

Abstract
Background
Apoptosis has been demonstrated to be an important upstream event in the pathogenesis of chronic obstructive pulmonary disease (COPD). Cyclooxygenase-2 (COX-2) seems to be biologically relevant in COPD. However, the role of COX-2 in the apoptosis in vascular endothelial cells induced by cigarette smoke extract (CSE) remains to be elucidated. Our recent study found that the prostacyclin, one of the COX products in the microvascular endothelium, inhibited apoptosis in the emphysematous lungs of rats induced by CSE. In order to clarify the role of COX-2 in the apoptosis of vascular endothelial cells induced by CSE, we performed the present experiment to elucidate it.
Methods
Twenty surgical lung specimens were obtained from 6 patients with COPD, 7 smoking controls and seven nonsmoking controls. The apoptotic index (AI) and COX-2 protein expression were detected in lung tissues. To further investigate the effects of CSE on the apoptosis and COX-2 expression in a human vascular endothelial cell line, the apoptosis rate and COX-2 expression were examined in human umbilical vein endothelial cells (ECV304) under exposure to varied concentrations of CSE as well as under exposure to 5.0% CSE for varied durations. Repeatedly, the apoptosis rate and COX-2 expression in ECV304 cells under 5.0% CSE were examined after exposing to varied concentrations of celecoxib, a highly selective COX-2 inhibitor.
Results
Significantly increased AI and expression of COX-2 were found both in the lungs of patients with COPD and smoking controls compared with nonsmoking controls. The CSE induced apoptosis in ECV304 cells in means of both dose-dependent and time-dependent manners. The COX-2 was slightly expressed in the cells after exposing to 5% CSE for 3 and 6 h, and markedly expressed after the exposure time for 9 and 12 h, but vanished after 24 h of the exposure. Of interest, with the completely block of the COX-2 expression by celecoxib at 50.0 µmol/L, the apoptosis rate was markedly increased again in ECV304 cells under exposure to 5.0% CSE.
Conclusions
Endothelial cell apoptosis and the expression of COX-2 protein were increased in both COPD patients and CSE-induced vascular endothelial cells. Of interest, it seems that the COX-2 probably had a protective role against the apoptosis in the vascular endothelial cells induced by cigarette smoking.
Keywords: Cyclooxygenase-2 (COX-2), vascular endothelial cell, apoptosis, cigarette smoking extract (CSE), celecoxib
Introduction
Chronic obstructive pulmonary disease (COPD) is a major and increasing global health problem which was the third leading cause of death worldwide in 2010 and the prevalence and burden of COPD are predicted to increase in the coming decades (1,2). In addition to the well-known mechanisms in which the chronic inflammation of airways, extracellular matrix destruction and oxidative stress are thought to contribute to the pathogenesis of COPD (3), the apoptosis of structural cells in the lung is recently suggested to be an important upstream event in the pathogenesis of COPD (4-9). Our previous studies demonstrated that the apoptosis was involved in both alveolar epithelial cells and endothelial cells in the parenchyma of COPD and that the apoptotic cells were increased in emphysematous lung parenchyma in both animal model and patients (10-13).
The major risk factor in the development of COPD is cigarette smoking that contains thousands of chemical components, including more than 10 (14) reactive species in the gas phase alone (15). Cigarette smoking induces oxidative stress, which might induce apoptosis via the intrinsic or mitochondrial pathways (14,15). Our previous study revealed that the apoptotic alveolar cells were increased through caspase-3 activation in emphysematous lung of rats induced by injection of cigarette smoking extract (CSE) (10). In addition, it is reported that CSE could trigger apoptosis of pulmonary endothelial cells by decreasing the expression of vascular endothelial growth factor (16). However, the exact mechanisms regarding the apoptosis in the role of cigarette smoking-induced emphysema have not yet been elucidated thus far.
Cyclooxygenase is the rate-limiting enzyme in the metabolic pathway that transforms arachidonic acid into prostaglandins (PGs). Cyclooxygenase-2 (COX-2) is an inducible isoform of COX and up-regulated by a variety of stimuli including growth factors, cytokines, and tumor promoters (17-19). The expression of COX-2 is induced by acrolein (a key component of cigarette smoke) in rat lung epithelial cells through NF-κB pathway (20). Our laboratory previously found that the COX-2 and its product, PG E2, were increased in sputum and involved in airway inflammation and also possibly contributed to the severity of airflow limitation during the progression of COPD (21). Moreover, CSE was reported to increase the expression of COX-2 in human pulmonary microvascular endothelial cells, which was proposed to be associated with the intensity of endothelial cell apoptosis (22). However, our recent study demonstrated prostacyclin, the major COX product of microvascular endothelium, protect against the development of emphysema by decelerating apoptosis (10).
The purpose of this study is to observe COX-2 expression in the lung tissues of COPD patients. In addition, we investigated the role of COX-2 in the apoptosis induced by CSE through analyzing the effects of CSE on the apoptosis and COX-2 expression with and without celecoxib, a highly selective COX-2 inhibitor, in a human vascular endothelial cell line.
