated ATPase, gelsolin was added to actin at a ratio of 1:500. Gelsolin at this concentration was used to decrease the viscosity of the actin and to thereby decrease pipetting error and allow higher concentrations of actin to be used. Actin-activated ATPase assays were then performed as previously described using a colorimetric readout (65). Briefly, sS1 was diluted to a final concentration of 0.07 to 0.15 μM (with four times as much for the basal myosin ATPase control in the absence of actin to amplify the signal), with 2 mM ATP and actin at concentrations ranging from 0 to 125 μM. The final buffer conditions were 10 mM imidazole (pH 7.5), 5 mM KCl, 4 mM MgCl2, and 1 mM DTT. The reaction was performed at 23°C with shaking using a Thermo Scientific Multiskan GO, and four to five time points were taken for each concentration. The sS1 activity was linear over the time period of the assay, and hence, an ATP-regenerating system was not necessary. Basal activity (<0.2 s−1) was subtracted to get actin-activated ATPase activity. The Michaelis-Menten equation was fit to the data to determine the maximal activity (kcat) and the associated actin constant for myosin (Km) using Prism 6 (GraphPad Software Inc.). All of the experiments were done using sS1 with a FLAG tag on the ELC, as previously described (10, 12), except for R723G which was assayed with and without the FLAG tag to check whether the presence of the FLAG tag had any effect on the kcat of the sS1. The FLAG tag was removed during the purification process by adding TEV protease to cleave the myosin that was bound to the FLAG resin. The sS1 was then purified by high-performance liquid chromatography. No significant difference was seen between the sS1 with the FLAG tag and the sS1 without the FLAG tag (fig. S2).
Unloaded and loaded in vitro motility
The basic method followed our previously described motility assay (66), with some modifications. Coverslips (VWR micro cover glass) were coated with a mixture of 0.2% nitrocellulose (Ernest Fullam Inc.) and 0.2% collodion (Electron Microscopy Sciences) dissolved in amyl acetate (Sigma) and air-dried for a few hours before use. Permanent double-sided tape (Scotch) was used to construct four channels in each slide (Gold Seal), and four different experiments were performed on the same slide. In general, both WT and mutant protein(s) were studied on the same slide to minimize variability in slide preparation for measurement of unloaded motility. For loaded in vitro motility assays, two to three slides were used under 8 to 12 different concentrations of utrophin to obtain a full curve with each run of an experiment.
Tenfold molar excess of F-actin was added in the presence of 4 mM ATP, incubated for 10 min, and sedimented at 350,000g for 20 min (termed “deadheading”) to reduce the number of partially inactivated myosin heads in S1 preparations. MgCl2 was added to 50 mM (to form F-actin paracrystals) and incubated for 20 min (with mixing by pipetting at 10 min), and the mixture was resedimented at 350,000g for 30 min to eliminate sS1 that remained bound to the actin and the residual actin in the supernatant. The supernatant was collected, and the sS1 concentration was measured using the Bradford reagent (Bio-Rad). A mock cleanup procedure containing storage buffer, actin, and MgCl2 without sS1 was also performed simultaneously and was used as blank for more accurate concentration determination of sS1. The quality of the sS1 cleanup was assessed by the percentage of stuck filaments under unloaded conditions. We repeated the sS1 cleanup procedure until the percentage of stuck filaments dropped below 10%. The amount of deadheads removed by this procedure was not significantly different between the WT and mutant motors. Before any experiments, deadheaded sS1 was diluted in 10% ABBSA {assay buffer (AB) [25 mM imidazole (pH 7.5), 25 mM KCl, 4 mM MgCl2, 1 mM EGTA, and 1 mM DTT] with bovine serum albumin (BSA) (0.1 mg/ml) diluted in AB}, unless otherwise stated.
