

Abstract
BACKGROUND
We reported that high-fat diet (HFD)-induced metabolic syndrome (MetS) exacerbates lipopolysaccharide (LPS)-stimulated periodontitis and palmitate, the major saturated fatty acid in the HFD, amplified LPS-stimulated gene expression in vitro. Since CD36 is a major receptor for fatty acids, we investigated periodontal CD36 expression in mice with periodontitis and MetS, and the role of CD36 in inflammatory genes in macrophages stimulated by palmitate.
METHODS
MetS and periodontitis were induced in mice by HFD and periodontal injection of LPS, respectively. The periodontal CD36 expression and its relationship with alveolar bone loss were studied using immunohistochemistry, real-time PCR, and correlation analysis. The role of CD36 in upregulation of inflammatory mediators by LPS and palmitate in macrophages was assessed using pharmacological inhibitor and small interfering RNA.
RESULTS
Periodontal CD36 expression was higher in mice with both MetS and periodontitis than that in mice with periodontitis or MetS alone and was correlated with osteoclastogenesis and alveolar bone loss. In vitro studies showed that CD36 expression in macrophages was upregulated by LPS and palmitate, and targeting CD36 attenuated palmitate-enhanced gene expression.
CONCLUSION
CD36 expression is upregulated in mice with periodontitis and MetS, and involved in gene expression in macrophages stimulated by palmitate and LPS.
Keywords: CD36, metabolic syndrome, periodontitis, lipopolysaccharide
Introduction
Metabolic syndrome (MetS) is a cluster of cardiovascular risk factors including central obesity, atherogenic dyslipidemia, hypertension, insulin resistance, and proinflammatory state (Grundy et al., 2004). It has been estimated that 34% of adult population in USA have MetS and the prevalence is increasing with age (Amihaesei & Chelaru, 2014). MetS is associated with type 2 diabetes since insulin resistance, an important element of MetS (Shanik et al., 2008), is a powerful predictor of type 2 diabetes (Grundy, 2012, Grundy et al., 2004, Dandona et al., 2005).
It is well known that diabetes is associated with periodontitis (Yalda et al., 1994). Hyperglycemia, a key diabetes-associated metabolic disorder, is considered as a major factor contributing to diabetes-related periodontitis (Lalla et al., 2000). Interestingly, a large number of studies have indicated that MetS is also associated with periodontitis (Watanabe & Cho, 2014, Bullon et al., 2009, Thanakun et al., 2014). A meta-analysis on 20 studies with 36,337 subjects confirmed a positive relationship between MetS and periodontitis (Nibali et al., 2013). Since most patients with MetS have dyslipidemia, but not hyperglycemia, dyslipidemia such as increased triglycerides and fatty acids is likely to play a key role in MetS-exacerbated periodontitis. However, the impact of dyslipidemia on periodontitis has not been well defined.
To study the relationship between dyslipidemia and periodontitis, we recently developed a mouse model with high-fat diet (HFD)-induced MetS and LPS-induced periodontitis (Li et al., 2015). The metabolic analysis showed that the mice had MetS since they had obesity, dyslipidemia including high triglycerides and high free fatty acids, and insulin resistance. The studies using histology and micro-computed tomography (μCT) showed that MetS was associated with an increased periodontal inflammation and alveolar bone loss (Li et al., 2015). To explore the underlying mechanisms by which MetS boosted LPS-induced periodontal inflammation, we focused on the role of saturated fatty acids (SFAs) in periodontitis since the HFD used to induce MetS in our animal studies was enriched with SFAs, which are known to play an important role in the pathogenesis of MetS (Phillips et al., 2012) and MetS-associated systemic inflammation (Kennedy et al., 2009). Interestingly, our in vitro study showed that palmitate, the most abundant SFA (Mayer & Belsham, 2010), markedly enhanced LPS-induced expression of inflammatory mediators such as interleukin (IL)-6, IL-1α, IL-1β, tumor necrosis factor (TNF)α, monocyte chemoattractant protein (MCP)-1, and colony-stimulating factor (CSF) (Li et al., 2015), which are known to play pivotal roles in the development of periodontitis (Garlet, 2010, Souza & Lerner, 2013, Marques-Rocha et al., 2015).
It is known that dietary SFAs are not only essential fuels, but also ligands for both membrane and nuclear receptors that are involved in proinflammatory signaling activation, leading to cell stress, apoptosis, insulin resistance and tissue inflammation (Cascio et al., 2012). The major membrane receptors for free fatty acids include CD36, GPR40 and GPR120 (Febbraio et al., 2001, Hara et al., 2011). However, these receptors have different tissue distributions and fatty acid preference. CD36, also called fatty acid translocase (FAT), is expressed by a variety of cells including monocytes, macrophages, endothelial cells, platelets, cardiac and skeletal muscle, adipocytes and epithelial cells, and involved in innate immunity, inflammation, atherosclerosis, oxidative stress and lipid metabolism (Febbraio et al., 2001, Park, 2014). GPR40 is mainly expressed by pancreatic β cells to mediate free fatty acid-promoted insulin secretion (Ichimura et al., 2014), and is undetected in macrophages (Oh et al., 2010). Although GPR120 is expressed by macrophages and many other types of cells, it serves as functional receptor for omega-3 unsaturated fatty acids and mediates anti-inflammatory response (Hara et al., 2011). Therefore, CD36 is a potential receptor responsible for SFA-enhanced inflammatory signaling in macrophages.
In the current study, we determined if periodontal CD36 expression is associated with periodontitis in mice with MetS. We also determined how LPS and palmitate regulate CD36 expression and if CD36 is involved in the stimulation of inflammatory mediators by palmitate and LPS in macrophages.
