

Abstract
Two categories of joint overloading cause post-traumatic osteoarthritis (PTOA): single acute traumatic loads/impactions and repetitive overloading due to incongruity/instability. We developed and refined three classes of complementary models to define relationships between joint overloading and progressive cartilage loss across the spectrum of acute injuries and chronic joint abnormalities: explant and whole joint models that allow probing of cellular responses to mechanical injury and contact stresses, animal models that enable study of PTOA pathways in living joints and pre-clinical testing of treatments, and patient-specific computational models that define the overloading that causes OA in humans. We coordinated methodologies across models so that results from each informed the others, maximizing the benefit of this complementary approach. We are incorporating results from these investigations into biomathematical models to provide predictions of PTOA risk and guide treatment. Each approach has limitations, but each provides opportunities to elucidate PTOA pathogenesis. Taken together, they help define levels of joint overloading that cause cartilage destruction, show that both forms of overloading can act through the same biologic pathways, and create a framework for initiating clinical interventions that decrease PTOA risk.
Keywords: Post-Traumatic Osteoarthritis, Chondrocytes, Joint Trauma, Animal Models, Computational Models
Introduction
Osteoarthritis (OA), the deterioration of synovial joints marked by joint pain and dysfunction,1 is the most common joint disease, a major cause of disability for middle age and older people, and an important driver of health care costs. No intervention has been proven to prevent the onset and progression of OA. Factors associated with increased risk of OA include increasing age, chronic joint overloading, joint dysplasia, joint injury, and neurologic disorders. Although acute and chronic joint overloading increase the risk of OA in all joints and in all populations, the thresholds of joint overloading that cause OA and the mechanisms responsible for OA due to excessive contact stresses remain obscure. To discover joint overloading-progressive cartilage loss relationships we have studied post-traumatic osteoarthritis (PTOA), the OA that develops following joint injury.2-6 This strategy has provided opportunities to better understand the sequence of events that lead to OA originating from a specific point in time.
Joint injuries that lead to PTOA include dislocations, ligament and capsular tears, meniscal injuries, articular surface impactions, and intra-articular fractures.5 Both the acute tissue damage and post-traumatic residual joint abnormalities, primarily instability or surface incongruity, lead to progressive loss of articular cartilage, bone remodeling, and changes in the joint soft tissues collectively recognized as PTOA.5 Joint dysplasia can also lead to chronic joint overloading that puts the joint at risk for early onset OA. Clinicians studying and caring for patients recognize that severity of joint injury and degree of articular surface incongruity or joint instability increases the risk of PTOA. Yet, because they have been unable to quantify the severity of injury or the degree of incongruity or instability that predictably cause cartilage loss, and because the relationships between excessive articular surface contact stresses and progressive joint destruction have not been defined, it has been difficult to devise and sensibly test new therapies that may decrease the burden of PTOA.
Current optimized treatments of joint injuries often fail to prevent PTOA. In the case of intra-articular fractures, as many as one in four patients develop OA after fractures of the acetabulum, between 23% and 44% of patients develop knee OA after intra-articular fractures of the knee and more than 50% of patients with fractures of the distal tibial articular surface develop OA.4 Patients who suffer anterior cruciate ligament (ACL) and meniscal injuries of the knee have a ten-fold increased risk of OA compared to individuals who do not have a knee injury.7-10
Breakthroughs in the treatment of joint injuries to forestall PTOA require elucidation of the mechanisms responsible for cartilage loss following joint trauma, the capacity to reliably predict PTOA risk, and methods of testing interventions that have the potential to decrease the risk of the disease. We developed three classes of complementary models to address these needs: in vitro explant and whole joint models, in vivo animal models, and patient-specific computational models of acute joint trauma and chronic overloading (detailed in Tables S-1, S-2, and S-3). Methodology choices have been coordinated across models to maximize the benefit of this complementary approach (Table 1). The high-throughput of rodent models is a valuable asset in drug discovery studies that require sorting through dozens of treatment alternatives.11 Moreover, transgenic mouse models have yielded unprecedented mechanistic insights into the pathogenesis of PTOA.12 Though these models are excellent for exploratory work, the main substance of our work has been to develop large animal models for advanced pre-clinical treatment testing. A distinct advantage of larger species over rodents is that their cartilage is thick enough to be imaged by MRI in vivo. Furthermore, cartilage in most large-animal joints is present in quantities sufficient for detailed physiological and biochemical analyses. In the case of our porcine intra-articular fracture model, the human-like size of the joint enables surgical treatments identical to those used in the clinical setting, a level of realism that cannot be matched in rodent versions of the model.