For motility experiments, reagents were sequentially flowed through the channels in the following order: (i) 10 μl of 3 μM SNAP-PDZ18 diluted in AB and incubated for 2 min; (ii) 30 ml of ABBSA to block the surface from nonspecific attachments, incubated for 2 min; (iii) 10 μl of a mixture of eight-residue (RGSIDTWV)–tagged human cardiac S1 (~0.05 to 0.1 mg/ml for actin motility and 0.1 to 0.2 mg/ml for RTF motility) and utrophin at desired concentrations, incubated for 5 min (before mixing sS1 and utrophin, sS1 and utrophin dilutions were prepared in 10% ABBSA; for unloaded motility, utrophin was skipped in this step); (iv) 30 μl of ABBSA to wash any unattached proteins; and finally, (v) 10 μl of the GO solution {1 to 5 nM bovine actin labeled with tetramethylrhodamine (TMR)–phalloidin (Invitrogen), 2 mM ATP (Calbiochem), an oxygen-scavenging system [0.2% glucose, glucose oxidase (0.11 mg/ml; Calbiochem), and catalase (0.018 mg/ml; Calbiochem)], and an ATP regeneration system [1 mM phosphocreatine (Calbiochem) and creatine phosphokinase (0.1 mg/ml; Calbiochem)]} in ABBSA.
For RTF motility experiments, a higher salt concentration was necessary to avoid aggregation of RTF in solution. We also attached the RTF with myosin on the surface in the absence of ATP, and then myosin was activated by the addition of a saturating concentration of ATP. Thus, after attaching the myosin at step (iii), the motility surface was washed with AB that contained 100 mM KCl, followed by the addition of RTF dilution in ABBSA containing 100 mM KCl, 5 mM CaCl2, 100 nM excess tropomyosin and troponin complex, and 1 to 5 nM TMR-phalloidin–labeled RTF. After 5 min of RTF binding, the surface was washed with ABBSA with 25 mM KCl to bring the salt concentration down. Final GO solution included AB (25 mM KCl), 5 mM CaCl2, 100 nM excess tropomyosin/troponin complex, and an oxygen-scavenging system as described above, and thus, the motility was done at the same salt concentration as in the actin experiment. The RTF mixture was made at least 1 hour before the motility was measured by mixing TMR-phalloidin–labeled bovine actin/bovine tropomyosin/human troponin complex in a 7:2:2 ratio [3.5 mM actin, 1 mM tropomyosin, 1 mM troponin, and BSA (1 mg/ml)].
For all experiments, movies were obtained at 23°C, at a frame rate of 1 Hz using a Nikon Ti-E inverted microscope with the Andor iXon+ EMCCD camera model DU885. All experiments were repeated with at least four different fresh protein preparations. At each condition, at least three different movies with a duration of 30 s were recorded. Filament tracking and analysis of movies, both under unloaded and loaded conditions, were performed by a recently published method, FAST, as described by Aksel et al. (11).
Single-molecule optical trap measurements
The dual-beam optical trap instrumentation is described in detail elsewhere (10, 12). The experimental condition is similar to the one described in the motility assay. Here, the myosin construct had an eGFP tag at the C-terminal end for surface attachment through binding with anti-GFP antibody. Deadheads of purified protein were eliminated as described before (10). All the experiments were performed at 23°C. The nitrocellulose-coated glass surface of the sample chamber was also coated with 1.5-μm-diameter silica beads that acted as platforms. The sequence of steps for preparing the chamber is as follows: (i) anti-GFP antibody (~0.01 mg/ml) (Abcam) was flowed through the chamber; (ii) AB buffer containing BSA (1 mg/ml) (ABBSA buffer) was flowed to block the exposed surface; (iii) the surface with myosin was sparsely coated with ~50 to 200 pM of human β-cardiac sS1; (iv) the chamber was washed with AB buffer; and finally, (v) ABBSA buffer containing 250 to 500 pM ATP, TMR-phalloidin–labeled biotin-actin filaments, neutravidin-coated polystyrene beads (Polysciences), and the oxygen-scavenging and ATP regeneration systems described above was flowed through the chamber. The chamber was sealed with vacuum grease to stop evaporation of the solution. Neutravidin-coated polystyrene beads (1 μm in diameter) bound to each end of a TMR-phalloidin– and biotin-labeled actin filament were trapped in two different laser beams. The bead-actin-bead assembly is known as a dumbbell, which was stretched to remove compliance in the actin filament and brought close to the bead pedestal on the surface for interaction with myosin. A detailed isometric force measurement procedure is described elsewhere (10, 12). A trap stiffness of ~0.1 pN/nm was used.