Methods
Animal treatments
As shown in Fig. 1, 6 weeks old male C57BL/6 mice (Taconic Farms, Hudson, New York, USA) were fed regular chow (D12450B, 10% kcal% fat) or high-fat diet (HFD) (D12492, 60 kcal% fat) (Research Diets, Inc., New Brunswick, NJ) for 16 weeks (14 mice fed regular chow; 14 mice fed HFD). The D12492 diet contains high SFA (81.5 g/kg) as compared to the D12450B diet (9.9 g/kg) (Research Diets, Inc.). The mice were housed with a 12-hour light/12-hour dark cycle and had free access to water and food. All animal-related work was performed in accordance with the National Institute of Health Guidelines. All experimental protocols were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina. Since our study focuses on the role of LPS in periodontitis in mice with or without MetS, we used a mouse model with LPS-induced periodontitis (Yu et al., 2011, Rogers et al., 2007, Jin et al., 2014). During the last 4 weeks of regular chow or HFD feeding, 7 mice fed regular chow and 7 mice fed HFD were injected with LPS isolated from A. actinomycetemcomitans (strain Y4, serotype B) (20 μg of LPS per mouse) through both left and right sides of the palatal gingiva between the maxillary 1st and 2nd molars, 3 times per week. For control, 7 mice fed regular chow and 7 mice fed HFD were injected with phosphate-buffered saline (PBS), the vehicle for LPS.
Figure 1.

Time course of animal feeding and treatment. Six weeks old mice were fed regular chow or HFD for 16 weeks. During the last 4 weeks of regular chow or HFD feeding, half of the mice received periodontal injection of LPS isolated from A. actinomycetemcomitans and another half of mice received periodontal injection of PBS, the vehicle for LPS.
Metabolic measurements
Blood samples were obtained under the fasted condition and glucose, cholesterol, triglycerides, free fatty acids (FFAs) and insulin were assayed as described previously (Li et al., 2015). Fasting whole body insulin sensitivity was estimated with the homeostasis model assessment of insulin resistance (HOMA-IR) by using the following formula: fasting plasma glucose (mg/dl) × fasting plasma insulin (μU/ml)]/405 (Agil et al., 2012).
Immunohistochemical analysis of CD36 expression in mouse periodontal tissue
Gingival tissues were fixed, immersed in paraffin and processed for sectioning as described previously (Jin et al., 2014). The sections were then incubated with 5% normal goat serum in 0.01 M PBS for 1 hour to block nonspecific binding and incubated with rabbit anti-CD36 antibody (1:500) (Novus biological, Littleton, CO) overnight at 4°C. Sections were incubated with secondary biotinylated-antibody (1:250) from the ABC Elite kit (Vector Laboratories, Burlingame, CA) for 1 hour and then the ABC reagent (Vector Laboratories) for 30 minutes. Counterstaining was performed with hematoxylin. Photomicrographs were taken using an Olympus BX53 digital microscope with Cellsens digital image software (Olympus American Inc., Center Valley, PA). Quantification of cells with CD36 staining was performed with software Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD).
Micro-computed tomography (μCT) and bone volume fraction (BVF) analysis
The micro-computed tomography (μCT) and bone volume fraction (BVF) analysis were performed as described previously (Li et al., 2015).
Tartrate-resistant acid phosphatase (TRAP) staining and quantification of osteoclasts
Tartrate-resistant acid phosphatase (TRAP) staining and quantification of osteoclasts were performed as described previously (Li et al., 2015). The region for counting TRAP positive cells is the entire region under the crown of the first molar and the second molar for each section. Three sections per sample were counted.
RNA isolation and quantitative real-time polymerase chain reaction
RNA was extracted from gingival tissue surrounding the injection sites and quantitative real-time polymerase chain reaction (PCR) was performed as described previously (Li et al., 2015). The IL-6 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers for real time PCR were purchased from Qiagen [IL-6, catalog #: PPM03015A-200; GAPDH, catalog #: PPM02946E-200]. The CD36 primers (forward: TGCTGGAGCTGTTATTGG TG; reverse: CATGAGAATGCCTCCAAA CA) were synthesized (Integrated DNA Technologies, Inc., Coralville, IA). Data were analyzed with the iCycler iQ™ software (Bio-Rad Laboratories). The average starting quantity (SQ) of fluorescence units was used for analysis. Quantification was calculated using the SQ of targeted cDNA relative to that of GAPDH cDNA in the same sample.
Cell culture
RAW264.7 macrophages were purchased (American Type Culture Collection, Manassas, VA) and grown in DMEM (American Type Culture Collection) supplemented with 10% heat-inactivated fetal calf serum (HyClone, Logan, UT). The cells were maintained in a 37 °C, 90% relative humidity, 5% CO2 environment.
Palmitate preparation
To prepare palmitate from palmitic acid for cell treatments, palmitic acid was dissolved in 0.1 N NaOH and 70% ethanol at 70°C to make 50 mM. The solution was kept at 55°C for 10 min, mixed and brought to room temperature.
Enzyme-linked immunosorbent assay (ELISA)
IL-6 in medium were quantified using sandwich ELISA kits according to the protocol provided by the manufacturer (Biolegend, San Diego, CA).
RNA interference
RAW264.7 cells were transfected with 10 nM of targeting mouse CD36 Stealth siRNA or the scrambled control siRNA (Stealth RNAi™ siRNA Negative Controls) using Lipofectamine® RNAiMAX transfection reagent (Invitrogen, Grand Island, NY) for 48 h according to the manufacturer’s instructions. After the transfection, RAW264.7 cells were treated with or without 1 ng/ml LPS, 100 μM palmitate or LPS plus palmitate for 24 h.
