

Abstract
Clonal cytogenetic abnormalities are found in 20-30% of patients with chronic myelomonocytic leukemia (CMML), while gene mutations are present in >90% of cases. Patients with low risk cytogenetic features account for 80% of CMML cases and often fall into the low risk categories of CMML prognostic scoring systems, but the outcome differs considerably among them. We performed targeted deep sequencing of 83 myeloid-related genes in 56 CMML patients with low risk cytogenetic features or uninformative conventional cytogenetics (CC) at diagnosis, with the aim to identify the genetic characteristics of patients with a more aggressive disease. Targeted sequencing was also performed in a subset of these patients at time of acute myeloid leukemia (AML) transformation. Overall, 98% of patients harbored at least one mutation. Mutations in cell signaling genes were acquired at time of AML progression. Mutations in ASXL1, EZH2 and NRAS correlated with higher risk features and shorter overall survival (OS) and progression free survival (PFS). Patients with SRSF2 mutations associated with poorer OS, while absence of TET2 mutations (TET2wt) was predictive of shorter PFS. A decrease in OS and PFS was observed as the number of adverse risk gene mutations (ASXL1, EZH2, NRAS and SRSF2) increased. On multivariate analyses, CMML-specific scoring system (CPSS) and presence of adverse risk gene mutations remained significant for OS, while CPSS and TET2wt were predictive of PFS. These results confirm that mutation analysis can add prognostic value to patients with CMML and low risk cytogenetic features or uninformative CC.
Keywords: chronic myelomonocytic leukemia, normal karyotype, gene mutations, targeted deep sequencing, prognostic factors
INTRODUCTION
Chronic myelomonocytic leukemia (CMML) is a hematopoietic stem cell disorder with features from both myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN) [1]. The original French-American-British (FAB) criteria identifies two variants based on leukocyte count (myelodysplastic [<13×109/L], MD-CMML, and myeloproliferative [>13×109/L], MP-CMML), while the 2008 World Health Organization (WHO) classification distinguishes two categories (CMML-1 and CMML-2) according to blast percentage in bone marrow (BM) or peripheral blood (PB) [1, 2]. Very recently, the 2016 revision of the WHO criteria has proposed the inclusion of a third CMML subtype, CMML-0, based on recent findings that demonstrate that these three CMML subtypes have different clinical outcomes [3, 4].
Clonal cytogenetic abnormalities are not frequent in CMML (20-30%), whereas gene mutations have been reported in >90% of patients at diagnosis [5–7]. Prognostic impact of cytogenetic alterations in CMML was first explored by the Spanish MDS group and recently reviewed by the Mayo Clinic-French Consortium [5, 8]. According to both studies, up to 80% of CMML patients present with low risk cytogenetic features (normal karyotype, isolated -Y or sole der(3q)).
During the past years, several studies have reported recurrent gene mutations in CMML, being mutations in TET2 (50-60%), ASXL1 (40-50%) and SRSF2 (40-50%) the most frequent [7, 9, 10]. Less frequent mutations (10-30%) have also been described in RUNX1, CBL, K/NRAS, EZH2, UTX, DNMT3A and JAK2 genes [6, 7, 10–12]. Prognostic relevance of mutations in ASXL1, TET2, RUNX1, CBL and NRAS has been demonstrated on univariate survival analyses on CMML [7, 13, 14], but only ASXL1 mutations seem to retain this impact on multivariate models [15, 16].
Several prognostic scoring systems have been proposed for CMML in the past years. The CMML-specific scoring system (CPSS) was developed by the Spanish MDS group and includes CMML-2, MP-CMML, transfusion dependency and cytogenetic risk stratification as independent adverse prognostic factors [17]. Other novel CMML-specific scoring systems, like the Groupe Francophone des Myélodysplasies (GFM) CMML model [15] and the Molecular Mayo model [16], include similar biological parameters but exclude cytogenetic abnormalities. These two models introduce for the first time the use of molecular criteria, such as the presence of mutations in ASXL1.
A significant subset of CMML patients fall into the low risk cytogenetic category and most of them present with normal karyotype, but the median overall survival (OS) and the risk of AML progression differ considerably among them [17]. With the aim to identify a subgroup of patients with a more aggressive disease, we performed targeted deep sequencing in 56 patients with CMML and low risk cytogenetic features or no metaphases and explored the prognostic value of gene mutations.
RESULTS
Characteristics of CMML patients
A total of 56 patients with CMML and low risk cytogenetic features or uninformative conventional cytogenetics (CC) were included in the study. Median follow-up of alive patients was 36 months (range, 5.8 to 83.5 months). Main clinical and biological characteristics of patients are summarized in Table 1. Median age at diagnosis was 72 years and the series included 37 (66%) males and 19 (34%) females. Following the FAB criteria [2], 46 (82%) patients were classified as MD-CMML and 10 (18%) as MP-CMML, while according to the 2008 WHO classification [1], 49 (87%) cases corresponded to CMML-1 and 7 (13%) to CMML-2. Progression to AML was observed in 16 (29%) patients. Risk stratification of patients was based on the CPSS [17] and the GFM CMML model [15] (Table 1).
Table 1. Main clinical and hematological characteristics of CMML patients at diagnosis (n=56).