Table 1.
Cell- and organ-level effects of various joint injuries.
| Models of Joint Injury | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Class I | Class II | Class III | |||||||||
|
| |||||||||||
| Cell/Organ Effect | Explant (4,14-24) | Whole Joint (26-30) | Rabbit (32,33) | Goat | Mini-Pig 37 | Patients (40-47) | |||||
|
| |||||||||||
| CEI | RAC | IAF | GrI | CEI | MEN | GrI | CEI+MEN | IAF | IAF | PAO | |
|
| |||||||||||
| Acute oxidative damage | X | n/d | X | n/d | X | n/d | n/d | n/d | X | n/d | n/a |
| Acute oxidative stress | X | X | X | n/d | X | X | n/d | n/d | X | n/d | n/a |
| Mitochondrial loss | X | X | X | n/d | X | X | n/d | X | X | n/d | n/a |
| Acute injury severity | X | n/a | X | n/a | X | X | X | X | X | X | X |
| Chronic contact stress | n/a | X | n/a | X | n/d | X | X | X | X | X | X |
| Synovitis | n/a | n/a | n/a | n/a | X | X | X | X | X | X | X |
| PTOA | n/a | n/a | n/a | n/a | X | X | X | ? | X | X | X |
CEI = controlled-energy impact; RAC = repeated axial compression; IAF = intra-articular fracture; MEN = meniscus destabilization; n/a = not applicable; n/d = not determined; GrI = graded instability; PAO = peri-acetabular osteotomy
In the studies included in this manuscript, articular injuries were uniformly delivered specifically to functional load-bearing surfaces of the joints. Loading conditions were comparably calibrated and validated. Consistent imaging methods, cell viability measures, and histologic staining techniques were used. This has enabled us to build in silico biomathematical models based on the wealth of information derived from these complementary models. Such models are being used to generate novel ideas for interventions in PTOA. In one example, we simulated the balance between pro- and anti-inflammatory factors in injured cartilage to predict if modest lesions would worsen over time.13 The problem was treated as a conventional wound healing scenario, with TNF-α and erythropoietin (EPO) as the main co-regulating signals. Validation experiments in an explant impact injury model confirmed the biomathematical model's prediction that locally produced EPO promotes cartilage recovery after mechanical insults, findings that warrant testing of EPO in an in vivo setting. A major goal for the future is to develop a whole-joint model in which synovitis and subchondral bone changes are simulated.
Class I: Explant and Whole Joint Models (Table S-1)
Viable Osteochondral Explants
We subjected fresh intact human and animal articular cartilage specimens, maintained in environments simulating in vivo conditions, to a range of acute impacts, repetitive loadings, and direct mechanical injuries. Investigations using these explants have revealed that injurious mechanical loads stimulate biologic responses that cause progressive cell death and activate chondrocyte progenitor cells (CPCs).14-20
Impaction of articular surfaces led to consistent immediate release of chondrocyte mitochondrial reactive oxygen species (ROS), an event followed by chondrocyte death. Inhibition of mitochondrial electron transport prevented ROS release and preserved chondrocyte viability.4,14-18,21-24 Anti-oxidants also preserved chondrocyte viability. Disruption of the chondrocyte cytoskeleton and matrix adhesion proteins prevented ROS release and helped maintain chondrocyte viability, suggesting that articular surface contact stresses pass through the matrix, across the cell membrane, and through the cytoskeleton to the mitochondria.
Repetitive physiologic cyclic loading of articular surfaces promoted lower levels of mitochondrial ROS production than those caused by injurious impacts. These lower ROS levels stimulated chondrocyte metabolism as measured by ATP production. Absence of physiologic cyclic loading, inhibition of mitochondrial electron transport, and the introduction of anti-oxidants all led to decreased chondrocyte ATP production, suggesting that chondrocytes may adapt to repetitive supra-physiologic but non-injurious levels of cyclic loading by increasing their anabolic activity.
Impaction and mechanical injury of articular surfaces stimulated release of alarmins from injured chondrocytes, and these molecules activated a population of superficial zone CPCs that proliferated and migrated to regions of chondrocyte death.19,20 These cells differed from both normal chondrocytes and mesenchymal stem cells in their expression of genes responsible for production of inflammatory mediators and matrix degrading proteases. CPC activation depended on high mobility group box 1 (HMGB1) binding to the receptor for advanced glycation end-products (RAGE), one of a small family of alarmin-responsive innate immune receptors that include toll-like receptors, which may play a role in osteoarthritis.25
The advantages of the viable explant models include their utility in studying cellular responses to mechanical loads and injuries and in testing methods of blocking those responses in intact articular cartilage. The limitations of these models include a reliance on the use of artificial impactors to deliver mechanical loads and the inability to replicate the complex biologic events and loading patterns of synovial joints in vivo.