PCR array
Mouse toll-like receptor (TLR) signal pathway PCR array (Qiagen, Santa Clarita, CA) was used to profile gene expression according to the instructions from the manufacturer. RNA isolated from duplicate wells was combined and converted to cDNA and then amplified by PCR. Data analysis is based on the delta delta threshold cycle (ΔΔCt) method as described in the instructions from the manufacturer. Briefly, the ΔCt was calculated by subtracting Ct for GAPDH from Ct for genes of interest and the ΔΔCt was calculated by subtracting the ΔCt for control cells from ΔCt for treated cells. The fold change was calculated as 2−ΔΔCt.
Statistic analysis
GraphPad Instat statistical software (Version 3.1a) (GraphPad Software, Inc. La Jolla, CA) was used for statistical analysis. One-way ANOVA was performed to determine the statistical significance of differences of alveolar bone loss, osteoclast number and gene expression level among three experimental groups, and data were presented as mean ± SD. The correlation of CD36 with osteoclasts or BVF was calculated using Pearson correlations. A value of P< 0.05 was considered significant.
Results
High-fat diet induces MetS in C57BL/6 mice
Bodyweight and fasting glucose were determined at 12 weeks before LPS treatment and at 16 weeks after LPS treatment. Results showed that before LPS treatment, mice fed HFD had significantly increased bodyweight and fasting glucose as compared with those fed regular chow (Fig. 2A and B). After LPS treatment, bodyweight of mice fed HFD remained increased as compared with that of mice fed regular chow. The metabolic parameters other than bodyweight and fasting glucose were determined at 16 weeks after LPS treatment. As shown in Fig. 2, mice fed HFD had significant increases in insulin (C), insulin resistance index HOMA-IR (D), cholesterol (E), triglycerides (F) and free fatty acids (G) as compared to mice fed regular chow, indicating that HFD induced MetS since these mice had obesity, hyperglycemia, dyslipidemia, hyperinsulinemia, and insulin resistance.
Figure 2.

The effect of high-fat diet on bodyweight (A), fasting glucose (B), fasting insulin (C), insulin resistance index (HOMA-IR) (D), cholesterol (E), triglycerides (F) and free fatty acids (G) in mice. The bodyweight and fasting glucose were measured before and after LPS treatment and all other parameters were determined at the end of the study. Glucose, insulin and lipids were assayed after 5 h fasting. The data presented are mean ± SD (n=7).
Increased expression of periodontal CD36 in mice with both MetS and periodontitis
We performed immunohistochemistry to detect CD36 protein in periodontal tissue samples. As shown in Fig. 3A, the gingival epithelial cells, fibroblasts, and infiltrated leukocytes all expressed CD36, but the latter were the predominant cells expressing high level of CD36. Results from CD36 protein quantification (Fig. 3B) showed that CD36 expression was increased in mice with either MetS or periodontitis alone as compared to the control mice and was significantly higher in mice with both MetS and periodontitis than that in mice with MetS or periodontitis alone.
Figure 3.

The periodontal CD36 protein expression in mice without MetS and periodontitis, with MetS or periodontitis alone, and with both MetS and periodontitis. C57BL/6 mice with or without HFD-induced MetS were injected with PBS or LPS in periodontal tissue. After the treatment, CD36 protein expression was determined by immunohistochemistry as described in Methods. Representative images of CD36 immunostaining in the subepithelial tissue were shown (A). The areas with positive CD36 staining were quantified (B) (n=7).
In addition to CD36 protein, we also quantified CD36 mRNA in periodontal tissues using quantitative real-time PCR. Results showed that CD36 mRNA level was increased in mice with MetS or periodontitis alone as compared to the control mice and was significantly higher in mice with both MetS and periodontitis than that in mice with MetS or periodontitis alone (Fig. 4), which is consistent with the findings from CD36 protein quantification (Fig. 3).
Figure 4.

The periodontal CD36 mRNA expression in mice without MetS and periodontitis, with MetS or periodontitis alone, and with both MetS and periodontitis. C57BL/6 mice with or without HFD-induced MetS were injected with PBS or LPS in periodontal tissue. After the treatment, RNA was isolated from the periodontal tissues and CD36 mRNA expression was quantified by real-time PCR as described in Methods. The data presented are mean ± SD (n=7).
CD36 expression in macrophages is upregulated by LPS and palmitate
The Fig. 3 and Fig. 4 showed that periodontal CD36 expression is upregulated in mice after HFD feeding and LPS injection. To investigate the mechanisms by which CD36 expression is upregulated by HFD feeding, we hypothesized that SFA in the HFD plays a role in upregulating CD36 since SFA is rich in the HFD and known to play an important role in the pathogenesis of MetS (Phillips et al., 2012). As we found that the infiltrated leukocytes had high periodontal CD36 expression in mice with both MetS and periodontitis (Fig. 3), we employed mouse RAW264.7 macrophages and treated them with palmitate, the most abundant SFA (Mayer & Belsham, 2010), LPS or palmitate in combination with LPS. We quantified CD36 mRNA expression after the treatment and results showed that while LPS or palmitate alone increased CD36 mRNA level, the combination of LPS and palmitate increased CD36 mRNA expression to a higher level (Fig. 5).
Figure 5.
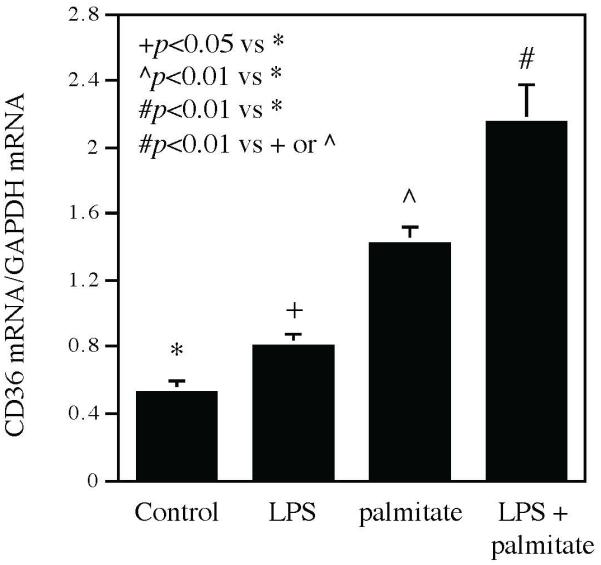
The CD36 mRNA expression in RAW264.7 macrophages treated with LPS, palmitate or LPS plus palmitate. RAW264.7 macrophages were treated with 1 ng/ml of LPS, 100 μM of palmitate or LPS plus palmitate for 24 h and CD36 mRNA was then quantified using real-time PCR. The data presented are mean ± SD of three experiments.