| Variable | Median (range) | N (%) |
|---|---|---|
| Age, years | 72 (48-89) | 22/56 (39) |
| <70 | 34/56 (61) | |
| ≥70 | ||
| Gender | 37/56 (66) | |
| Male | 19/56 (34) | |
| Female | ||
| FAB classification | 46/56 (82) | |
| Myelodysplastic (CMML-MD) | 10/56 (18) | |
| Myeloproliferative (CMML-MP) | ||
| WHO classification | 49/56 (87) | |
| CMML-1 | 7/56 (13) | |
| CMML-2 | ||
| Hemoglobin level, g/dL | 12.1 (7.2-16.1) | 8/56 (14) |
| <10 | 48/56 (86) | |
| ≥10 | ||
| Leukocyte count, x109/L | 8.0 (3.2-45.0) | 46/56 (82) |
| <13 | 10/56 (18) | |
| ≥13 | ||
| Platelet count, x109/L | 139.0 (25.0-481.0) | 18/56 (32) |
| <100 | 38/56 (68) | |
| ≥100 | ||
| Neutrophil count, x109/L | 4.1 (0.8-30.2) | 9/56 (16) |
| <1.8 | 47/56 (84) | |
| ≥1.8 | ||
| Blasts in BM, % | 2.0 (0.0-15.0) | 53/56 (95) |
| <10 | 3/56 (5) | |
| ≥10 | ||
| RBC transfusion dependency | 50/56 (89) | |
| No | 6/56 (11) | |
| Yes | ||
| Splenomegaly | 34/43 (79) | |
| No | 9/43 (21) | |
| Yes | ||
| Cytogenetics | 51/56 (91) | |
| Normal karyotype | 3/56 (5) | |
| Isolated -Y | 2/56 (4) | |
| Uninformative CC | ||
| CPSS risk group [17] | 42/56 (75) | |
| Low | 9/56 (16) | |
| Intermediate-1 | 5/56 (9) | |
| Intermediate-2 | ||
| GFM CMML model [15] | 37/56 (66) | |
| Low | 17/56 (30) | |
| Intermediate | 2/56 (4) | |
| High | ||
| Progression to AML | 40/56 (71) | |
| No | 16/56 (29) | |
| Yes |
BM: bone marrow; RBC: red blood cell; Uninformative CC: cases with no metaphases; AML: acute myeloid leukemia
Conventional cytogenetics
Conventional cytogenetics was performed in all patients at diagnosis (n=56) and in 12 patients at the time of AML transformation. All patients had low risk cytogenetic features at diagnosis (51 with normal karyotype and three with isolated -Y) except for two cases in which no metaphases were obtained, therefore being considered as uninformative for CC (Table 1). At the time of AML progression, 6 (50%) patients still presented with normal karyotype, while the other 6 (50%) cases had acquired chromosomal aberrations. In 4 out of these 6 patients, these corresponded to high risk cytogenetic abnormalities according to CPSS [5].
Targeted deep sequencing
Targeted deep sequencing was performed in a total of 64 samples, with a mean depth per base per sample of 1256-fold (1256x). More than 95% of the target sequences were analyzed with >100 independent reads and >99% with at least 30 reads. After excluding sequencing and mapping errors a mean of 299 single nucleotide variants (SNVs) and insertions/deletions (indels) were called per sample. After filtering non-silent variants and excluding known polymorphisms, a mean of 4 variants per sample were called as high-probability somatic changes.
Spectrum of gene mutations at diagnosis
Across the entire cohort, 98% (55/56) of patients harbored at least one mutation. Details of all the variants detected in the series can be seen in Supplementary Table S1. Overall, 2 (4%) patients had 1 mutation, 5 (9%) had 2 concurrent mutations, 12 (21%) had 3, 15 (27%) had 4, 11 (20%) had 5, 8 (14%) had 6, and 2 (4%) had 8 (Supplementary Figure S1A). Distribution of the detected mutations across the CMML patients are described in Figure 1. Most frequently affected genes (in >10% of patients) were TET2 (71%), ASXL1 (43%) and SRSF2 (36%); followed by RUNX1 (23%), ZRSR2 (16%), CBL (13%) and NRAS (13%). Mutations detected in 5-10% of patients were found in the following genes: EZH2, CREBBP, UMODL1, SETBP1, SH2B3, NF1, IDH2, SF3B1, KMT2D, CSF3R, JAK2, PTPN11, SMC1A, U2AF1 and DNMT3A (Supplementary Figure S1B). The list of all the affected genes can be seen in Supplementary Table S2, and the mutation type distribution according to each affected gene can be seen in Supplementary Figure S1C. Most of these genes are involved in cell signaling, epigenetic mechanisms and spliceosome machinery (Supplementary Figure S1D). We then examined the correlation between gene mutations in order to identify possible functional interactions across the different affected genes. All genes were included in all statistical analyses, but to ensure a minimum statistical accuracy, from now on we will focus on mutations detected in at least 5 patients. Mutations in ASXL1 frequently co-occurred with mutations in NRAS (P=0.035) and EZH2 (P=0.011). A positive correlation was also found between RUNX1 and CBL (P=0.043). Finally, mutations in SRSF2 correlated with mutations in CBL (P=0.043), but were mutually exclusive with mutations in ZRSR2 (P=0.019).
Figure 1. Distribution of the affected genes across the 56 CMML patients at diagnosis.
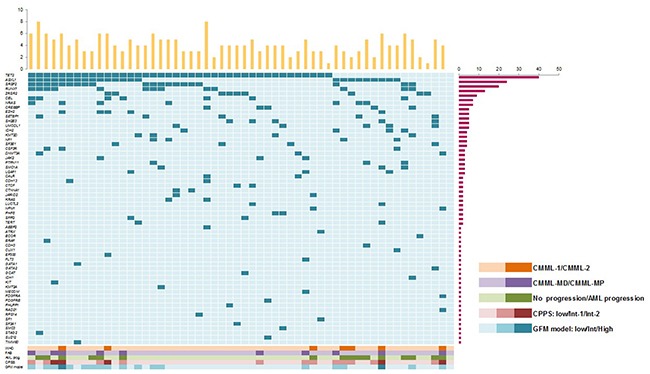
One gene is represented in each line and one patient in each column. Bars at the right represent the number of mutations present in each gene, while columns at the top represent the number of mutations per patient. At the bottom correlations of mutations with WHO, FAB, AML progression, CPSS and CFM model.
Coexistence of gene mutations and loss of heterozygosity
Most of the patients in this cohort (n=48/56, 85.7%) had been previously studied by our group using single nucleotide polymorphism arrays (SNP-A) [18]. Therefore, we investigated whether some of the mutations detected in the present study correlated with the alterations previously detected by SNP-A. Interstitial copy number neutral loss of heterozygosity (CNN-LOH) was detected in 14 of these patients, 10 of which also presented with one mutation affecting a gene located in the region with CNN-LOH. All patients (n=4) with CNN-LOH in 4q24q35 region harbored a TET2 mutation; all patients (n=3) with CNN-LOH in 11q13.3q25 had a mutation in CBL; one patient with CNN-LOH in 7q22.1q36.3 showed a EZH2 mutation, another one with CNN-LOH in 12q21.2q24.33 had a KRAS mutation and one patient with CNN-LOH in 17q25.3 harbored a SRSF2 mutation (Supplementary Table S3). Interstitial CNN-LOH from the four remaining patients affected regions that did not include any of the studied genes (Supplementary Table S3).
Acquisition of mutations during AML progression
Targeted deep sequencing was performed at time of AML transformation in seven patients and at time of CMML-2 progression in one patient. The spectrum of mutations detected per patient was different between diagnosis and AML progression for all except from one patient. In the case that evolved from CMML-1 to CMML-2 it did not differ (Table 2). Number of mutations per patient was higher at time of AML progression in 5/7 (71.4%) patients. Considering alterations detected by both CC and sequencing, median number of alterations at time of progression was higher than at diagnosis (5 alterations at progression vs. 3 alterations at diagnosis, P=0.017). Mutations acquired in all but one patient that progressed to AML affected genes involved in cell signaling pathways that affect cell division, growth, differentiation and survival; such as BRAF, FLT3, KRAS, PTPN11 and NRAS. Of note, the remaining patient that progressed to AML did not acquire any additional mutation, but presented with the intermediate cytogenetic abnormality t(8;16)(p11;13), detected by CC at time of AML progression.