Methods
Lung tissue study
The lung tissue of patients with COPD and controls
This study was approved by the Institutional Review Board of Central South University and performed in accordance with the recommendations of the Helsinki Declaration of 1975 (23). Written informed consent was obtained from all subjects. We recruited consecutive patients from June 2007 to April 2010. We obtained lung tissues from twenty patients that were suspected primary lung cancer of stage 1 (UICC 2009) and had the surgery of pulmonary segmentectomy or lobectomy. According to the COPD diagnostic criteria (24) and smoking history, the patients were divided into three groups including non-smokers without COPD, smokers without COPD and smokers with COPD (Table 1). All the patients with COPD were clinically stable for 4 weeks without acute pulmonary infection, did not receive chemotherapy half a year before the study, and were absence of obstructive atelectasis, metastasis, other pulmonary diseases and severe diseases in other systems. The lung tissues were taken from areas distant from the cancerous lesions during resection operation. The pathological examination confirmed that these samples presented lung structure without metastasis or inflammation. The fresh lung tissues from patients were stored under –70 °C for later tissue examinations of apoptosis by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) and COX-2 protein expression.
Table 1. Lung function of the study subjects.
| Characteristics | Nonsmoking controls (n=7) | Smoking controls (n=7) | COPD (n=6) |
|---|---|---|---|
| Male/Female, n | 3/4 | 4/3 | 4/2 |
| Age, years | 51.5±14.3 | 57.7±8.2 | 59.8±15.8 |
| Smoking, p.y | – | 18.8±4.8* | 48.7±25.3*# |
| FEV1pred, % | 110.1±12.1 | 96.5±6.6 | 75.5±9.7*# |
| FEV1/FVC, % | 81.8±1.7 | 75.8±2.9 | 60.3±5.0*# |
Continues data are expressed as the mean ± SD. *, P<0.05 in comparison to the nonsmoking control group; #, P<0.05 in comparison to the smoking control group. SD, standard deviation.
Detection of apoptosis in lung tissues of patients with COPD and controls
TUNEL was performed to label the DNA damaged cells in human tissues by using in Situ Cell Death Detection Kit, POD (Roche GER) following the manufacturer’s instructions. The apoptotic index (AI) was calculated as the percentage of TUNEL-positive nuclei in a total of more than 3,000 nuclei randomly counted for each lung at ×400 magnification.
Detection of COX-2 expression in lung tissues of patients with COPD and controls
We assessed the expression of COX-2 protein in lung tissues by Western blotting (21). The total protein in cells was extracted and measured using the Bradford assay. The protein (60 µg) was boiled for 5 min with an equal volume of ×2 gel loading buffer (100 mol/L Tris-HCl, 10% β-mercaptoethanol, 4% SDS, 0.2% bromophenol blue, and 20% glycerol) and separated by electrophoresis (8% SDS-PAGE) at 100 V for 2 h. The protein bands were transferred to the hybond-enhanced chemiluminescence (ECL) membrane, and then probed with goat anti-human COX-2 antibody (1:250) (Santa Cruz CA, USA) at 4 °C overnight. The blots were then incubated with a horseradish peroxidase-linked anti-goat secondary antibody (1:2000) (Santa Cruz CA, USA). Antibody labeling was detected by ECL (Santa Cruz CA, USA). Band detection was performed using Quantity One 4.62 Analysis Software (Bio-Rad Laboratories, Hercules, CA, USA).
Cell study
ECV304 cells culture
ECV304 cells were obtained from Xiangya central experiment lab (Central South University, Changsha, China). ECV304 cells were grown in RPMI 1640 (containing 10% fetal calf serum, 1% L-glutamine, 100 U/mL penicillin, and 100 U/ml streptomycin, purchased from Gibco BBL (NY, USA) to confluence at 37 °C in a humidified atmosphere of 5% CO2.
Preparation of CSE
CSE was prepared as previously reported with modification (22). Briefly, one cigarette without filter was burned and the smoke passed through 10 mL of phosphate-buffered saline (PBS) using a vacuum pump. This 100% CSE was adjusted to a pH of 7.4 and filtered through a 0.22 µm pore filter to remove large particles and bacteria, and the CSE was diluted to the concentration of 0%, 0.5%, 1%, and 5% in each and added to endothelial cells within 30 minutes of preparation. Cigarettes of a domestic brand were obtained from Changde Tobacco (Changde, Hunan, China).
Design of the cells study
There were three experiments in this part of study: (I) experiment to observe dose-dependent effect of CSE on apoptosis and COX-2 expression: ECV304 cells were cultured with varied concentrations of CSE at 0.0%, 0.5%, 1.0%, and 5.0% for 12 h individually; (II) experiment to observe time-dependent effect of 5% CSE on apoptosis and COX-2 expression: ECV304 cells were cultured with 5.0% CSE for varied durations of 0, 3, 6, 9, 12 and 24 h individually; (III) experiment to observe the effects of celecoxib on apoptosis and COX-2 expression: ECV304 cells under exposure to 5.0% CSE were cultured with varied concentrations of celecoxib, a selective COX-2 inhibitor, at 0.0, 2.5, 5.0, 10.0, 20.0, and 50.0 µmol/L for 9 h individually. Celecoxib was purchased from Hefei Scenery Chemical Co., Ltd. (Hefei, China) and was dissolved in 0.5% dimethyl sulfoxide (DMSO).