CD36 expression is correlated with osteoclast formation and alveolar bone loss
Although the above studies showed that periodontal CD36 expression is upregulated by HFD feeding and LPS injection, it remains unknown if CD36 is involved in HFD-exacerbated periodontitis. Since we have reported previously that MetS is associated with an increased osteoclastogenesis and alveolar bone loss in mice with LPS-induced periodontitis (Li et al., 2015), we determine if periodontal CD36 expression is associated with osteoclast formation and alveolar bone loss. Results showed that LPS induced alveolar bone loss by 4.9% in mice fed regular chow and HFD feeding induced alveolar bone loss by 6.5%. Strikingly, the combination of LPS and HFD led to bond loss by 19.4% (Table 1). Results also showed that periodontal CD36 expression had a positive correlation with the number of osteoclasts and a negative correlation with the alveolar bone volume fraction that reflects remaining bone in the maxillae samples after the treatment (Fig. 6). These results reveal a close association between periodontal CD36 expression and alveolar bone loss, suggesting that CD36 is involved in the alveolar bone resorption.
Table 1.
MetS Exacerbates LPS-induced Alveolar Bone Loss
| No MetS | MetS | |
|---|---|---|
| No periodontitis | 0.386 ± 0.014* | 0.361 ± 0.013+ |
| Periodontitis | 0.367 ± 0.017^ | 0.311 ± 0.028# |
C57BL/6 mice with or without MetS were injected with phosphate-buffered saline (PBS) or LPS in periodontal tissue to induce periodontitis. After the treatment, the maxillae were scanned by micro±computed tomography and bone volume fractions were quantified. The data are presented as means ± SD.
p<0.01 vs *
p<0.01 vs *
p<0.01 vs *, + or ^
Figure 6.

Correlation between periodontal CD36 protein expression and osteoclast formation and alveolar bone volume fraction (BVF). C57BL/6 mice with or without HFD-induced MetS were injected with PBS or LPS in periodontal tissue to induce periodontitis. After the treatment, CD36 protein expression was quantified using immunohistochemistry, osteoclasts were detected using tartrate-resistance acid phosphatase (TRAP) staining, and alveolar BVF were quantified using μCT. The correlations between CD36 expression and osteoclast formation (A) or alveolar BVF (B) were statistically analyzed.
CD36 is involved in the enhancement by palmitate of LPS-induced IL-6 expression
Our previous in vitro study has shown that palmitate enhances LPS-stimulated expression of proinflammatory mediators (Li et al., 2015), which are known to contribute to periodontal inflammation and alveolar bone loss (Souza & Lerner, 2013). To understand how CD36 is involved in the alveolar bone loss, we determine if CD36 contributes to the enhancement of proinflammatory mediator expression by palmitate using pharmacological inhibitor and siRNA. Results showed that while palmitate markedly enhanced LPS-induced IL-6 secretion from macrophages, sulfosuccinimidyl oleate (SSO), a selective CD36 inhibitor (Coort et al., 2002), robustly attenuated the enhancement by palmitate of LPS-induced IL-6 secretion (Fig. 7). Interestingly, SSO also inhibited the induction of IL-6 secretion by LPS (Fig. 7). Similarly, the study targeting CD36 expression using siRNA (Fig. 8A) showed that CD36 knockdown also significantly attenuated the enhancement by palmitate of LPS-induced IL-6 mRNA expression (Fig. 8B) and secretion (Fig. 8C).
Figure 7.
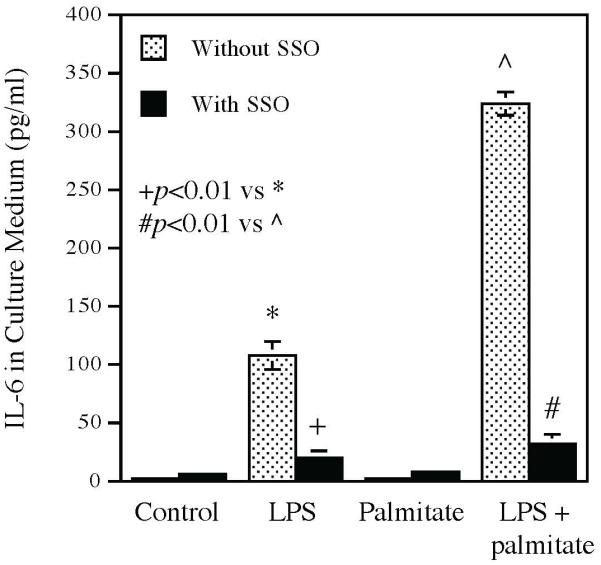
The inhibitory effect of CD36 inhibitor SSO on the enhancement by palmitate of LPS-induced IL-6 secretion. RAW264.7 macrophages were treated with 1 ng/ml of LPS, 100 μM of palmitate or LPS plus palmitate in the absence or presence of 100 μM of SSO for 24 h and IL-6 secreted into culture medium was then quantified using ELISA. The data presented are mean ± SD of three experiments.
Figure 8.