Table 2. List of affected genes in CMML patients that were studied at diagnosis and at time of AML or CMML-2 progression (n=8).
| Data at diagnosis | Data at progression | ||||||
|---|---|---|---|---|---|---|---|
| Diagnosis | N of genes | Gene | Mutation Freq. (%) | Progression | N of genes | Gene | Mutation Freq. (%) |
| CMML-1 | 5 | SETBP1 | 45 | CMML-2 | 5 | SETBP1 | 49 |
| UMODL1 | 46 | UMODL1 | 47 | ||||
| SH2B3 | 43 | SH2B3 | 41 | ||||
| SF3B1 | 48 | SF3B1 | 45 | ||||
| GATA2 | 46 | GATA2 | 41 | ||||
| CMML-1 | 5 | ASXL1 | 49 | AML | 5 | ASXL1 | 47 |
| UMODL1 | 50 | UMODL1 | 51 | ||||
| CDH3 | 40 | CDH3 | 20 | ||||
| NRAS | 15 | NRAS | 6 | ||||
| PTPN11 | 6 | ||||||
| BRAF | 9 | ||||||
| CMML-1 | 2 | RUNX1 | 52 | AML | 4 | RUNX1 | 52 |
| TET2 | 45 | TET2 | 43 | ||||
| SRSF2 | 24 | ||||||
| FLT3 | 22 | ||||||
| CMML-1 | 3 | TET2 | 47 | AML | 6 | TET2 | 48 |
| CBL | 36 | CBL | 5 | ||||
| ASXL1 | 43 | ASXL1 | 46 | ||||
| KMT2D | 51 | ||||||
| KRAS | 42 | ||||||
| AEBP2 | 32 | ||||||
| CMML-2 | 3 | JARID2 | 51 | AML | 4 | JARID2 | 48 |
| TET2 | 44 | TET2 | 43 | ||||
| NPM1 | 31 | NPM1 | 30 | ||||
| GNAS | 20 | ||||||
| CMML-2 | 6 | ASXL1 | 52 | AML | 6 | ASXL1 | 49 |
| CSF3R | 51 | CSF3R | 47 | ||||
| SRSF2 | 41 | SRSF2 | 12 | ||||
| TET2 | 44 | TET2 | 46 | ||||
| NRAS | 47 | NRAS | 47 | ||||
| EZH2 | 39 | EZH2 | 43 | ||||
| CMML-2 | 2 | ASXL1 | 43 | AML | 4 | ASXL1 | 48 |
| ZRSR2 | 19 | ZRSR2 | 22 | ||||
| KMT2D | 22 | ||||||
| PTPN11 | 9 | ||||||
| CMML-2 | 3 | ASXL1 | 34 | AML | 4 | ASXL1 | 39 |
| RUNX1 | 45 | RUNX1 | 50 | ||||
| NF1 | 6 | NF1 | 8 | ||||
| NRAS | 5 | ||||||
Genes that are only affected at time of diagnosis or progression are highlighted in bold
Correlations between gene mutations and clinical variables
We investigated the correlation between mutations detected at diagnosis and main clinical and biological parameters of the patients, including age, sex, CMML FAB and WHO subtypes, BM and PB cell counts, RBC transfusion dependency, presence of splenomegaly, CPSS and GFM models and progression to AML. Mutations in EZH2 gene associated with WHO 2008 CMML-2 subtype (P=0.011), FAB CMML-MP subtype/leukocyte count (P=0.035) and higher risk groups according to CPSS (P<0.001) and GFM (P=0.001) models. Mutations in NRAS correlated with FAB CMML-MP subtype/leukocyte count (P=0.015), presence of splenomegaly (P>0.001) and age <70 years (P=0.012). ASXL1 mutations associated with AML progression (P=0.034), age <70 years (P=0.015) and higher risk groups according to the CPSS (P=0.014) and GFM (P=0.001) models. SRSF2 mutations correlated with platelet count <100 x109/L and higher risk groups according to GFM model (P=0.025). Even though JAK2 mutations were only present in three patients, it is worth highlighting, because it has been previously reported [19], that they associated with FAB CMML-MP subtype/leukocyte count (P=0.004). Interestingly, mutations in TET2 gene were the only ones associated with good prognosis features, such as Hemoglobin>10g/dL (P=0.005), not progression to AML (P=0.008) and lower risk groups according to CPSS (P=0.036).
Univariate survival analyses
We then explored the impact of clinical, biological and genetic data on patients’ outcome (Table 3). Median OS and progression free survival (PFS) of the cohort were 47 months (IC95% 15-79) and 128 months (NA), respectively. The following clinical and biological variables were predictive of both OS and PFS: CMML WHO subtype, CMML FAB subtype, transfusion dependency, presence of splenomegaly, hemoglobin level, leukocyte count, CPSS risk group, alternative CPSS risk group and GFM CMML model. In addition, BM blast percentage and age were also predictive of PFS. Regarding genetic features, total number of mutations was predictive of OS and PFS when patients were stratified into the following subgroups: 0-3 mutations, 4-5 mutations, >5 mutations (Table 3, Supplementary Figure S2A). Focusing on specific genes, mutations in ASXL1, NRAS and EZH2 associated with both shorter OS and PFS. Furthermore, mutations in SRSF2 only associated with inferior OS, while absence of TET2 mutations (TET2wt) associated with inferior PFS but did not correlate with OS (Table 3, Supplementary Figure S2B). Overall, 34/56 (61%) of patients presented with at least one adverse risk gene mutation (ASXL1, EZH2, NRAS and SRSF2). Presence of a mutation in one of these genes correlated with both shorter OS and PFS (Table 3, Figure 2A). Moreover, a decrease in OS and PFS was observed as the number of adverse risk mutations increased. Patients were classified in three groups according to the number of mutations in these genes (0, 1, ≥2), which associated with poorer OS and PFS (Table 3, Figure 2B). Even when patients were classified in four groups (0, 1, 2, ≥3), the statistical association was maintained (Table 3, Supplementary Figure S2C). Recently, Patnaik et al. reported a prognostic interaction between ASXL1 and TET2 mutations in CMML [6], confirming the negative impact in OS imparted by ASXL1 mutations and suggesting a favorable impact from TET2 mutations in the absence of ASXL1 mutations. We investigated this interaction in our cohort of patients and observed that the different combinations between ASXL1 and TET2 mutations were able to stratify patients in subgroups with significantly different OS (Table 3, Supplementary Figure S2D). Regarding PFS, patients with mutations only in TET2 presented a better outcome, but we observed a high overlap between the rest of the categories (Table 3, Supplementary Figure S2D). In order to delineate the benefit of TET2 mutations related to other adverse mutations, we investigated the prognostic interaction between TET2 mutations and adverse risk genes excluding ASXL1, which was able to separate the patients in four distinct prognostic groups with clear different OS and PFS (Table 3, Supplementary Figure S2E). Overall, focusing on patients with mutations in one of these genes (EZH2, NRAS or SRSF2), regardless of TET2 status, they had an unfavorable prognosis, even though patients with TET2 mutations showed a better prognosis in this subset. On the other hand, in the absence of adverse risk gene mutations, patients with TET2 mutations again had a better outcome than patients without, suggesting a protective role for TET2 mutations.