Ex Vivo Human Whole Joints
To study injuries that closely resemble those resulting from clinically significant impacts, we developed a whole joint model of intra-articular fracture.26 Fresh human ankle joints were obtained from amputation specimens. Within four hours of surgery, we subjected the joints to acute loads that reproducibly caused intra-articular fractures. We used confocal microscopy to measure chondrocyte viability immediately and at 24 and 48 hours following injury. Immediately following injury, 20% of the superficial zone cells within 500 microns of the fracture lines died. Over the next 48 hours, the proportion of dead cells in these regions of the joint increased to 60%. In articular cartilage more than 500 microns from the fracture lines, few cells died. Thus, even severe joint injuries caused little immediate chondrocyte death, suggesting that, as the explant studies had indicated, chondrocyte dysfunction and mediators released from injured cartilage cause progressive tissue damage and that treatment of injured joints within hours, or perhaps even days, of injury could preserve chondrocyte viability and restore joint health.
Ex Vivo Human Whole Joints
Ligament and capsular tears, including joint sprains and dislocations, also increase the risk of PTOA, presumably because of residual joint instability and resulting abnormal joint kinematics that expose the joint to repetitive overloading. An in vitro study of cadaveric human ankle joints, using a methodology that measured instantaneous joint surface contact stresses, showed that joint instability increased peak contact stress by 20% to 25%, and increased the peak temporal rate of change of contact stress by 115%.27-30
The principal advantage of human whole joint models, as compared with explant models, is that the former make it possible to study a range of impacts transmitted across the entire normal opposing articular surfaces and to analyze the effects of incongruity and instability on articular surface contact stresses. Limitations include the lack of bleeding and synovial responses associated with in vivo injuries, as well as the scarcity of available specimens.
Class II: In vivo animal models (Table S-2)
As we developed in vivo animal models of joint injury, a critical focus was placed on ensuring that any injuries produced involved the habitual weight-bearing surface of that animal's joint.31 Specific consideration was given to determining the habitual posture of a given animal species (e.g., rabbit knees are deeply flexed during their habitual posture of squatting), which then had implications in choosing the appropriate surgical approaches (e.g., posterior (popliteal) rather than anterior (medial para-patellar) in the rabbit) and delivering mechanical insults upon the weight-bearing associated portion of the joint surface.
Rabbit Medial Condyle Impacts
Joint surface contusions associated with dislocations or ligament and capsular tears commonly do not cause intra-articular fractures. To simulate these types of injuries we surgically exposed the weight-bearing region of rabbit medial femoral condyles, and then applied controlled energy impacts of 2 or 4 Joules/cm2 with a 5 mm diameter metal platen. Like joint dislocations and ligament and capsular tears, the arthrotomy caused bleeding and synovial inflammation. In five animals the impaction injury led to marked loss of staining for matrix proteolgycans and fissuring and thinning of the articular cartilage at the impact site at eight weeks following injury.
Based on the evidence that articular surface impact causes release of mitochondrial ROS that leads to progressive chondrocyte death in vitro,16,18,22 we hypothesized that drugs that block steps in the mechano-transduction pathway leading to ROS production would help maintain chondrocyte viability and metabolism as measured by chondrocyte density and ATP production. To test this hypothesis we subjected rabbit medial femoral condyles to 2 Joule/cm2 impactions and immediately injected three different agents following injury: amobarbital, a drug that reduces ROS by blocking mitochondrial electron flow, and cytochlasin B and nocodazole, which disrupt the chondrocyte cytoskeleton thereby blocking transmission of mechanical signals to the mitochondria, a strategy that had proven effective in preserving cell viability in the in vitro experiments.
At seven days after a 2 Joule/cm2 impact femoral condyle matrix proteogly can staining was reduced slightly and damage to cartilage was limited to scattered superficial fissures. All three treatments improved both chondrocyte viability and ATP production compared to untreated controls. Although cytochalasin B and nocodazole were less effective than amobarbital, they also tended to ameliorate impact effects (Figure 1). These data show that at least some of the progressive cartilage damage that follows mechanical insults is caused by cellular reactions to injury, which are subject to intervention after the fact, and confirms that the mechano-transduction pathway we found in in vitro studies operates in vivo.