The inhibitory effect of CD36 knockdown on the enhancement by palmitate of LPS-induced IL-6 expression and secretion. RAW264.7 macrophages were transfected with 10 nM control or CD36 siRNA for 48 h and then treated with 1 ng/ml of LPS, 100 μM of palmitate or both LPS and palmitate for 24 h. After the treatment, IL-6 mRNA expression and secretion was quantified using ELISA. A: CD36 expression in cells after transfection with control or CD36 siRNA. B. IL-6 mRNA expression in cells treated with LPS, palmitate or LPS plus palmitate after transfection with either control or CD36 siRNA. C. IL-6 secretion by cells treated with LPS, palmitate or LPS plus palmitate after transfection with either control or CD36 siRNA. The data presented are mean ± SD of three experiments.
CD36 inhibition attenuates the enhancement by palmitate of LPS-induced expression of proinflammatory molecules
In addition to IL-6, we further investigated how CD36 inhibition affects the enhancement of other inflammatory molecule expression by palmitate using a PCR array system. As shown in Table 2, while palmitate enhanced LPS-stimulated expression of several genes such as IL1β, IL8, MAPK2K3, MYD88, and RIPK2, CD36 inhibition by SSO effectively attenuated the enhancement by palmitate of LPS-stimulated gene expression in macrophages.
Table 2.
The effect of CD36 inhibitor SSO on the expression of pro-inflammatory molecules stimulated by LPS, palmitate or LPS in combination with palmitate
| Fold increase by LPS | Fold increase by palmitate | Fold increase by LPS in combination with palmitate |
|||||||
|---|---|---|---|---|---|---|---|---|---|
|
Without SSO |
With SSO |
% Inhibition by SSO |
Without SSO |
With SSO |
% Inhibition by SSO |
Without SSO |
With SSO |
% Inhibition by SSO |
|
| MCP-1 | 12.92 | 3.36 | 74% | 7.72 | 1.75 | 77% | 24.65 | 8.05 | 67% |
| CLEC4E | 5.75 | 2.42 | 58% | 1.13 | 0.88 | 23% | 8.47 | 3.45 | 59% |
| IL1B | 91.86 | 19.24 | 79% | 11.47 | 3.75 | 67% | 206.33 | 100.23 | 52% |
| IL8 | 140.42 | 26.90 | 81% | 31.42 | 6.69 | 79% | 330.95 | 105.59 | 68% |
| MAP2K3 | 2.05 | 1.18 | 42% | 3.26 | 1.68 | 48% | 5.92 | 3.31 | 44% |
| MYD88 | 2.57 | 1.03 | 60% | 1.46 | 0.69 | 53% | 5.12 | 1.63 | 68% |
| RIPK2 | 2.16 | 1.03 | 52% | 1.71 | 0.98 | 42% | 5.29 | 2.02 | 62% |
MCP-1: Monocyte chemoattractant protein-1; CLEC4E: C-type lectin domain family 4; IL1B: Interleuk 1p; IΛ8: Interleukin 8; MAP2K3: Mitogen-activated protein kinase kinase 3; MYD88: Myeloid differentiation primary response gene 88; RIPK2: Receptor-interacting serine/threonin-protein kinase 2.
Discussion
We have reported recently that HFD-induced MetS exacerbates LPS-stimulated periodontal inflammation and alveolar bone loss in C57BL/6 mice (Li et al., 2015). Since our research focus in this study is the role of the interaction between LPS and lipid metabolites in the development of periodontitis in vivo, we utilized an established mouse model in which periodontitis is induced by periodontal injection of LPS isolated from A. actinomycetemcomitans (Yu et al., 2011, Rogers et al., 2007, Jin et al., 2014). A. actinomycetemcomitans LPS, but not P. gingivalis LPS, was used since A. actinomycetemcomitans LPS is a more potent inducer of bone resorption than P. gingivalis LPS (Nishida et al., 2001).
In the present study, we showed that mice with periodontitis and MetS had an increased periodontal CD36 expression. This finding is in agreement partially with a recent report by Shikama et al. that gingival fibroblasts of mice with HFD-induced type 2 diabetes had increased CD36 expression (Shikama et al., 2015). In addition to gingival fibroblasts, our study showed that the infiltrated leukocytes are the predominant cells expressing high level of CD36 in the periodontal tissue of mice with periodontitis and MetS (Fig. 3).
To elucidate the potential mechanisms whereby periodontal CD36 expression was upregulated in mice with MetS and periodontitis, we performed in vitro studies using macrophages since macrophages are part of the infiltrated leukocytes and play an important role in the pathogenesis of periodontitis (Charon et al., 1981, Huang et al., 2016, Kukita et al., 2012). We tested our hypothesis that SFA, which is rich in the HFD used in our animal study and involved in the pathogenesis of MetS (Phillips et al., 2012), acts with LPS to upregulate CD36 expression in macrophages. The finding from this study supports our hypothesis as it showed that while either palmitate or LPS stimulated CD36 expression, which is in agreement with the previous reports (Li et al., 2013, Pararasa et al., 2014, Chabowski et al., 2013), the combination of palmitate and LPS stimulated CD36 expression to a higher level. Since it is known that SFA is increased in MetS patients (Rivellese et al., 2002, Tuomilehto, 2005) and MetS mice (Morselli et al., 2014), and LPS is increased in patients with periodontitis, palmitate and LPS are likely to act in concert to stimulate CD36 expression in patients or mice with both MetS and periodontitis.