Individual force events were collected from several single molecules of multiple protein purifications. The number of total force events from individual molecules on average was 30 to 400. We have always observed that for all of our sS1 constructs, the force distribution is accompanied with a long tail (10, 12). This phenomenon is commonly reported in the single-molecule force measurements of myosin (67–69), although the exact reason is not known. It is important to take account of the smaller population of higher-force events in the analysis. Hence, we chose a double Gaussian function to fit the force histogram data, and the major first peak of the fit yielded the intrinsic force of an individual molecule reported here. These intrinsic force values of multiple molecules were used to calculate the mean force value. Additionally, we have used cumulative probability distribution analysis (12) to compare the force-producing ability of different myosins. This method of analysis takes account of all the events to generate the function. The probability distribution at any particular force value is calculated by adding up the number of events to that force value divided by the total number of events of all force values. This distribution starts from a value close to 0 at the lowest measurable force to 1 at the maximum force. If the force-producing ability between two proteins is different, then the probability value at any force value will be different.
Development of human β-cardiac myosin protein models
We developed our models on the basis of known human β-cardiac myosin S1 motor domain structural data (56) as described previously (18). Briefly, we retrieved the protein sequence of human β-cardiac myosin and the human cardiac light chains from the UniProt database (70): MyHC motor domain (MYH7)–P12883, myosin ELC (MYL3)–P08590, and myosin regulatory light chain (MYL2)–P10916. We used a multitemplate homology modeling approach to build the structural coordinates of MYH7 (residues 1 to 840), MYL3 (residues 1 to 195), MYL2 (residues 1 to 166), and S2 (residues 841 to 1280). The templates used to model the poststroke structure were obtained from the human β-cardiac myosin motor domain (PDB ID: 4P7H; no nucleotide in the active site) solved by Winkelmann et al. (56), supplemented with the rigor structure from the squid myosin motor domain (PDB ID code 3I5G, no nucleotide in the active site) (71) to model the converter domain, lever arm, and light chains. The S2 region is a long coiled-coil structure; hence, we used the template from the Myosinome database (72). Figure 5 shows a homology-modeled folded-back structure of human β-cardiac myosin from the 3D-reconstructed images of tarantula skeletal muscle thick filaments by Alamo et al. (49). Modeling was done using the MODELLER package. Visualizations were performed using PyMOL version 1.7.4 (www.pymol.org).
Supplementary Material
Acknowledgments
We thank A. Houdusse for helpful structure-function discussions, S. Nag and R. Sommese for the preparation of the tropomyosin and troponin complex, and A. Borrayo for his technical help in maintaining virus and cell stocks. An early version of this work by M.K., S.S.S., S.S., K.M.R., and J.A.S. was deposited in BioRxiv last July (https://doi.org/10.1101/065649). Funding: This project was funded by NIH grants GM33289 and HL117138 to J.A.S. M.K. was a recipient of the National Research Service Award Postdoctoral Fellowship F32HL124883. Author contributions: M.K. performed myosin and actin preparations, with help from S.S. and K.M.R., and steady-state ATPase and in vitro motility assays. S.S.S. conducted the intrinsic force measurements. M.K. and J.A.S. wrote the manuscript. M.K., S.S.S., K.M.R., and J.A.S. contributed to the data analysis/interpretation and editing of the manuscript. Competing interests: J.A.S. is a founder of and owns shares in Cytokinetics Inc. and MyoKardia Inc., biotech companies that are developing therapeutics that target the sarcomere. J.A.S. is on the Scientific Advisory Board of MyoKardia and has a paid consultancy relationship. K.M.R. is also on the Scientific Advisory Board of MyoKardia Inc. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. A complete version of the FAST program (11) for motility analysis is available for download at http://spudlab.stanford.edu.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/2/e1601959/DC1
fig. S1. sS1 purification and ATPase measurements.
fig. S2. Comparison of kcat (relative to WT) for R723G human β-cardiac sS1 with and without a FLAG tag on the ELC.
fig. S3. Unloaded MVEL measurements for actin and RTF.
fig. S4. Structure of the part of the converter domain showing G741R (green), R719W (blue), and R723G (red), neighboring residues whose conflict with the G741R conversion (residues I736 and P731; magenta) and whose interactions with R719 may be lost by the R719W conversion.
table S1. Summary of kcat and Km values from actin-activated ATPase assays.