Detection of apoptosis in ECV304 cells
TUNEL staining
TUNEL was performed to detect the characteristic features of apoptotic nuclei in ECV304 cells as we described in the lung tissue study.
Flow cytometry
The experimental procedure was described previously (25). Trypsinized adherent ECV304 cells were collected by centrifugation at 1,000 r/min for 5 min, washed in PBS, fixed with ice-cold 70% ethanol for at least 24 h and then treated with 20 mg/L RNase for 30 min. Propidium iodide was added to a final concentration of 50 mg/L. The samples were analyzed using an EPICS-XL MCL cytometer (Beckman-Coulter Inc., Fullerton, CA, USA) and the Mcycle program (Coulter) for evaluating cell death.
Detection of COX-2 expression in ECV304 cells
Western blotting
We assessed the expression of COX-2 protein in ECV304 cells by Western blotting as we described in the lung tissue study.
Immunocytochemistry
The expression of COX-2 was further determined and quantified by immunocytochemistry according to manufacturer’s instruction. The slide was fixed for 15 min with 4% paraformaldehyde for 90 min. Avidine and biotin block was performed, and endogenous peroxidase was quenched by 3% hydrogen peroxide. After blocking with 5% normal goat serum, goat anti-human COX-2 antibody IgG (1:50) was applied overnight at 4 °C. Cells treated with 5 mg/L lipopolysaccharide (LPS) were as positive control and PBS substituted for the COX-2 antibody was as a negative control. The sections were washed with PBS and incubated with anti-goat secondary antibody. After washing, the sections were stained with DAB. Ipp6.0 software (Media Cybernetics, Inc., Bethesda, MD, USA) was used to determine the integrated optical density (IOD) that quantified the expression of COX-2 in ECV304 cells.
Statistical Analysis
Each experiment was replicated three times. A software package (SPSS 13.0, SPSS Inc., Chicago, IL, USA) was used to perform all statistical analyses and the Students’ t-test was used to assess significant differences in continuous data with Gaussian distribution, while the Mann-Whitney U-test was used for non-normal distribution. Continuous data were expressed as the mean ± standard deviation (SD). Any differences among the groups within each experiment were evaluated by a One-Way ANOVA. A P value less than 0.05 was considered statistically significance.
Result
Apoptosis in vascular endothelial cells in lung tissues of patients with COPD and controls
Twenty surgical lung tissues were obtained from where patients with suspected primary lung cancer of stage 1 (UICC 2009) and had the surgery of pulmonary segmentectomy or lobectomy, the patients were divided into three groups including non-smokers without COPD, smokers without COPD and smokers with COPD (Table 1). TUNEL positive cells were localized in the vascular endothelial cells in both patients with COPD, smoking controls and nonsmoking controls (Figure 1). AI of medium-sized vessels was significantly increased in the lungs of patients with COPD (13.8%±1.9%) compared with both that in the lungs of smoking controls (9.6%±0.8%, P<0.05) and nonsmoking controls (5.9%±1.0%, P<0.05). Additionally, the AI was significantly higher in the smoking controls than that in the nonsmoking controls (P<0.05) in the lung tissues.
Figure 1.

Terminal deoxynucleotide transferase-mediated dUTP nick-end labelling (TUNEL) staining of vascular endothelial cells in human lung tissue (magnification, ×400). There were fewer positive signals in nonsmoking controls (A) and medium-sized vessels of smoking controls (B). The positive signals for TUNEL staining (arrows) were predominantly detected in medium-sized vessels (C) in patients with chronic obstructive pulmonary disease (COPD). Apoptotic index of each group was calculated (D).
COX-2 protein expression in lung tissues of patients with COPD and controls
As showed in Figure 2, the Western blotting showed that the COX-2 protein expression was significantly increased in both the lung tissue of patients with COPD and smoking controls compared with nonsmoking smoking controls. In addition, the expression of COX-2 is significantly higher in the patients with COPD than that of smoking controls.
Figure 2.
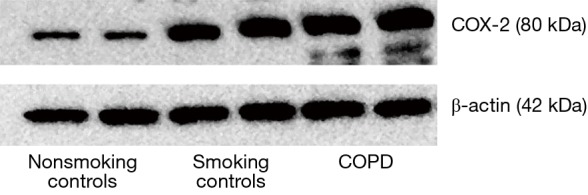
The level of cyclooxygenase-2 (COX-2) protein expression in the lung tissue increased both in the patients with chronic obstructive pulmonary disease (COPD) and the smoking controls in comparison to that in the nonsmoking controls.