Table 3. Overall survival analyses and progression free survival according to the main clinical, hematological and genetic characteristics of CMML patients at diagnosis (n=56).
| Variable | Overall survival (OS) | Progression free survival (PFS) | ||
|---|---|---|---|---|
| 3 year % OS (95% CI) | Log-rank P value | 3 year % PFS (95% CI) | Log-rank P value | |
| WHO classification | ||||
| CMML-1 | 57 (41, 73) | 0.020 | 75 (60, 90) | <0.001 |
| CMML-2 | 29 (0, 63) | 19 (0, 52) | ||
| FAB classification | ||||
| MD-CMML | 61 (45, 77) | 0.007 | 74 (59, 89) | 0.036 |
| MP-CMML | 23 (5, 51) | 38 (1, 75) | ||
| Sex | ||||
| Male | 50 (32, 68) | 0.246 | 68 (50, 86) | 0.916 |
| Female | 61 (34, 84) | 68 (44, 92) | ||
| Age (years) | ||||
| <70 years | 37 (14, 60) | 0.322 | 42 (18, 67) | 0.003 |
| ≥70 years | 64 (47, 81) | 85 (71, 99) | ||
| Hemoglobin level | ||||
| <10 g/dL | 17 (0, 46) | 0.045 | 42 (2, 82) | 0.009 |
| ≥10 g/dL | 60 (45, 75) | 72 (57, 87) | ||
| Leukocyte count | ||||
| <13×109/L | 61 (45, 77) | 0.007 | 74 (59, 89) | 0.036 |
| ≥13×109/L | 23 (5, 51) | 38 (1, 75) | ||
| Platelet count | ||||
| <100×109/L | 53 (27, 79) | 0.797 | 58 (31, 85) | 0.302 |
| ≥100×109/L | 54 (37, 71) | 72 (55, 89) | ||
| Neutrophil count | ||||
| <1,8×109/L | 65 (33, 97) | 0.880 | 73 (41, 100) | 0.956 |
| ≥1,8×109/L | 52 (36, 58) | 66 (50, 82) | ||
| Blasts in BM | ||||
| <5% | 58 (42, 74) | 0.499 | 76 (61, 91) | 0.008 |
| ≥5% | 40 (10, 70) | 40 (10, 70) | ||
| Splenomegaly | ||||
| Absent | 68 (52, 84) | 0.001 | 78 (62, 94) | 0.004 |
| Present | 0 (NA) | 0 (NA) | ||
| Transfusion requirement | ||||
| Independent | 65 (50, 80) | 0.001 | 74 (59, 89) | 0.010 |
| Dependent | 0 (NA) | 0 (NA) | ||
| CPSS | ||||
| Low | 63 (47, 79) | <0.001 | 81 (67, 95) | <0.001 |
| Intermediate-1 | 50 (15, 85) | 39 (5, 73) | ||
| Intermediate-2 | 0 (NA) | 0 (NA) | ||
| Alternative CPSS | ||||
| Low | 70 (53, 87) | 0.005 | 82 (67, 97) | <0.001 |
| Intermediate-1 | 26 (2, 50) | 39 (6, 72) | ||
| Intermediate-2 | 25 (0, 58) | 25 (0, 68) | ||
| GFM CMML model | 67 (50, 84) | <0.001 | 79 (64, 94) | <0.001 |
| Low | 31 (6, 56) | 42 (8, 76) | ||
| Intermediate | 0 (NA) | 0 (NA) | ||
| Number of mutations | ||||
| 0-3 | 70 (48, 92) | <0.001 | 75 (53, 97) | 0.001 |
| 4-5 | 61 (39, 83) | 80 (61, 99) | ||
| >5 | 10 (0, 29) | 15 (0, 42) | ||
| ASXL1 | ||||
| Wild-type | 69 (52, 86) | 0.027 | 81 (66, 96) | 0.015 |
| Mutated | 30 (2, 52) | 45 (13, 77) | ||
| NRAS | ||||
| Wild-type | 62 (47, 77) | <0.001 | 74 (59, 89) | 0.005 |
| Mutated | 0 (NA) | 0 (NA) | ||
| EZH2 | ||||
| Wild-type | 58 (43, 73) | 0.002 | 72 (57, 87) | 0.004 |
| Mutated | 20 (0, 55) | 27 (0, 71) | ||
| SRSF2 | ||||
| Wild-type | 64 (47, 81) | 0.049 | 69 (52, 86) | 0.835 |
| Mutated | 35 (12, 58) | 65 (37, 93) | ||
| TET2 | ||||
| Wild-type | 41 (14, 68) | 0.476 | 41 (14, 68) | 0.005 |
| Mutated | 59 (42, 76) | 80 (65, 95) | ||
| Presence of adverse risk gene mutationsa | ||||
| No | 81 (64, 98) | 0.008 | 85 (69, 100) | 0.031 |
| Yes | 35 (16, 54) | 52 (29, 75) | ||
| Number of adverse risk gene mutationsa | ||||
| 0 | 81 (64, 98) | <0.001 | 85 (69, 100) | 0.030 |
| 1 | 53 (25, 81) | 16 (0, 36) | ||
| ≥2 | 62 (32, 92) | 48 (19, 77) | ||
| Number of adverse risk gene mutationsa | ||||
| 0 | 81 (64, 98) | 0.001 | 85 (69, 100) | 0.047 |
| 1 | 53 (25, 81) | 62 (32, 92) | ||
| 2 | 20 (0, 44) | 54 (22, 86) | ||
| ≥3 | 0 (NA) | 0 (NA) | ||
| Combination of TET2 and ASXL1 mutations | ||||
| TET2mut-ASXL1wt | 78 (61, 95) | 0.001 | 96 (16, 100) | 0.004 |
| TET2wt-ASXL1mut | 47 (10, 84) | 47 (10, 84) | ||
| TET2wt-ASXL1wt | 34 (0, 72) | 36 (0, 75) | ||
| TET2mut-ASXL1mut | 20 (0, 44) | 42 (3, 81) | ||
| Combination of TET2 and AR gene mutations (excluding ASXL1) | ||||
| TET2mut-ARwt | 83 (65, 100) | 0.001 | 88 (72, 100) | <0.001 |
| TET2wt-ARwt | 64 (31, 97) | 64 (31, 97) | ||
| TET2mut-ARmut | 35 (12, 58) | 76 (55, 97) | ||
| TET2wt-ARmut | 0 (NA) | 0 (NA) | ||
ASXL1, EZH2, NRAS, SRSF2; BM: bone marrow
CI: confidence interval; NA: Not applicable; AR genes (excluding ASXL1): adverse risk genes EZH2, NRAS and SRSF2. Variables with significant impact (P<0.05) in overall survival or progression free survival are highlighted in bold.