The upregulation of CD36 by its ligand palmitate in macrophages (Pararasa et al., 2014) reveals a positive feedback mechanism for CD36 expression since CD36 upregulation by palmitate allows more palmitate binding, resulting in a subsequent higher level of CD36 expression. Obviously, the positive feedback mechanism on CD36 expression leads to a strong CD36-mediated inflammatory signaling and cellular innate immune response as evidenced by our findings that palmitate amplified LPS-triggered inflammatory molecule expression (Figs. 7-8 and Table 2). The data from the PCR array (Table 2) showed that palmitate not only enhanced LPS-induced expression of inflammatory cytokines such as MCP-1, IL-1β, IL-8 and IL-6, but also augmented LPS-stimulated expression of signaling molecules such as mitogen-activated protein kinase kinase 3 (MAP2K3) and receptor-interacting serine-threonine kinase 2 (RIPK2), which are involved in LPS-activated p38 mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NFκB) signaling pathways (Rossa et al., 2006, Lupfer et al., 2013).
In addition to the finding from our in vitro study that CD36 expression was upregulated by palmitate and LPS, we also showed the biological function of CD36 in the enhancement of LPS-induced inflammatory molecules by palmitate. In this study, we employed both the pharmacological inhibitor SSO and CD36 siRNA to target CD36 and found that either CD36 inhibition with SSO or CD36 knockdown with siRNA markedly attenuated the effect of palmitate on the enhancement of LPS-induced expression of IL-6 and other inflammatory molecules (Figs. 7, 8 and Table 2), suggesting a role of CD36 in the innate immune response to palmitate in macrophages. Interestingly, Pillon et al. reported recently that CD36 inhibition in human adipose microvascular endothelial cells did not inhibit the palmitate-induced expression of inflammatory genes (Pillon et al., 2015), suggesting a specific involvement of CD36 in certain types of cells in palmitate-stimulated gene expression.
Another interesting finding from our study is that CD36 inhibition attenuates inflammatory molecule expression mediated by not only CD36, but also TLR4 (Figs. 7, 8 and Table 2). Since CD36 inhibitor SSO does not antagonize TLR4, this finding supports the postulation by the previous studies that CD36 and TLR4 form a complex on the cell surface and targeting either one will affect the other (Chavez-Sanchez et al., 2014, Stewart et al., 2010, Baranova et al., 2012). Chavez-Sanchez et al. reported that TLR4 block using monoclonal antibodies significantly reduced the stimulation by oxidized LDL (oxLDL), a ligand for CD36 (Stewart et al., 2010), of IL-1β and IL-6 secretion from human macrophages (Chavez-Sanchez et al., 2014). Baranova et al. demonstrated that induction of CD36 expression in HeLa cells increased LPS binding and subsequent cytokine secretion (Baranova et al., 2012). The same study also showed that macrophages with CD36 deficiency had 40-50% reduction of IL-6 secretion in response to LPS. Furthermore, Stewart et al. reported that CD36 was co-precipitated with TLR4 and TLR6 from THP-1 monocytes after treatment with oxLDL (Stewart et al., 2010). However, the exact interaction between CD36 and TLR4 and how the interaction boosts LPS-mediated host inflammatory response are not well understood and more studies are warranted.
In this study, we demonstrated that the CD36 protein expression in the periodontal tissue was highly correlated with osteoclast formation and alveolar bone loss (Fig. 6). While our correlation study and the study using immunohistochemistry suggest a potential role of CD36 in MetS-exacerbated periodontitis, the study using loss-of-function mouse model may provide stronger evidence to define the role of CD36. Indeed, studies have applied CD36-deficient mice or cells in defining the role of CD36 in pathological processes. For examples, Nozaki et al. used CD36-deficient macrophages to show the role of CD36 in the uptake of oxidized LDL (Nozaki et al., 1995) and Brundert et al. used CD36-deficient mice to show the role of CD36 in the uptake of high density lipoproteins (Brundert et al., 2011).
In conclusion, we demonstrated in this study for the first time that periodontal CD36 expression was associated with MetS-exacerbated periodontitis. We also demonstrated that CD36 is involved in the enhancement by palmitate of LPS-induced inflammatory molecule expression in macrophages.
Acknowledgements
This work was supported by National Institutes of Health grant DE016353 and the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs (to Y.H.). This project utilized facilities, resources, and/or technical assistance of the Laboratory of the Center for Oral Health Research that is supported by the National Institute of General Medicine grant P30 GM103331.
Footnotes
Author contributions
Zhongyang Lu, Yanchun Li and Colleen W. Brinson performed the experiments, analyzed the results and contributed to the writing of the manuscript. Keith L. Kirkwood and Maria F. Lopes-Virella analyzed the results and contributed to the writing of the manuscript. Yan Huang designed the study, analyzed the results and wrote and edited the manuscript.
Conflict of interest
The authors declare no conflict of interest with respect to the authorship and/or publication of this article.