REFERENCES AND NOTES
- 1.Semsarian C., Ingles J., Maron M. S., Maron B. J., New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Elliott P. M., Gimeno J. R., Thaman R., Shah J., Ward D., Dickie S., Tome Esteban M. T., McKenna W. J., Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 92, 785–791 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geisterfer-Lowrance A. A. T., Kass S., Tanigawa G., Vosberg H.-P., McKenna W., Seidman C. E., Seidman J. G., A molecular basis for familial hypertrophic cardiomyopathy: A β cardiac myosin heavy chain gene missense mutation. Cell 62, 999–1006 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Wang L., Seidman J. G., Seidman C. E., Narrative review: Harnessing molecular genetics for the diagnosis and management of hypertrophic cardiomyopathy. Ann. Intern. Med. 152, 513–520 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maron B. J., Maron M. S., Semsarian C., Genetics of hypertrophic cardiomyopathy after 20 years. J. Am. Coll. Cardiol. 60, 705–715 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Seidman J. G., Seidman C., The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell 104, 557–567 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Ho C. Y., Sweitzer N. K., McDonough B., Maron B. J., Casey S. A., Seidman J. G., Seidman C. E., Solomon S. D., Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation 105, 2992–2997 (2002). [DOI] [PubMed] [Google Scholar]
- 8.C. E. Seidman, J. G. Seidman, in The Metabolic and Molecular Bases of Inherited Disease, C. R. Scriver, A. L. Beaudet, D. Valle, W. S. Sly, B. Childs, K. W. Kinzler, B. Vogelstein, Eds. (McGraw-Hill, 2000), pp. 5433–5452. [Google Scholar]
- 9.J. Sung, S. Sivaramakrishnan, A. R. Dunn, J. A. Spudich, Single-molecule dual-beam optical trap analysis of protein structure and function, in Methods in Enzymology (Elsevier Inc., ed. 1, 2010), pp. 321–375. [DOI] [PubMed] [Google Scholar]
- 10.Sommese R. F., Sung J., Nag S., Sutton S., Deacon J. C., Choe E., Leinwand L. A., Ruppel K., Spudich J. A., Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc. Natl. Acad. Sci. U.S.A. 110, 12607–12612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aksel T., Yu E. Choe, Sutton S., Ruppel K. M., Spudich J. A., Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep. 11, 910–920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nag S., Sommese R. F., Ujfalusi Z., Combs A., Langer S., Sutton S., Leinwand L. A., Geeves M. A., Ruppel K. M., Spudich J. A., Contractility parameters of human β-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci. Adv. 1, e1500511 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spudich J. A., Hypertrophic and dilated cardiomyopathy: Four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 106, 1236–1249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debold E. P., Schmitt J. P., Patlak J. B., Beck S. E., Moore J. R., Seidman J. G., Seidman C., Warshaw D. M., Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse α-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 293, H284–H291 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Tyska M. J., Hayes E., Giewat M., Seidman C. E., Seidman J. G., Warshaw D. M., Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 86, 737–744 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Lowey S., Lesko L. M., Rovner A. S., Hodges A. R., White S. L., Low R. B., Rincon M., Gulick J., Robbins J., Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an α-or β-myosin heavy chain backbone. J. Biol. Chem. 283, 20579–20589 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adhikari A. S., Kooiker K. B., Sarkar S. S., Liu C., Bernstein D., Spudich J. A., Ruppel K. M., Early-onset hypertrophic cardiomyopathy mutations significantly increase the velocity, force, and actin-activated ATPase activity of human β-cardiac myosin. Cell Rep. 17, 2857–2864 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homburger J. R., Green E. M., Caleshu C., Sunitha M. S., Taylor R. E., Ruppel K. M., Metpally R. P. R., Colan S. D., Michels M., Day S. M., Olivotto I., Bustamante C. D., Dewey F. E., Ho C. Y., Spudich J. A., Ashley E. A., Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc. Natl. Acad. Sci. U.S.A. 113, 6701–6706 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colegrave M., Peckham M., Structural implications of β-cardiac myosin heavy chain mutations in human disease. Anat. Rec. 297, 1670–1680 (2014). [DOI] [PubMed] [Google Scholar]