Figure 2. Prognostic impact of gene mutations.
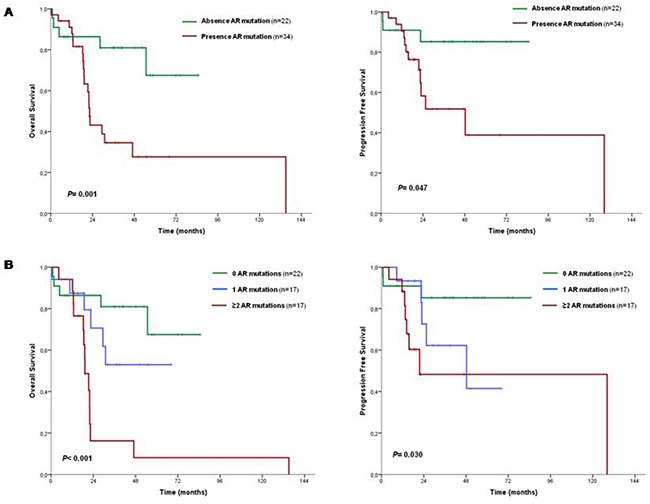
A. OS and PFS curves according to presence or absence of an adverse risk gene; B. OS and PFS curves according to number of mutations in an adverse risk gene. See Table 3 for 3-year percentage overall survival and progression free survival and confidence intervals. AR mutations: adverse risk gene mutations (ASXL1, EZH2, NRAS, SRSF2).
Multivariate survival analyses
Finally, we performed an adjusted multivariate analysis including clinical, biological and genetic features that were statistically significant in the univariate analyses (Table 4). For OS, the variables that remained significant in the multivariate model, taking as baseline the low risk group, were the CPSS scoring system and the presence of at least one adverse risk gene mutation (ASXL1, EZH2, NRAS, SRSF2). Regarding PFS multivariate analysis, the following variables remained significant in the model: CPSS scoring system and the absence of a TET2 mutation (TET2wt).
Table 4. Multivariate model including clinical, biological and genetic characteristics of CMML patients at diagnosis (n=56).
| Overall survival | Progression free survival | ||||||
|---|---|---|---|---|---|---|---|
| Variable | HR | 95% CI | P | Variable | HR | 95% CI | P |
| CPSSa: | 1.2 | 0.4 to 3.4 | 0.005 | CPSSa: | 2.7 | 0.8 to 8.7 | 0.002 |
| Int-1 | 6.2 | 2.0 to 18.8 | 0.695 | Int-1 | 16.5 | 3.4 to 79.4 | 0.093 |
| Int-2 | 0.001 | Int-2 | <0.001 | ||||
| Presence of adverse risk gene mutationsb,c | 2.9 | 1.0 to 8.2 | 0.042 | TET2 wtd | 4.1 | 1.3 to 12.8 | 0.013 |
Reference category: Low risk
ASXL1, EZH2, NRAS, SRSF2
Reference category: No mutations
Reference category: TET2 mutation
CI: confidence interval
DISCUSSION
Over the past few years, next-generation sequencing (NGS) has led to a revolution in the study of hematological malignancies, with remarkable efforts to characterize the genetic basis of these disorders. In the field of CMML recent studies have reported mutations in >90% of patients, affecting genes mainly involved in the following mechanisms: epigenetic regulation (TET2, ASXL1, EZH2, DNMT3A, IDH1/2), spliceosome machinery (SRSF2, ZRSR2, SF3B1, U2AF1), cell signaling and transcription factor regulation (NRAS, KRAS, CBL, JAK2, RUNX1) [6, 7, 11, 15, 20, 21]. Mutations in ASXL1, SRSF2, CBL, IDH2, EZH2, DNMT3A, NRAS and RUNX1 have been associated, in some of these studies, with poorer OS or increased risk of AML progression [6, 7, 11, 14, 15, 21–24]. However, up to date, the only gene that has shown to correlate with worse outcome on multivariate models is ASXL1 [15, 16].
Cytogenetic abnormalities are not common in CMML (20-30%), but when present they confer a significant adverse outcome, except for isolated -Y [5, 8]. Patients with low risk cytogenetic features (normal karyotype and isolated -Y) account for approximately 80% of CMML cases and often fall into the low risk categories of CMML prognostic scores, but the OS and risk of AML transformation differs considerably among them [17]. Therefore, we have focused our study on 56 CMML patients with low risk cytogenetic abnormalities or no metaphases, since CC does not provide prognostic information in all these cases. Our aim is to identify, in this cytogenetically homogeneous cohort, the genetic characteristics of the subset of patients that present with a more aggressive disease.
By performing targeted deep sequencing using a panel of 83 myeloid-related genes, we have detected mutations in 98% of CMML patients at diagnosis. Spectrum of gene mutations does not differ from the ones reported in more heterogeneous CMML cohorts [6, 7, 11, 15]. This study confirms the molecular heterogeneity of the disease. In addition, in the current study, we report for the first time recurrent mutations (5-10%) in CMML in the genes CREBBP, KMT2D and UMODL1, which have been previously reported in lymphoid neoplasms or solid tumors [25, 26]. Studies in larger cohorts should provide more insights in the involvement of these genes in the pathogenesis of CMML. On the other hand, although multiple mutations in different genes can be detected in most of CMML patients, it is interesting to point out that 91% of patients are characterized by harboring a mutation in at least one of the three most recurrent genes in CMML (TET2, ASXL1, SRSF2), which can be used for diagnostic purposes.