References
- Agil A, Rosado I, Ruiz R, Figueroa A, Zen N, Fernandez-Vazquez G. Melatonin improves glucose homeostasis in young Zucker diabetic fatty rats. J Pineal Res. 2012;52:203–10. doi: 10.1111/j.1600-079X.2011.00928.x. [DOI] [PubMed] [Google Scholar]
- Amihaesei IC, Chelaru L. Metabolic syndrome a widespread threatening condition; risk factors, diagnostic criteria, therapeutic options, prevention and controversies: an overview. Rev Med Chir Soc Med Nat Iasi. 2014;118:896–900. [PubMed] [Google Scholar]
- Baranova IN, Vishnyakova TG, Bocharov AV, Leelahavanichkul A, Kurlander R, Chen Z, Souza AC, Yuen PS, Star RA, Csako G, Patterson AP, Eggerman TL. Class B scavenger receptor types I and II and CD36 mediate bacterial recognition and proinflammatory signaling induced by Escherichia coli, lipopolysaccharide, and cytosolic chaperonin 60. J Immunol. 2012;188:1371–80. doi: 10.4049/jimmunol.1100350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundert M, Heeren J, Merkel M, Carambia A, Herkel J, Groitl P, Dobner T, Ramakrishnan R, Moore KJ, Rinninger F. Scavenger receptor CD36 mediates uptake of high density lipoproteins in mice and by cultured cells. J Lipid Res. 2011;52:745–58. doi: 10.1194/jlr.M011981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullon P, Morillo JM, Ramirez-Tortosa MC, Quiles JL, Newman HN, Battino M. Metabolic syndrome and periodontitis: is oxidative stress a common link? J Dent Res. 2009;88:503–18. doi: 10.1177/0022034509337479. [DOI] [PubMed] [Google Scholar]
- Cascio G, Schiera G, Di Liegro I. Dietary fatty acids in metabolic syndrome, diabetes and cardiovascular diseases. Curr Diabetes Rev. 2012;8:2–17. doi: 10.2174/157339912798829241. [DOI] [PubMed] [Google Scholar]
- Chabowski A, Zendzian-Piotrowska M, Konstantynowicz K, Pankiewicz W, Miklosz A, Lukaszuk B, Gorski J. Fatty acid transporters involved in the palmitate and oleate induced insulin resistance in primary rat hepatocytes. Acta Physiol (Oxf) 2013;207:346–57. doi: 10.1111/apha.12022. [DOI] [PubMed] [Google Scholar]
- Charon J, Toto PD, Gargiulo AW. Activated macrophages in human periodontitis. J Periodontol. 1981;52:328–35. doi: 10.1902/jop.1981.52.6.328. [DOI] [PubMed] [Google Scholar]
- Chavez-Sanchez L, Garza-Reyes MG, Espinosa-Luna JE, Chavez-Rueda K, Legorreta-Haquet MV, Blanco-Favela F. The role of TLR2, TLR4 and CD36 in macrophage activation and foam cell formation in response to oxLDL in humans. Hum Immunol. 2014;75:322–9. doi: 10.1016/j.humimm.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Coort SL, Willems J, Coumans WA, van der Vusse GJ, Bonen A, Glatz JF, Luiken JJ. Sulfo-N-succinimidyl esters of long chain fatty acids specifically inhibit fatty acid translocase (FAT/CD36)-mediated cellular fatty acid uptake. Mol Cell Biochem. 2002;239:213–9. [PubMed] [Google Scholar]
- Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation. 2005;111:1448–54. doi: 10.1161/01.CIR.0000158483.13093.9D. [DOI] [PubMed] [Google Scholar]
- Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108:785–91. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89:1349–63. doi: 10.1177/0022034510376402. [DOI] [PubMed] [Google Scholar]
- Grundy SM. Pre-diabetes, metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol. 2012;59:635–43. doi: 10.1016/j.jacc.2011.08.080. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Brewer HB, Jr., Cleeman JI, Smith SC, Jr., Lenfant C, American Heart A, National Heart L and Blood I Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–8. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- Hara T, Hirasawa A, Ichimura A, Kimura I, Tsujimoto G. Free fatty acid receptors FFAR1 and GPR120 as novel therapeutic targets for metabolic disorders. J Pharm Sci. 2011;100:3594–601. doi: 10.1002/jps.22639. [DOI] [PubMed] [Google Scholar]
- Huang X, Yu T, Ma C, Wang Y, Xie B, Xuan D, Zhang J. Macrophages Play a Key Role in the Obesity Induced Periodontal Innate Immune Dysfunction Via NLRP3 Pathway. J Periodontol. 2016:1–18. doi: 10.1902/jop.2016.160102. [DOI] [PubMed] [Google Scholar]
- Ichimura A, Hasegawa S, Kasubuchi M, Kimura I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front Pharmacol. 2014;5:236. doi: 10.3389/fphar.2014.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Zhang X, Lu Z, Li Y, Lopes-Virella MF, Yu H, Haycraft CJ, Li Q, Kirkwood KL, Huang Y. Simvastatin inhibits lipopolysaccharide-induced osteoclastogenesis and reduces alveolar bone loss in experimental periodontal disease. J Periodontal Res. 2014;49:518–26. doi: 10.1111/jre.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy A, Martinez K, Chuang CC, LaPoint K, McIntosh M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. J Nutr. 2009;139:1–4. doi: 10.3945/jn.108.098269. [DOI] [PubMed] [Google Scholar]
- Kukita A, Ichigi Y, Takigawa I, Watanabe T, Kukita T, Miyamoto H. Infection of RANKL-primed RAW-D macrophages with Porphyromonas gingivalis promotes osteoclastogenesis in a TNF-alpha-independent manner. PLoS One. 2012;7:e38500. doi: 10.1371/journal.pone.0038500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalla E, Lamster IB, Drury S, Fu C, Schmidt AM. Hyperglycemia, glycoxidation and receptor for advanced glycation endproducts: potential mechanisms underlying diabetic complications, including diabetes-associated periodontitis. Periodontol 2000. 2000;23:50–62. doi: 10.1034/j.1600-0757.2000.2230104.x. [DOI] [PubMed] [Google Scholar]