CSE induces apoptosis in ECV304 cells in means of dose-dependent and time-dependent manners
After exposing ECV304 cells to varied CSE concentrations in 0.0%, 0.5%, 1.0%, and 5.0% for 12 h individually, both TUNEL staining and flow cytometry showed that the apoptosis rate initially increased at the 1.0% CSE and furthered at the 5.0% CSE, suggesting a dose-dependent effect of CSE on the apoptosis in ECV304 cells (Figures 3,4). Specifically, the apoptosis rate of ECV304 cells was significantly increased at the 5.0% CSE (32.3%±3.22% by TUNEL staining and 5.40%±0.39% by flow cytometry) in comparison to that at the 0.0% CSE (6.5%±0.5% and 1.61%±0.24%, respectively), 0.5% CSE (7.6%±0.4% and 2.01%±0.36%, respectively), and 1.0% CSE (13.5%±1.8% and 2.31%±0.26%, respectively) (P<0.05 in all). Meanwhile, the apoptosis rate of ECV304 cells exposed to 5.0% CSE was gradually increased along with the exposure time. In the flow cytometry, the apoptosis rate of ECV304 cells exposed to 5.0% CSE was 1.73%±0.16% for 0 h, 2.69%±0.28% for 3 h, 2.86%±0.15% for 6 h, 3.66%±0.40% for 9 h, 5.39%±0.57% for 12 h and 8.87%±0.41% for 24 h. The apoptosis rate was significantly increased after 24 h of the exposure in comparison to those less than 12 h of the exposure (Figure 5).
Figure 3.

The transferase-mediated dUTP nick-end labelling (TUNEL) staining shows the apoptosis increased in ECV304 cells under exposure to 5% of cigarette smoking extract (CSE) compared with that under 0.0% CSE (A,B) for 12 h (magnification, ×400). The flow cytometry (C-F) shows the apoptosis occurred in ECV304 cells under exposure to varied concentrations of CSE at 0.0% (C), 0.5% (D), 1.0% (E), and 5.0% (F) for 12 h individually.
Figure 4.
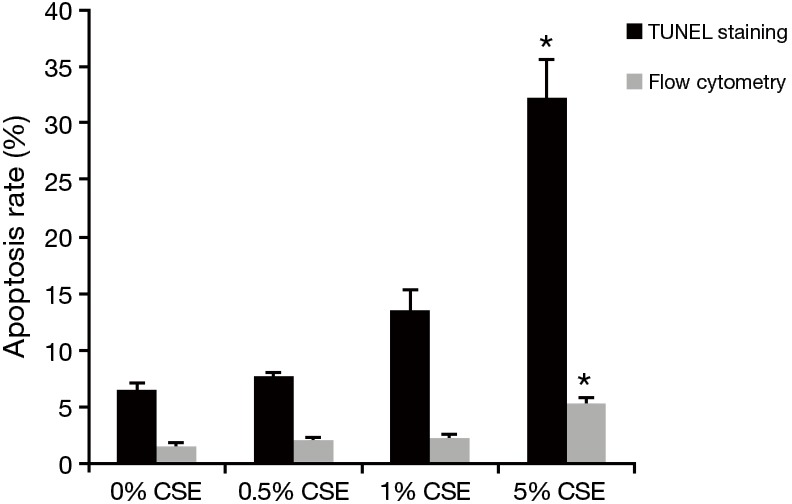
The apoptosis rate of ECV304 cells under exposure to varied concentrations of cigarette smoking extract (CSE) at 0.0%, 0.5%, 1.0%, and 5.0% for 12 h individually measured by transferase-mediated dUTP nick-end labelling (TUNEL) staining (black bar) and flow cytometry (gray bar). *, P<0.05 in comparison to others.
Figure 5.

The flow cytometry shows the apoptosis of ECV304 cells under exposure to 5.0% cigarette smoking extract (CSE) for 0, 3, 6, 9, 12, and 24 h (A). The apoptosis rate of ECV304 cells under exposure to 5.0% CSE for 0, 3, 6, 9, 12, and 24 h measured by flow cytometry (B). *, P<0.05 in comparison to 0 h of exposure; †, P<0.05 in comparison to 0, 3, 6, 9 h of exposure; §, P<0.05 in comparison to 0, 3, 6, 9, 12 h of exposure.
COX-2 expression was vanished in ECV304 cells after 24 h of exposure to 5.0% CSE
As shown in Figure 6A, the COX-2 was slightly expressed in ECV304 cells with 1.0% CSE after 9 h, and the expression was markedly increased in the cells with 5.0% CSE, suggesting a dose-dependent effect of CSE on the COX-2 expression in ECV304 cells. We thus selected the 5.0% CSE as an exposure concentration to observe the COX-2 expression in ECV304 cells by the time trend. We found that the COX-2 was slightly expressed in the cells after 3 and 6 h of the exposure, and then markedly expressed in the cells after 9 and 12 h, however, no COX-2 expression after 24 h of the exposure (Figure 6B).
Figure 6.