Across the entire cohort, 14/48 (29.2%) patients presented with interstitial CNN-LOH, as previously reported by our group [18]. All patients with CNN-LOH in 4q24 harbored a TET2 mutation and all cases with CNN-LOH in 11q23.3 presented with a CBL mutation, confirming the association between both types of molecular events [27, 28]. Similarly, single patients with EZH2, NRAS and SRSF2 mutations also presented CNN-LOH in 7q35-q36, 12p12.1 and 17q25.3, respectively, suggesting that detection of CNN-LOH in CMML indicates the presence of homozygous mutations in genes located in the affected region. The negative impact of CNN-LOH observed in CMML [18] may be influenced or even enhanced by the presence of mutations in genes located in these regions.
Targeted sequencing was also performed in seven patients at time of AML progression and revealed the acquisition of additional mutations in 6/7 (85.7%) patients, all of them with at least one mutation in a gene involved in cell signaling. Five of these patients acquired mutations in components or regulators of the RAS signaling pathway (KRAS, NRAS, PTPN1, BRAF and FLT3), suggesting that activation of RAS pathway is probably involved in the evolution of CMML in some patients. Mutations in these genes have been previously associated with CMML-MP [14, 23]. The remaining patient acquired a mutation in GNAS, a gene involved in the GPCR signaling pathway. Somatic activating mutations in GNAS are common in solid tumors but were recently identified for the first time in hematological neoplasms, in 1% of MDS [29]. Implication of GNAS mutations in MDS or related myeloid neoplasms has not been further investigated. In our study, mutations in ASXL1 and TET2wt at diagnosis correlated with AML progression. This association has already been reported for ASXL1 mutations [14, 15]. Thus, even though acquisition of cell signaling mutations involved in AML progression cannot be anticipated, presence of other mutations at diagnosis may predict AML transformation and serve as prognostic markers for a closer monitoring of these patients or to be considered as candidates to a more aggressive treatment.
Correlation analyses between clinical and biological features and gene mutations revealed interesting associations. Of note, adverse risk genes such as ASXL1, EZH2 and TET2wt, were in many cases associated with high risk parameters, such as CMML-2, lower Hg levels, higher leukocyte counts, higher risk groups of CPSS and GFM models or progression to AML. In addition, as expected, myeloproliferative features such as CMML-MP variant, leukocyte count or presence of splenomegaly, correlated with genes involved in cell signaling, such as NRAS and JAK2.
The prognostic value of individual gene mutations was explored by performing survival analyses and investigating correlation with clinical features. Mutations in ASXL1, EZH2, NRAS and SRSF2 correlated with different clinical or biological features that are known to be associated with worse outcome in CMML [15–17]. Mutations in these four genes were associated with shorter OS in univariate survival analyses and all, except from SRSF2, correlated with shorter PFS as well. Some of these associations have already been reported, but ASXL1 is the only marker that has been shown to be prognostically detrimental on multivariate models [6, 15, 24]. Therefore, impact on presenting one or more adverse risk gene mutations (ASXL1, EZH2, NRAS, SRSF2) was investigated. Presence of mutations in at least one of these genes was predictive of both OS and PFS and was the only variant that remained significant in OS multivariate model in addition to CPSS. Of note, number of adverse risk gene mutations was also predictive of OS and PFS when patients were classified in three (0, 1, ≥2) and four (0, 1, 2, ≥3) subgroups, which suggests an additive negative impact of presenting mutations in these genes. Interestingly, survival outcomes were also affected by the number of total concurrent mutations, meaning that each acquired mutation confers an additional detrimental value, as has been reported in MDS and MPN [12, 30]. Focusing on PFS, TET2wt was also predictive of shorter PFS, supporting the favorable impact of TET2 mutations that has been previously reported but remains controversial [5, 13, 27, 28]. Of note, we detected TET2 mutations at a higher frequency than the one reported in other studies [31-33]. Considering that TET2 mutations are associated with good prognosis features and may play a protective role in CMML, this higher frequency could be explained due to the fact that our cohort of patients is focused on lower risk CMML. Patnaik et al recently reported a prognostic interaction between ASXL1 and TET2 mutations, and suggested a favorable impact from TET2 mutations in the absence of ASXL1 mutations [6]. We investigated this interaction in our cohort and observed that the classification of patients according to the status of these two genes was associated with OS and PFS. Even though the four survival curves did not match the ones reported by Patnaik et al, probably due to the limited number of patients in our cohort in the middle subgroups (TET2wt-ASXL1mut and TET2wt-ASXL1wt), we did confirm the significant negative impact of ASXL1 mutations and the favorable impact of TET2 mutations in the absence of ASXL1 mutations. Interestingly, the group of TET2wt-ASXL1wt had a quite unfavorable prognosis, worse than the TET2-wt/ASXL1-mut, which is what Patnaik et al described and what would be expected. This could be explained because some of the patients in the TET2wt-ASXL1wt group (n=7) carry mutations in other adverse risk genes. Specifically, two of them carried SRSF2 mutations and two carried RUNX1 mutations, which also have been reported to have an adverse prognostic impact, even though this was not confirmed in our cohort [15]. The prognostic interaction between TET2 and other adverse risk gene mutations was also analyzed. The analysis confirmed that the negative impact of mutations in adverse risk genes prevails over TET2 mutations, although patients with TET2 mutations have a better outcome compared to patients without, suggesting a protective role for TET2 mutations. The analysis also revealed that in the absence of adverse risk gene mutations, TET2 mutations confer the best outcome. This beneficial impact of TET2 mutations was even more noticeable in the PFS analysis, which Patnaik et al did not report. Furthermore, TET2wt was the only significant variant in the PFS multivariate model in addition to CPSS.
Although our series is limited by the number of samples compared to other series, it is the only one mainly focused on CMML patients with low risk cytogenetic features. In addition, the results of multivariate analyses for both OS and PFS suggest that our findings may be applicable to larger series of patients. Our multivariate model confirms the prognostic impact of the CPSS scoring system for both OS and PFS and implies that molecular studies can add prognostic value to this model, especially in patients with low risk cytogenetic features (normal karyotype, isolated -Y) or uninformative CC.
In summary, we report mutations in nearly all patients with CMML and low risk cytogenetic features, some of which have a negative impact on the outcome of patients. With NGS technologies being more accessible each day, we would recommend to perform targeted molecular analysis of ASXL1 and, if possible, EZH2, NRAS, SRSF2 and TET2 in patients with CMML and low risk cytogenetic features or uninformative CC. This may allow to identify patients that are more likely to present with an aggressive disease evolution and that could benefit from closely monitoring and more intensive treatments.