- 20.García-Giustiniani D., Arad M., Ortíz-Genga M., Barriales-Villa R., Fernández X., Rodríguez-García I., Mazzanti A., Veira E., Maneiro E., Rebolo P., Lesende I., Cazón L., Freimark D., Gimeno-Blanes J. R., Seidman C., Seidman J., McKenna W., Monserrat L., Phenotype and prognostic correlations of the converter region mutations affecting the β myosin heavy chain. Heart 101, 1047–1053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Köhler J., Winkler G., Schulte I., Scholz T., McKenna W., Brenner B., Kraft T., Mutation of the myosin converter domain alters cross-bridge elasticity. Proc. Natl. Acad. Sci. U.S.A. 99, 3557–3562 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Littlefield K. P., Swank D. M., Sanchez B. M., Knowles A. F., Warshaw D. M., Bernstein S. I., The converter domain modulates kinetic properties of Drosophila myosin. Am. J. Physiol. Cell Physiol. 284, C1031–C1038 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Kronert W. A., Melkani G. C., Melkani A., Bernstein S. I., Mutating the converter–relay interface of Drosophila myosin perturbs ATPase activity, actin motility, myofibril stability and flight ability. J. Mol. Biol. 398, 625–632 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swank D. M., Knowles A. F., Suggs J. A., Sarsoza F., Lee A., Maughan D. W., Bernstein S. I., The myosin converter domain modulates muscle performance. Nat. Cell Biol. 4, 312–316 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Ramanath S., Wang Q., Bernstein S. I., Swank D. M., Disrupting the myosin converter-relay interface impairs Drosophila indirect flight muscle performance. Biophys. J. 101, 1114–1122 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anan R., Greve G., Thierfelder L., Watkins H., McKenna W. J., Solomon S., Vecchio C., Shono H., Nakao S., Tanaka H., Mares A., Towbin J. A., Spirito P., Roberts R., Seidman J. G., Seidman C. E., Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J. Clin. Invest. 93, 280–285 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enjuto M., Francino A., Navarro-López F., Viles D., Paré J.-C., Ballesta A. M., Malignant hypertrophic cardiomyopathy caused by the Arg723Gly mutation in β-myosin heavy chain gene. J. Mol. Cell. Cardiol. 32, 2307–2313 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Fananapazir L., Dalakas M. C., Cyran F., Cohn G., Epstein N. D., Missense mutations in the β-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 90, 3993–3997 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seebohm B., Matinmehr F., Köhler J., Francino A., Navarro-Lopéz F., Perrot A., Özcelik C., McKenna W. J., Brenner B., Kraft T., Cardiomyopathy mutations reveal variable region of myosin converter as major element of cross-bridge compliance. Biophys. J. 97, 806–824 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teekakirikul P., Eminaga S., Toka O., Alcalai R., Wang L., Wakimoto H., Nayor M., Konno T., Gorham J. M., Wolf C. M., Kim J. B., Schmitt J. P., Molkentin J. D., Norris R. A., Tager A. M., Hoffman S. R., Markwald R. R., Seidman C. E., Seidman J. G., Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J. Clin. Invest. 120, 3520–3529 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirschner S. E., Becker E., Antognozzi M., Kubis H.-P., Francino A., Navarro-López F., Bit-Avragim N., Perrot A., Mirrakhimov M. M., Osterziel K.-J., McKenna W. J., Brenner B., Kraft T., Hypertrophic cardiomyopathy-related β-myosin mutations cause highly variable calcium sensitivity with functional imbalances among individual muscle cells. Am. J. Physiol. Heart Circ. Physiol. 288, H1242–H1251 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Kraft T., Witjas-Paalberends E. R., Boontje N. M., Tripathi S., Brandis A., Montag J., Hodgkinson J. L., Francino A., Navarro-Lopez F., Brenner B., Stienen G. J. M., van der Velden J., Familial hypertrophic cardiomyopathy: Functional effects of myosin mutation R723G in cardiomyocytes. J. Mol. Cell. Cardiol. 57, 13–22 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Lankford E. B., Epstein N. D., Fananapazir L., Sweeney H. L., Abnormal contractile properties of muscle fibers expressing beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J. Clin. Invest. 95, 1409 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller G., Maycock J., White E., Peckham M., Calaghan S., Heterologous expression of wild-type and mutant β-cardiac myosin changes the contractile kinetics of cultured mouse myotubes. J. Physiol. 548, 167–174 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kronert W. A., Melkani G. C., Melkani A., Bernstein S. I., Alternative relay and converter domains tune native muscle myosin isoform function in Drosophila. J. Mol. Biol. 416, 543–557 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bick A. G., Flannick J., Ito K., Cheng S., Vasan R. S., Parfenov M. G., Herman D. S., DePalma S. R., Gupta N., Gabriel S. B., Funke B. H., Rehm H. L., Benjamin E. J., Aragam J., Taylor H. A. Jr, Fox E. R., Newton-Cheh C., Kathiresan S., O’Donnell C. J., Wilson J. G., Altshuler D. M., Hirschhorn J. N., Seidman J. G., Seidman C., Burden of rare sarcomere gene variants in the Framingham and Jackson Heart Study cohorts. Am. J. Hum. Genet. 91, 513–519 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toyoshima Y. Y., Kron S. J., McNally E. M., Niebling K. R., Toyoshima C., Spudich J. A., Myosin subfragment-1 is sufficient to move actin filaments in vitro. Nature 328, 536–539 (1987). [DOI] [PubMed] [Google Scholar]