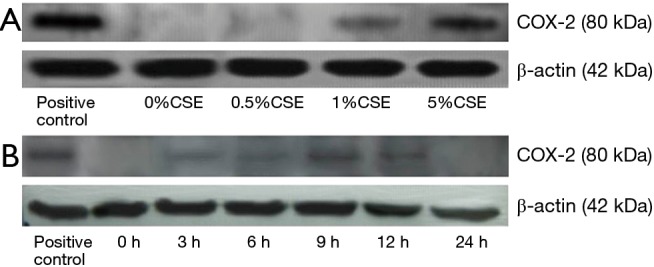
The Western blotting shows the expression of cyclooxygenase-2 (COX-2) protein in ECV304 cells under exposure to varied concentrations of cigarette smoking extract (CSE) at 0%, 0.5%, 1.0%, and 5.0% (A), as well as under exposure to 5.0% CSE for 0, 3, 6, 9, 12 and 24 h (B).
Quantifying the COX-2 expression by IOD in immunocytochemistry, as shown in Figure 7, it was indeed that the COX-2 expression showed dose-dependent increase in ECV304 cells under exposures of varied CSE concentration of 0.0%, 0.5%, 1.0%, and 5.0% for 9 h. The IOD of COX-2 in ECV304 cells was 36.8%±2.1% under exposure to 0.0% CSE, 51.4%±7.4% under 0.5% CSE, 83.1%±12.1% under 1.0% CSE, and 206.1%±15.5% under 5.0% CSE in each exposure to different CSE concentration. The IOD of COX-2 was significantly higher in ECV304 cells under exposures to 1.0% and 5.0% CSE than to 0.0% and 0.5% CSE (P<0.05).
Figure 7.

The immunocytochemistry determined the expression of cyclooxygenase-2 (COX-2) in ECV304 cells under exposure to controls [negative control (A) and positive control (B)] and varied concentrations of cigarette smoking extract (CSE) at 0% (C), 0.5% (D), 1.0% (E), and 5.0% (F) for 9 h (magnification, ×200). The integrated optical density (IOD) indicates the quantities of COX-2 protein in ECV304 cells under exposure to varied concentrations of CSE at 0%, 0.5%, 1.0%, and 5.0% (G). *, P<0.05 in comparison to 0% and 0.5% CSE exposure, †, P<0.05 in comparison to 0%, 0.5% and 1% CSE exposure.
Effects of celecoxib, a selective COX-2 inhibitor, on COX-2 expression and CSE-induced apoptosis in ECV304 cells
In order to observe the role of COX-2 in the apoptosis of ECV304 cells induced by CSE, a selective COX-2 inhibitor, celecoxib, at varied concentrations of 0.0, 2.5, 5.0, 10.0, 20.0, and 50.0 µmol/L was applied to ECV304 cells with an exposure to 5.0% CSE for 9 h individually. As shown in Figure 8, the COX-2 expression was gradually reduced with the increasing concentration of celecoxib, and the expression was completely blocked by 20.0 µmol/L, and 50.0 µmol/L of celecoxib. Surprisingly, the apoptosis rate of the cells was dramatically increased from 4.69%±0.50% at 20 µmol/L to 32.60%±5.51% at 50.0 µmol/L of celecoxib (Figure 9). It seems that the apoptosis rate was markedly increased when the COX-2 expression was completely blocked by the celecoxib at 50.0 µmol/L in ECV304 cells exposed to 5.0% CSE. However, such apoptosis rate did not show significant alterations between the treatment of celecoxib under 20.0 µmol/L and the treatment without celecoxib (Figure 9). The apoptosis rate was 3.56%±0.39% with the treatment of celecoxib at 2.5 µmol/L, 3.13%±0.38% at 5 µmol/L, 2.92%±0.21% at 10 µmol/L, and 3.90%±0.27% without the treatment of celecoxib in ECV304 cells exposed to 5.0% CSE. This experiment suggested that the inhibition of COX-2 expression prominently accelerated the progression of apoptosis in ECV304 cells exposed to 5.0% CSE. COX-2 seems to play a protective role in the progression of the apoptosis in the vascular endothelial cells.
Figure 8.

The Western blotting shows the expression of cyclooxygenase-2 (COX-2) protein in ECV304 cells exposed to 5.0% cigarette smoking extract (CSE) with varied concentrations of celecoxib, a selective COX-2 inhibitor, at 0.0, 2.5, 5.0, 10.0, 20.0, and 50.0 µmmol/L for 9 h individually.
Figure 9.

The flow cytometry shows the apoptosis of ECV304 cells exposed to 5.0% cigarette smoking extract (CSE) with varied concentrations of celecoxib at 0.0, 2.5, 5.0, 10.0, 20.0, and 50.0 µmmol/L for 9 h individually (A). The apoptosis rate of ECV304 cells exposed to 5.0% CSE with varied concentrations of celecoxib at 0.0, 2.5, 5.0, 10.0, 20.0, and 50.0 µmmol/L for 9 h individually measured by flow cytometry (B). *, P<0.05 in comparison to others.