MATERIALS AND METHODS
Patients and samples
A retrospective study was performed on a total of 56 patients with CMML from Institut Català d’Oncologia (ICO). Patients were diagnosed according to the FAB [2] and 2008 WHO [1] classifications. Cases with CMML and low risk cytogenetic features (normal karyotype or isolated -Y) or no metaphases at diagnosis were included in the study. Study approval was obtained from ICO-Hospital Germans Trias i Pujol Ethics Committee. Informed consent was obtained from all patients, in accordance with the Declaration of Helsinki.
Cytogenetics
Conventional G-banding cytogenetics was performed on bone marrow samples at diagnosis following standard procedures [5]. We analyzed 20 metaphases per sample (n=54) except in two cases in which no metaphases were obtained. Karyotypes were described according to the International System for Human Cytogenetic Nomenclature 2013 [34].
DNA samples
Samples were collected at diagnosis for all patients and at time of AML progression in 7 patients and CMML-2 progression in one case. Whole bone marrow samples (n= 58) or peripheral blood (n=6) were used. Genomic DNA was extracted with QiaAmp DNA Blood Mini kit (Qiagen, Hilden, Germany) and quantified using Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, CA, USA).
Targeted deep sequencing
Targeted deep-sequencing of a panel of 83 myeloid-related genes was performed in all samples (Supplementary Table S4). Indexed libraries were prepared with 1μg of double strand genomic DNA using the Kapa Library Preparation Kit (Kapa Biosystems, MA, USA). Custom target capture enrichment using the SeqCap EZ capture chemistry (Nimblegen, Roche, Basel, Switzerland) was performed on pools of 8 libraries. Multiplexed captured libraries were sequenced on an Illumina MiSeq following a 150bp paired-end reads standard protocol.
Targeted sequencing data analysis
Sequencing data were analyzed using an in-house pipeline. Reads were aligned against human genome build 19 (hg19) using BWA 0.7.12 [35]. Post-alignment including local indel realignment and base recalibration was performed using the tools in GATK 3.4.46 software package [36]. Packages SAMtools 1.2 and VarScan 2.4.0 were used for variant calling and ANNOVAR (version 2015Jun17) for variant annotation [37, 38]. High-probability oncogenic mutations were called by eliminating sequencing and mapping errors and by discarding variants located in highly variable regions or with low coverage, as well as SNPs described on the available databases and synonymous variants. Variants were also filtered according to the variant allele frequency (VAF): all variants with VAF ≥5% were reported, as well as variants with VAF<5% and at least 25 reads for the variant that are known hotspots and have been reported in hematological neoplasms.
Statistical analysis
Baseline characteristics were described as frequency and percentage for categorical variables and median and range for quantitative variables. Comparisons of categorical variables between patient subsets were compared using χ2 or Fisher's exact test, when appropriate, while median test was used to compare continuous variables. Comparisons of paired data for continuous variables were performed with the Wilcoxon test. OS was defined as time from diagnosis to the last follow-up or death from any cause and PFS as time from diagnosis to progression to AML (presence of ≥20% of blasts in bone marrow or peripheral blood) or death from CMML [39]. Survival curves were calculated using the Kaplan-Meier method and log-rank test was used for comparisons between groups. Multivariate analysis was performed using Cox proportional-hazards regression model, considering Wald Backward as selection method. Two-sided P values <0.05 were considered as statistically significant. The statistical package SPSS, version 23.0 (SPSS Inc., Chicago, IL, USA) was used for all analyses.
SUPPLEMENTARY MATERIALS FIGURES AND TABLES
Acknowledgments
The authors would like to thank Diana Domínguez, Isabel Granada, Paula Gómez, Maria Pilar Armengol and Anna Oliveira for their technical assistance.
Footnotes
CONFLICTS OF INTEREST
The authors have no potential conflicts of interest to disclose.
GRANT SUPPORT
This work was supported in part by a grant from the Ministerio de Educación Cultura y Deporte (FPU13/03770); Instituto de Salud Carlos III, Ministerio de Economia y Competividad, Spain (PI 11/02519; PI 11/02010; PI 14/00013); Red Temática de Investigación Cooperativa en Cáncer (RTICC, FEDER) (RD12/0036/0044); 2014 SGR225 (GRE) Generalitat de Catalunya; with economical support from Fundació Internacional Josep Carreras, Obra Social “la Caixa” and Celgene Spain
REFERENCES
- 1.Orazi A, Bennett JM, Germing U, Brunning RD, Bain BJ, Thiele J. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC; 2008. Chronic Myelomonocytic Leukemia. [Google Scholar]
- 2.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick H, Sultan C, Cox C. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br J Haematol. 1994;87:746–54. doi: 10.1111/j.1365-2141.1994.tb06734.x. [DOI] [PubMed] [Google Scholar]
- 3.Schuler E, Schroeder M, Neukirchen J, Strupp C, Xicoy B, Kündgen A, Hildebrandt B, Haas R, Gattermann N, Germing U. Refined medullary blast and white blood cell count based classification of chronic myelomonocytic leukemias. Leuk Res. 2014;38:1413–9. doi: 10.1016/j.leukres.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 5.Such E, Cervera J, Costa D, Solé F, Vallespí T, Luño E, Collado R, Calasanz MJ, Hernández-Rivas JM, Cigudosa JC, Nomdedeu B, Mallo M, Carbonell F, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96:375–83. doi: 10.3324/haematol.2010.030957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patnaik MM, Lasho TL, Vijayvargiya P, Finke CM, Hanson CA, Ketterling RP, Gangat N, Tefferi A. Prognostic interaction between ASXL1 and TET2 mutations in chronic myelomonocytic leukemia. Blood Cancer J. 2016;6:e385. doi: 10.1038/bcj.2015.113.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, Kohlmann A, Alpermann T, Yoshida K, Ogawa S, Koeffler HP, Kern W, Haferlach C, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML) Blood. 2012;120:3080–8. doi: 10.1182/blood-2012-01-404863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wassie EA, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, Ketterling RP, Solary E, Tefferi A, Patnaik MM. Molecular and prognostic correlates of cytogenetic abnormalities in chronic myelomonocytic leukemia: a Mayo Clinic-French Consortium Study. Am J Hematol. 2014;89:1111–5. doi: 10.1002/ajh.23846. [DOI] [PubMed] [Google Scholar]