- 38.Uyeda T. Q. P., Kron S. J., Spudich J. A., Myosin step size: Estimation from slow sliding movement of actin over low densities of heavy meromyosin. J. Mol. Biol. 214, 699–710 (1990). [DOI] [PubMed] [Google Scholar]
- 39.Uyeda T. Q., Abramson P. D., Spudich J. A., The neck region of the myosin motor domain acts as a lever arm to generate movement. Proc. Natl. Acad. Sci. U.S.A. 93, 4459–4464 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brenner B., Seebohm B., Tripathi S., Montag J., Kraft T., Familial hypertrophic cardiomyopathy: Functional variance among individual cardiomyocytes as a trigger of FHC-phenotype development. Front. Physiol. 5, 392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg M. J., Moore J. R., The molecular basis of frictional loads in the in vitro motility assay with applications to the study of the loaded mechanochemistry of molecular motors. Cytoskeleton 67, 273–285 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greenberg M. J., Kazmierczak K., Szczesna-Cordary D., Moore J. R., Cardiomyopathy-linked myosin regulatory light chain mutations disrupt myosin strain-dependent biochemistry. Proc. Natl. Acad. Sci. U.S.A. 107, 17403–17408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green E. M., Wakimoto H., Anderson R. L., Evanchik M. J., Gorham J. M., Harrison B. C., Henze M., Kawas R., Oslob J. D., Rodriguez H. M., Song Y., Wan W., Leinwand L. A., Spudich J. A., McDowell R. S., Seidman J. G., Seidman C. E., A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351, 617–621 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mann D. L., Mechanisms and models in heart failure: The biomechanical model and beyond. Circulation 111, 2837–2849 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Bernardo B. C., Weeks K. L., Pretorius L., McMullen J. R., Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 128, 191–227 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Spudich J. A., The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 43, 64–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.S. Nag, D. V. Trivedi, S. S. Sarkar, S. Sutton, K. M. Ruppel, J. A. Spudich, Beyond the myosin mesa: A potential unifying hypothesis on the underlying molecular basis of hyper-contractility caused by a majority of hypertrophic cardiomyopathy mutations. bioRxiv, 10.1101/065508 (2016). [Google Scholar]
- 48.Woodhead J. L., Zhao F.-Q., Craig R., Egelman E. H., Alamo L., Padrón R., Atomic model of a myosin filament in the relaxed state. Nature 436, 1195–1199 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Alamo L., Wriggers W., Pinto A., Bártoli F., Salazar L., Zhao F.-Q., Craig R., Padrón R., Three-dimensional reconstruction of tarantula myosin filaments suggests how phosphorylation may regulate myosin activity. J. Mol. Biol. 384, 780–797 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alamo L., Li X. E., Espinoza-Fonseca L. M., Pinto A., Thomas D. D., Lehman W., Padrón R., Tarantula myosin free head regulatory light chain phosphorylation stiffens N-terminal extension, releasing it and blocking its docking back. Mol. Biosyst. 11, 2180–2189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alamo L., Qi D., Wriggers W., Pinto A., Zhu J., Bilbao A., Gillilan R. E., Hu S., Padrón R., Conserved intramolecular interactions maintain myosin interacting-heads motifs explaining tarantula muscle super-relaxed state structural basis. J. Mol. Biol. 428, 1142–1164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naber N., Cooke R., Pate E., Slow myosin ATP turnover in the super-relaxed state in tarantula muscle. J. Mol. Biol. 411, 943–950 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hooijman P., Stewart M. A., Cooke R., A new state of cardiac myosin with very slow ATP turnover: A potential cardioprotective mechanism in the heart. Biophys. J. 100, 1969–1976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wendt T., Taylor D., Trybus K. M., Taylor K., Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc. Natl. Acad. Sci. U.S.A. 98, 4361–4366 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zoghbi M. E., Woodhead J. L., Moss R. L., Craig R., Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc. Natl. Acad. Sci. U.S.A. 105, 2386–2390 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winkelmann D. A., Forgacs E., Miller M. T., Stock A. M., Structural basis for drug-induced allosteric changes to human β-cardiac myosin motor activity. Nat. Commun. 