Discussion
In the present study, endothelial cell apoptosis and the expression of COX-2 protein were increased in both COPD patients and CSE-induced vascular endothelial cells. However, the inhibition of COX-2 expression by celecoxib unexpectedly led to an up-regulation of apoptosis in the ECV304 cells induced by CSE. The most remarkable finding in the present study was that a deficiency of COX-2 might play an important part in the apoptosis of vascular endothelial cells induced by CSE, and thus, the COX-2 might have a protective role in the progression of apoptosis in the vascular endothelial cells.
An increasing number of data have shown that exposure to cigarette smoking can initiate apoptosis in fibroblasts, macrophages, alveolar epithelial cell lines and vascular endothelial cells (26-31). Tobacco consumption plays a critical role in the pathogenesis of pulmonary vascular abnormalities in COPD, of which the pulmonary vascular endothelial dysfunction is characteristically exhibited (32). The impairment of endothelial cell-dependent vasodilatation, inflammation, apoptosis, and proliferation are important to the endothelial dysfunction observed in patients with COPD (32). Morphological changes such as denudation of endothelium and endothelial cell apoptosis as well as functional alterations have been observed in the pulmonary vasculature in patients with COPD and experimental emphysema animal models (11-13). Consistent with this concept, evidenced such proposals in cell level, the current study demonstrated that CSE indeed induced the apoptotic cell deaths in ECV304 cells in means of dose-dependent and time-dependent manners.
COX-2 expression can be induced rapidly by a variety of stimuli including growth factors, cytokines, and tumor promoters (18-20) and enhanced expression of COX-2 can be seen in cancer cells of several tissues including lung, which may interfere with our results. Concerning that, there is no significant difference among groups of our study subjects in cancerous protopathy diseases. COX-2 plays an important role in the pathophysiology of airway inflammatory diseases including asthma and COPD (22,33). Our previous studies found an increase of the COX-2 protein expression in the induced sputum in patients with COPD in vivo (22) and an up-regulation of the COX-2 expression by IL-1β in means of dose-dependent and time-dependent manners in vitro (34). In addition, Martey et al. reported that the COX-2 expression was induced in normal human lung fibroblasts by exposing to CSE (35). However, our recent study demonstrated that a prostacyclin analogue (beraprost sodium) had a novel protective effect in the development of cigarette smoke extract-induced emphysema through decelerating apoptosis of the alveolar epithelial cells (10). The series of arguable findings is a sign of that the role of COX-2 in the pathogenesis of COPD is uncertain. In this study, we found that the COX-2 expression in ECV304 cells was increased in parallel within 12 h with the apoptosis rate of the cells which was induced by exposing to CSE in means of dose-dependent and time-dependent manners. However, when the COX-2 expression was vanished after 24 h of exposure to 5% CSE (Figure 6B), the apoptosis rate was still kept the raise in ECV304 cells (Figure 7B). It seems that the COX-2 struggled to increase in the initial period of CSE exposure and tried to protect ECV304 cells from apoptosis induced by CSE until overcome by the CSE-induced apoptosis after the exposure over 12 h resulting in pathological outcomes.
COX-2 is known to have a significant role in apoptosis and cell survival in cancers (36). A higher number of apoptotic neutrophils in COX-2 deficient mice than COX-1 deficient mice suggested an anti-apoptotic role for COX-2 (37). Jinzhou et al. demonstrated that up-regulation of intrapulmonary COX-2 expression contributed to the suppression of polymorphonuclear neutrophil (PMN) apoptosis after acute lung injury (38). Moreover, overexpression of COX-2 was thought to be linked to down regulation of apoptosis in many cell types (39) and played an anti-apoptotic role in human bronchial epithelial cells (40). Furthermore, celecoxib, a highly selective COX-2 inhibitor, has a pro-apoptotic effect on synovial fibroblasts of rheumatoid arthritis (41). In the current study, it seems that the apoptosis rate was reduced when the COX-2 expression was incompletely blocked by the celecoxib with dose lower than 10 µmol/L. We assumed that partially inhibited COX-2 might have some other compensatory mechanisms to enhance its activity, such as alteration of spatial structure of the enzyme. With treatment with 50 µmol/L celecoxib, we found that celecoxib inhibited the COX-2 expression completely, leading to a dramatically progression of apoptosis in the vascular endothelial cells. The COX-2 might play a protective role against the progression of apoptosis in the vascular endothelial cells induced by CSE.
In summary, AI and expression of COX-2 were found increased both in the lungs of patients with COPD and smoking controls compared with nonsmoking controls. CSE induced apoptosis in vascular endothelial cells in dose-dependent and time-dependent manners. Paralleling with the increase of the apoptosis rate in the vascular endothelial cells induced by 5% CSE, the COX-2 expression was increased in the cells within 12 h of the CSE exposure, then the COX-2 expression was vanished after the exposure over 24 h but the apoptosis rate was kept the increase. Moreover, the inhibition of COX-2 expression by celecoxib led to an up-regulation of apoptosis in the vascular endothelial cells induced by CSE. This study evidences that the deficiency of COX-2 might accelerate the progress of apoptosis in the vascular endothelial cells induced by CSE, suggesting that COX-2 might have a potentially protective role against the progression of apoptosis. However, due to the less severe situation of COPD study subjects, limited samples and possible irrelevance of ECV304, the results need to be verified in severe COPD patients and physiologically relevant cells as well as larger samples to drag firmer conclusion. Further studies are necessary to figure out the route that the COX-2 expression is abolished by CSE and the mechanism by which the deficiency of COX-2 leads to CSE-induced apoptosis in the vascular endothelial cells.