- 9.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, Kosmider O, Le Couedic J-P, Robert F, Alberdi A, Lécluse Y, Plo I, Dreyfus FJ, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 10.Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, Chaffanet M, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- 11.Jankowska AM, Makishima H, Tiu RV, Szpurka H, Huang Y, Traina F, Visconte V, Sugimoto Y, Prince C, O’Keefe C, Hsi ED, List A, Sekeres MA, et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011;118:3932–41. doi: 10.1182/blood-2010-10-311019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, Shlien A, Groves MJ, Forbes SA, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–3627. doi: 10.1182/blood-2013-08-518886. quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tefferi A, Lim K-H, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, Hanson CA, Pardanani A, Gilliland DG, Levine RL. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23:1343–5. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gelsi-Boyer V, Trouplin V, Roquain J, Adélaïde J, Carbuccia N, Esterni B, Finetti P, Murati A, Arnoulet C, Zerazhi H, Fezoui H, Tadrist Z, Nezri M, et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol. 2010;151:365–75. doi: 10.1111/j.1365-2141.2010.08381.x. [DOI] [PubMed] [Google Scholar]
- 15.Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, Berthon C, Adès L, Fenaux P, Beyne-Rauzy O, Vey N, Braun T, Haferlach T, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31:2428–36. doi: 10.1200/JCO.2012.47.3314. [DOI] [PubMed] [Google Scholar]
- 16.Patnaik MM, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, Knudson RA, Ketterling RP, Tefferi A, Solary E. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014;28:2206–12. doi: 10.1038/leu.2014.125. [DOI] [PubMed] [Google Scholar]
- 17.Such E, Germing U, Malcovati L, Cervera J, Kuendgen A, Della Porta MG, Nomdedeu B, Arenillas L, Luño E, Xicoy B, Amigo ML, Valcarcel D, Nachtkamp K, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood. 2013;121:3005–15. doi: 10.1182/blood-2012-08-452938. [DOI] [PubMed] [Google Scholar]
- 18.Palomo L, Xicoy B, Garcia O, Mallo M, Ademà V, Cabezón M, Arnan M, Pomares H, José Larrayoz M, José Calasanz M, Maciejewski JP, Huang D, Shih L-Y, et al. Impact of SNP array karyotyping on the diagnosis and the outcome of chronic myelomonocytic leukemia with low risk cytogenetic features or no metaphases: SNP-a karyotyping impact on the diagnosis and outcome of patients with chronic myelomonocytic leukemia. Am J Hematol. 2016;91:185–92. doi: 10.1002/ajh.24227. [DOI] [PubMed] [Google Scholar]
- 19.Pich A, Riera L, Sismondi F, Godio L, Davico Bonino L, Marmont F, Francia di Celle P. JAK2V617F activating mutation is associated with the myeloproliferative type of chronic myelomonocytic leukaemia. J Clin Pathol. 2009;62:798–801. doi: 10.1136/jcp.2009.065904. [DOI] [PubMed] [Google Scholar]
- 20.Patnaik MM, Parikh SA, Hanson CA, Tefferi A. Chronic myelomonocytic leukaemia: a concise clinical and pathophysiological review. Br J Haematol. 2014;165:273–86. doi: 10.1111/bjh.12756. [DOI] [PubMed] [Google Scholar]
- 21.Cervera N, Itzykson R, Coppin E, Prebet T, Murati A, Legall S, Vey N, Solary E, Birnbaum D, Gelsi-Boyer V. Gene mutations differently impact the prognosis of the myelodysplastic and myeloproliferative classes of chronic myelomonocytic leukemia. Am J Hematol. 2014;89:604–9. doi: 10.1002/ajh.23702. [DOI] [PubMed] [Google Scholar]
- 22.Kuo M-C, Liang D-C, Huang C-F, Shih Y-S, Wu J-H, Lin T-L, Shih L-Y. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23:1426–31. doi: 10.1038/leu.2009.48. [DOI] [PubMed] [Google Scholar]
- 23.Ricci C, Fermo E, Corti S, Molteni M, Faricciotti A, Cortelezzi A, Lambertenghi Deliliers G, Beran M, Onida F. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res Off J Am Assoc Cancer Res. 2010;16:2246–56. doi: 10.1158/1078-0432.CCR-09-2112. [DOI] [PubMed] [Google Scholar]
- 24.Grossmann V, Kohlmann A, Eder C, Haferlach C, Kern W, Cross NCP, Haferlach T, Schnittger S. Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia. 2011;25:877–9. doi: 10.1038/leu.2011.10. [DOI] [PubMed] [Google Scholar]
- 25.Chung YR, Schatoff E, Abdel-Wahab O. Epigenetic alterations in hematopoietic malignancies. Int J Hematol. 2012;96:413–27. doi: 10.1007/s12185-012-1181-z. [DOI] [PubMed] [Google Scholar]
- 26.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H, Sekeres MA, Wang XF, McDevitt MA, Maciejewski JP. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68:10349–57. doi: 10.1158/0008-5472.CAN-08-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, O’Keefe CL, Ganetzky R, McDevitt MA, Maciejewski JP. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113:6403–10. doi: 10.1182/blood-2009-02-205690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guglielmelli P, Lasho TL, Rotunno G, Score J, Mannarelli C, Pancrazzi A, Biamonte F, Pardanani A, Zoi K, Reiter A, Duncombe A, Fanelli T, Pietra D, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28:1804–10. doi: 10.1038/leu.2014.76. [DOI] [PubMed] [Google Scholar]
- 31.Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gäken J, Lea NC, Przychodzen B, Mian SA, Nasser EE, Shooter C, Westwood NB, Strupp C, Gattermann N, et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood. 2010;116:3923–32. doi: 10.1182/blood-2010-03-274704. [DOI] [PubMed] [Google Scholar]
- 32.Kohlmann A, Grossmann V, Klein H-U, Schindela S, Weiss T, Kazak B, Dicker F, Schnittger S, Dugas M, Kern W, Haferlach C, Haferlach T. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28:3858–65. doi: 10.1200/JCO.2009.27.1361. [DOI] [PubMed] [Google Scholar]
- 33.Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, Quesnel B, Fenaux P, Bastie J-N, Beyne-Rauzy O, Stamatoulas A, Dreyfus F, Ifrah N, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676–81. doi: 10.3324/haematol.2009.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaffer LG, McGowan-Jordan J, Schmid M. ISCN2013: An International System for Human Cytogenetic Nomenclature. Basel, Switzerland: Karger; 2013. [Google Scholar]
- 35.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma Oxf Engl. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinforma Oxf Engl. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, Pinto A, Beran M, de Witte TM, Stone RM, Mittelman M, Sanz GF, Gore SD, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419–25. doi: 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.