6, 7974 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oshima K., Sugimoto Y., Irving T. C., Wakabayashi K., Pastore A., Head-head interactions of resting myosin crossbridges in intact frog skeletal muscles, revealed by synchrotron x-ray fiber diffraction. PLOS ONE 7, e52421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pardee J. D., Spudich J. A., Purification of muscle actin. Methods Enzymol. 85, 164–181 (1982). [DOI] [PubMed] [Google Scholar]
- 59.Way M., Gooch J., Pope B., Weeds A. G., Expression of human plasma gelsolin in Escherichia coli and dissection of actin binding sites by segmental deletion mutagenesis. J. Cell Biol. 109, 593–605 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smillie L. B., Preparation and identification of alpha- and beta-tropomyosins. Meth. Enzymol. 85, 234–241 (1982). [DOI] [PubMed] [Google Scholar]
- 61.Sommese R. F., Nag S., Sutton S., Miller S. M., Spudich J. A., Ruppel K. M., Kimura A., Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human β-cardiac myosin. PLOS ONE 8, e83403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheng Z., Pan B. S., Miller T. E., Potter J. D., Isolation, expression, and mutation of a rabbit skeletal muscle cDNA clone for troponin I. The role of the NH2 terminus of fast skeletal muscle troponin I in its biological activity. J. Biol. Chem. 267, 25407–25413 (1992). [PubMed] [Google Scholar]
- 63.Pan B. S., Potter J. D., Two genetically expressed troponin T fragments representing alpha and beta isoforms exhibit functional differences. J. Biol. Chem. 267, 23052–23056 (1992). [PubMed] [Google Scholar]
- 64.Szczesna D., Guzman G., Miller T., Zhao J., Farokhi K., Ellemberger H., Potter J. D., The role of the four Ca2+ binding sites of troponin C in the regulation of skeletal muscle contraction. J. Biol. Chem. 271, 8381–8386 (1996). [DOI] [PubMed] [Google Scholar]
- 65.Trybus K. M., Biochemical studies of myosin. Methods 22, 327–335 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Kron S. J., Toyoshima Y. Y., Uyeda T. Q. P., Spudich J. A., Assays for actin sliding movement over myosin-coated surfaces. Methods Enzymol. 196, 399–416 (1991). [DOI] [PubMed] [Google Scholar]
- 67.Allersma M. W., Gittes F., deCastro M. J., Stewart R. J., Schmidt C. F., Two-dimensional tracking of ncd motility by back focal plane interferometry. Biophys. J. 74, 1074–1085 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simmons R. M., Finer J. T., Chu S., Spudich J. A., Quantitative measurements of force and displacement using an optical trap. Biophys. J. 70, 1813–1822 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takagi Y., Homsher E. E., Goldman Y. E., Shuman H., Force generation in single conventional actomyosin complexes under high dynamic load. Biophys. J. 90, 1295–1307 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.UniProt Consortium, Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 41, D43–D47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang Y., Gourinath S., Kovács M., Nyitray L., Reutzel R., Himmel D. M., O’Neall-Hennessey E., Reshetnikova L., Szent-Györgyi A. G., Brown J. H., Cohen C., Rigor-like structures from muscle myosins reveal key mechanical elements in the transduction pathways of this allosteric motor. Structure 15, 553–564 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Syamaladevi D. P., Sunitha M. S., Kalaimathy S., Reddy C. C., Iftekhar M., Pasha S. N., Sowdhamini R., Myosinome: A database of myosins from select eukaryotic genomes to facilitate analysis of sequence-structure-function relationships. Bioinform. Biol. Insights 6, 247–254 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/2/e1601959/DC1
fig. S1. sS1 purification and ATPase measurements.
fig. S2. Comparison of kcat (relative to WT) for R723G human β-cardiac sS1 with and without a FLAG tag on the ELC.
fig. S3. Unloaded MVEL measurements for actin and RTF.
fig. S4. Structure of the part of the converter domain showing G741R (green), R719W (blue), and R723G (red), neighboring residues whose conflict with the G741R conversion (residues I736 and P731; magenta) and whose interactions with R719 may be lost by the R719W conversion.
table S1. Summary of kcat and Km values from actin-activated ATPase assays.