Acknowledgements
Funding: This work was supported by the National Natural Science Foundation of China (810700039, 81170036 and 81270100) and National key clinical specialty construction projects.
Ethical Statement: The study was approved by the Institutional Review Board of Central South University (No. 2006S181) and written informed consent was obtained from all patients.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 2004;364:613-20. 10.1016/S0140-6736(04)16855-4 [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2095-128. 10.1016/S0140-6736(12)61728-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003;22:672-88. 10.1183/09031936.03.00040703 [DOI] [PubMed] [Google Scholar]
- 4.Demedts IK, Demoor T, Bracke KR, et al. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res 2006;7:53. 10.1186/1465-9921-7-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang M, Chen P, Peng H, et al. Cigarette smoke extract induces aberrant cytochrome-c oxidase subunit II methylation and apoptosis in human umbilical vascular endothelial cells. Am J Physiol Cell Physiol 2015;308:C378-84. 10.1152/ajpcell.00197.2014 [DOI] [PubMed] [Google Scholar]
- 6.He ZH, Chen P, Chen Y, et al. Comparison between cigarette smoke-induced emphysema and cigarette smoke extract-induced emphysema. Tob Induc Dis 2015;13:6. 10.1186/s12971-015-0033-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokohori N, Aoshiba K, Nagai A, et al. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest 2004;125:626-32. 10.1378/chest.125.2.626 [DOI] [PubMed] [Google Scholar]
- 8.Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol 2003;28:555-62. 10.1165/rcmb.2002-0090OC [DOI] [PubMed] [Google Scholar]
- 9.Petrache I, Fijalkowska I, Zhen L, et al. A novel antiapoptotic role for alpha1-antitrypsin in the prevention of pulmonary emphysema. Am J Respir Crit Care Med 2006;173:1222-8. 10.1164/rccm.200512-1842OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Hanaoka M, Chen P, et al. Protective effect of beraprost sodium, a stable prostacyclin analog, in the development of cigarette smoke extract-induced emphysema. Am J Physiol Lung Cell Mol Physiol 2009;296:L648-56. 10.1152/ajplung.90270.2008 [DOI] [PubMed] [Google Scholar]
- 11.Zhang C, Cai S, Chen P, et al. Inhibition of tumor necrosis factor-alpha reduces alveolar septal cell apoptosis in passive smoking rats. Chin Med J (Engl) 2008;121:597-601. [PubMed] [Google Scholar]
- 12.Cai S, Chen P, Zhang C, et al. Oral N-acetylcysteine attenuates pulmonary emphysema and alveolar septal cell apoptosis in smoking-induced COPD in rats. Respirology 2009;14:354-9. 10.1111/j.1440-1843.2009.01511.x [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Cao J, Chen Y, et al. Intraperitoneal injection of cigarette smoke extract induced emphysema, and injury of cardiac and skeletal muscles in BALB/C mice. Exp Lung Res 2013;39:18-31. 10.3109/01902148.2012.745910 [DOI] [PubMed] [Google Scholar]
- 14.Celli BR, MacNee W, ATS/ERS Task Force Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 2004;23:932-46. 10.1183/09031936.04.00014304 [DOI] [PubMed] [Google Scholar]
- 15.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 2007;87:1047-82. 10.1152/physrev.00048.2006 [DOI] [PubMed] [Google Scholar]
- 16.Tuder RM, Wood K, Taraseviciene L, et al. Cigarette smoke extract decreases the expression of vascular endothelial growth factor by cultured cells and triggers apoptosis of pulmonary endothelial cells. Chest 2000;117:241S-2S. 10.1378/chest.117.5_suppl_1.241S [DOI] [PubMed] [Google Scholar]
- 17.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem 2000;69:145-82. 10.1146/annurev.biochem.69.1.145 [DOI] [PubMed] [Google Scholar]
- 18.Maier JA, Hla T, Maciag T. Cyclooxygenase is an immediate-early gene induced by interleukin-1 in human endothelial cells. J Biol Chem 1990;265:10805-8. [PubMed] [Google Scholar]
- 19.Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999;18:7908-16. 10.1038/sj.onc.1203286 [DOI] [PubMed] [Google Scholar]
- 20.Sarkar P, Hayes BE. Induction of COX-2 by acrolein in rat lung epithelial cells. Mol Cell Biochem 2007;301:191-9. 10.1007/s11010-007-9411-z [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Chen P, Hanaoka M, et al. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology 2008;13:1014-21. [DOI] [PubMed] [Google Scholar]