- Li XY, Wang C, Xiang XR, Chen FC, Yang CM, Wu J. Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP-binding cassette transporter A1 in macrophages. Oncol Rep. 2013;30:1329–36. doi: 10.3892/or.2013.2600. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu Z, Zhang X, Yu H, Kirkwood KL, Lopes-Virella MF, Huang Y. Metabolic syndrome exacerbates inflammation and bone loss in periodontitis. J Dent Res. 2015;94:362–70. doi: 10.1177/0022034514561658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, Xavier RJ, Green DR, Lamkanfi M, Dinarello CA, Doherty PC, Kanneganti TD. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14:480–8. doi: 10.1038/ni.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Rocha JL, Samblas M, Milagro FI, Bressan J, Martinez JA, Marti A. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 2015;29:3595–611. doi: 10.1096/fj.14-260323. [DOI] [PubMed] [Google Scholar]
- Mayer CM, Belsham DD. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance and apoptosis through adenosine 5' monophosphate-activated protein kinase activation. Endocrinology. 2010;151:576–85. doi: 10.1210/en.2009-1122. [DOI] [PubMed] [Google Scholar]
- Morselli E, Criollo A, Rodriguez-Navas C, Clegg DJ. Chronic High Fat Diet Consumption Impairs Metabolic Health of Male Mice. Inflamm Cell Signal. 2014;1:e561. doi: 10.14800/ics.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibali L, Tatarakis N, Needleman I, Tu YK, D'Aiuto F, Rizzo M, Donos N. Clinical review: Association between metabolic syndrome and periodontitis: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2013;98:913–20. doi: 10.1210/jc.2012-3552. [DOI] [PubMed] [Google Scholar]
- Nishida E, Hara Y, Kaneko T, Ikeda Y, Ukai T, Kato I. Bone resorption and local interleukin-1alpha and interleukin-1beta synthesis induced by Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis lipopolysaccharide. J Periodontal Res. 2001;36:1–8. doi: 10.1034/j.1600-0765.2001.00637.x. [DOI] [PubMed] [Google Scholar]
- Nozaki S, Kashiwagi H, Yamashita S, Nakagawa T, Kostner B, Tomiyama Y, Nakata A, Ishigami M, Miyagawa J, Kameda-Takemura K, et al. Reduced uptake of oxidized low density lipoproteins in monocyte-derived macrophages from CD36-deficient subjects. J Clin Invest. 1995;96:1859–65. doi: 10.1172/JCI118231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–98. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pararasa C, Bailey C, Griffiths H. Macrophage polarisation by fatty acids is PPARgamma-dependent. Free Radic Biol Med. 2014;75(Suppl 1):S31–2. doi: 10.1016/j.freeradbiomed.2014.10.764. [DOI] [PubMed] [Google Scholar]
- Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. 2014;46:e99. doi: 10.1038/emm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Goumidi L, Bertrais S, Field MR, McManus R, Hercberg S, Lairon D, Planells R, Roche HM. Dietary saturated fat, gender and genetic variation at the TCF7L2 locus predict the development of metabolic syndrome. J Nutr Biochem. 2012;23:239–44. doi: 10.1016/j.jnutbio.2010.11.020. [DOI] [PubMed] [Google Scholar]
- Pillon NJ, Azizi PM, Li YE, Liu J, Wang C, Chan KL, Hopperton KE, Bazinet RP, Heit B, Bilan PJ, Lee WL, Klip A. Palmitate-induced inflammatory pathways in human adipose microvascular endothelial cells promote monocyte adhesion and impair insulin transcytosis. Am J Physiol Endocrinol Metab. 2015;309:E35–44. doi: 10.1152/ajpendo.00611.2014. [DOI] [PubMed] [Google Scholar]
- Rivellese AA, De Natale C, Lilli S. Type of dietary fat and insulin resistance. Ann N Y Acad Sci. 2002;967:329–35. doi: 10.1111/j.1749-6632.2002.tb04288.x. [DOI] [PubMed] [Google Scholar]
- Rogers JE, Li F, Coatney DD, Rossa C, Bronson P, Krieder JM, Giannobile WV, Kirkwood KL. Actinobacillus actinomycetemcomitans lipopolysaccharide-mediated experimental bone loss model for aggressive periodontitis. J Periodontol. 2007;78:550–8. doi: 10.1902/jop.2007.060321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossa C, Ehmann K, Liu M, Patil C, Kirkwood KL, Rossa C, Jr., Liu M, Kirkwood KL. MKK3/6-p38 MAPK signaling is required for IL-1beta and TNF-alpha-induced RANKL expression in bone marrow stromal cells. J Interferon Cytokine Res. 2006;26:719–29. doi: 10.1089/jir.2006.26.719. [DOI] [PubMed] [Google Scholar]
- Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care. 2008;31(Suppl 2):S262–8. doi: 10.2337/dc08-s264. [DOI] [PubMed] [Google Scholar]
- Shikama Y, Kudo Y, Ishimaru N, Funaki M. Possible Involvement of Palmitate in Pathogenesis of Periodontitis. J Cell Physiol. 2015;230:2981–9. doi: 10.1002/jcp.25029. [DOI] [PubMed] [Google Scholar]
- Souza PP, Lerner UH. The role of cytokines in inflammatory bone loss. Immunol Invest. 2013;42:555–622. doi: 10.3109/08820139.2013.822766. [DOI] [PubMed] [Google Scholar]
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–61. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanakun S, Watanabe H, Thaweboon S, Izumi Y. Association of untreated metabolic syndrome with moderate to severe periodontitis in Thai population. J Periodontol. 2014;85:1502–14. doi: 10.1902/jop.2014.140105. [DOI] [PubMed] [Google Scholar]
- Tuomilehto J. Cardiovascular risk: prevention and treatment of the metabolic syndrome. Diabetes Res Clin Pract. 2005;68(Suppl 2):S28–35. doi: 10.1016/j.diabres.2005.03.018. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Cho YD. Periodontal disease and metabolic syndrome: a qualitative critical review of their association. Arch Oral Biol. 2014;59:855–70. doi: 10.1016/j.archoralbio.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalda B, Offenbacher S, Collins JG. Diabetes as a modifier of periodontal disease expression. Periodontology 2000. 1994;6:37–49. doi: 10.1111/j.1600-0757.1994.tb00025.x. [DOI] [PubMed] [Google Scholar]
- Yu H, Li Q, Herbert B, Zinna R, Martin K, Junior CR, Kirkwood KL. Anti-inflammatory effect of MAPK phosphatase-1 local gene transfer in inflammatory bone loss. Gene Ther. 2011;18:344–53. doi: 10.1038/gt.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]