- 22.Nana-Sinkam SP, Lee JD, Sotto-Santiago S, et al. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med 2007;175:676-85. 10.1164/rccm.200605-724OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.World Medical Association declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. JAMA 1997;277:925-6. 10.1001/jama.1997.03540350075038 [DOI] [PubMed] [Google Scholar]
- 24.Rabe KF, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007;176:532-55. 10.1164/rccm.200703-456SO [DOI] [PubMed] [Google Scholar]
- 25.Nicoletti I, Migliorati G, Pagliacci MC, et al. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 1991;139:271-9. 10.1016/0022-1759(91)90198-O [DOI] [PubMed] [Google Scholar]
- 26.He S, He Z, Chen Y, et al. C-Kit/c-Kit ligand interaction of bone marrow endothelial progenitor cells is influenced in a cigarette smoke extract-induced emphysema model. Exp Lung Res 2013;39:258-67. 10.3109/01902148.2013.802828 [DOI] [PubMed] [Google Scholar]
- 27.Aoshiba K, Tamaoki J, Nagai A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2001;281:L1392-401. [DOI] [PubMed] [Google Scholar]
- 28.Shetty SK, Bhandary YP, Marudamuthu AS, et al. Regulation of airway and alveolar epithelial cell apoptosis by p53-Induced plasminogen activator inhibitor-1 during cigarette smoke exposure injury. Am J Respir Cell Mol Biol 2012;47:474-83. 10.1165/rcmb.2011-0390OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carnevali S, Petruzzelli S, Longoni B, et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 2003;284:L955-63. 10.1152/ajplung.00466.2001 [DOI] [PubMed] [Google Scholar]
- 30.Aldonyte R, Brantly M, Block E, et al. Nuclear localization of active matrix metalloproteinase-2 in cigarette smoke-exposed apoptotic endothelial cells. Exp Lung Res 2009;35:59-75. 10.1080/01902140802406059 [DOI] [PubMed] [Google Scholar]
- 31.Yang YM, Liu GT. Damaging effect of cigarette smoke extract on primary cultured human umbilical vein endothelial cells and its mechanism. Biomed Environ Sci 2004;17:121-34. [PubMed] [Google Scholar]
- 32.Peinado VI, Barbera JA, Ramirez J, et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol 1998;274:L908-13. [DOI] [PubMed] [Google Scholar]
- 33.Profita M, Sala A, Bonanno A, et al. Increased prostaglandin E2 concentrations and cyclooxygenase-2 expression in asthmatic subjects with sputum eosinophilia. J Allergy Clin Immunol 2003;112:709-16. 10.1016/S0091-6749(03)01889-X [DOI] [PubMed] [Google Scholar]
- 34.Chen P, Cai Y, Yang ZG, et al. Involvement of PKC, p38 MAPK and AP-2 in IL-1beta-induced expression of cyclooxygenase-2 in human pulmonary epithelial cells. Respirology 2006;11:18-23. 10.1111/j.1440-1843.2006.00779.x [DOI] [PubMed] [Google Scholar]
- 35.Martey CA, Pollock SJ, Turner CK, et al. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol 2004;287:L981-91. 10.1152/ajplung.00239.2003 [DOI] [PubMed] [Google Scholar]
- 36.Fecker LF, Stockfleth E, Nindl I, et al. The role of apoptosis in therapy and prophylaxis of epithelial tumours by nonsteroidal anti-inflammatory drugs (NSAIDs). Br J Dermatol 2007;156 Suppl 3:25-33. 10.1111/j.1365-2133.2007.07856.x [DOI] [PubMed] [Google Scholar]
- 37.Langenbach R, Loftin CD, Lee C, et al. Cyclooxygenase-deficient mice. A summary of their characteristics and susceptibilities to inflammation and carcinogenesis. Ann N Y Acad Sci 1999;889:52-61. 10.1111/j.1749-6632.1999.tb08723.x [DOI] [PubMed] [Google Scholar]
- 38.Jinzhou Z, Tao H, Wensheng C, et al. Cyclooxygenase-2 suppresses polymorphonuclear neutrophil apoptosis after acute lung injury. J Trauma 2008;64:1055-60. 10.1097/TA.0b013e318047c07c [DOI] [PubMed] [Google Scholar]
- 39.Parente L, Perretti M. Advances in the pathophysiology of constitutive and inducible cyclooxygenases: two enzymes in the spotlight. Biochem Pharmacol 2003;65:153-9. 10.1016/S0006-2952(02)01422-3 [DOI] [PubMed] [Google Scholar]
- 40.Tang H, Sun Y, Xiu Q, et al. Cyclooxygenase-2 induction requires activation of nuclear factor of activated T-cells in Beas-2B cells after vanadium exposure and plays an anti-apoptotic role. Arch Biochem Biophys 2007;468:92-9. 10.1016/j.abb.2007.09.016 [DOI] [PubMed] [Google Scholar]
- 41.Kusunoki N, Yamazaki R, Kawai S. Pro-apoptotic effect of nonsteroidal anti-inflammatory drugs on synovial fibroblasts. Mod Rheumatol 2008;18:542-51. 10.3109/s10165-008-0102-8 [DOI] [PubMed] [Google Scholar]