Figure 1. Results of inhibiting mechanical strain-ROS pathway following joint injury: (A) viable chondrocytes/mm3 and (B) chondrocyte ATP (ng/mg) [CTB: cytochlasin B, NOC: nocodazole, AMO: amobarbital].
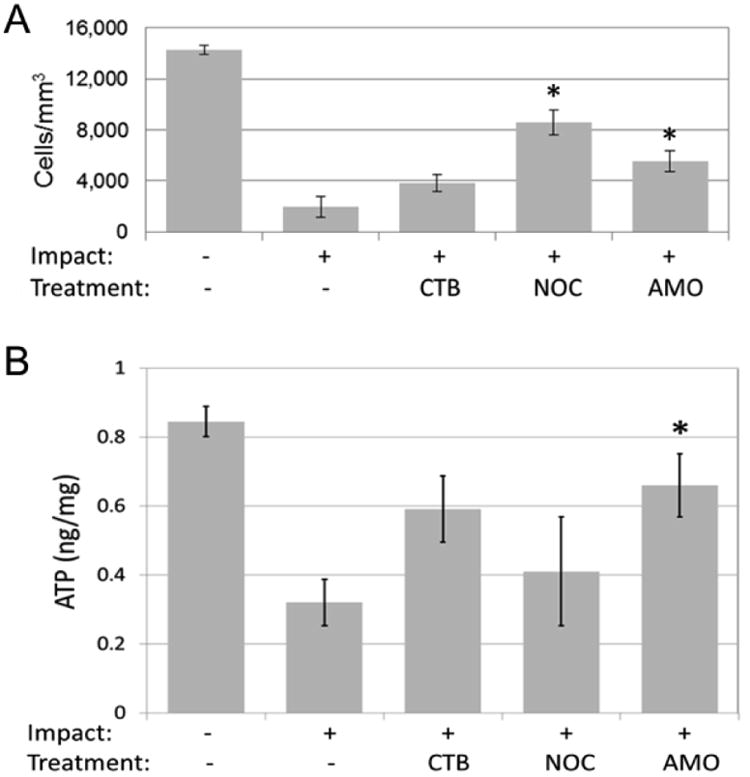
The rabbit joint impaction model is useful for testing acute treatments, but it may also be used to study the long-term consequences of these interventions, as cartilage degenerates in and around impact sites in untreated joints. The mild single chondral insult in this impact model complements more severely damaging intra-articular fracture models, as well as models that create chronically abnormal joint mechanics. One issue with this experiment is that we injected the agents immediately after injury; this will not be possible in clinical situations. Our in vitro experiments showed that a delay in the interruption of the ROS pathway for four hours after injury did not decrease the efficacy of the treatment.17 However, it will be important to determine if interruption of the ROS pathway is still effective for as long as a day after injury.
Rabbit Meniscal Destabilization
Ligamentous, capsular, and meniscal injuries in humans may not be associated with acute damage to the articular cartilage, but these types of injuries also increase the long-term risk of PTOA. The PTOA associated with these injuries is presumably a result of repeated joint surface overloading, possibly caused by loss of soft tissues that stabilize the joint and distribute loads, or by the movement of physiologic loading to areas of the articular surface that are not typically subjected to load. To model these more subtle injuries, we created a meniscal destabilization model in rabbits.32
By releasing the posterior horn of the medial meniscus in the rabbit knee, the load bearing and distribution capability of the meniscus is lost to the same degree as if it was totally removed. Medial meniscus destabilization increased peak articular contact stresses by 56% compared to normal, and the location of peak contact stress moved approximately 0.5 mm posterior-laterally. Meniscal destabilization and total meniscectomy both caused cartilage degeneration within eight weeks. An advantage of this model is that the surgery is straightforward and causes minimal collateral damage that would alter animal gait and limb usage. This approach provides a relatively economical animal model in which to investigate the progression of biological changes associated with chronic joint surface overloading and allows study of interventions intended to prevent its adverse consequences.
Rabbit Partial and Complete ACL Transection
To simulate in vivo graded instability we developed a rabbit ACL partial and complete transection model and measured the degree of joint laxity.33 Increasing degrees of instability correlated directly with increasing histologically-apparent articular cartilage damage at eight weeks following injury.33 Together, the results of in vitro whole joint study showing that ligamentous instability of human ankles increases articular surface contact stresses, the meniscal destabilization study, and this ACL transaction study support the clinical impression that joint instability increases joint contact stresses, which leads to OA. This ACL transection model allows studies of interventions intended to prevent or delay the onset of OA following injuries that cause joint instability without initial cartilage damage.
Goat Joint Impact and Meniscal Destabilization
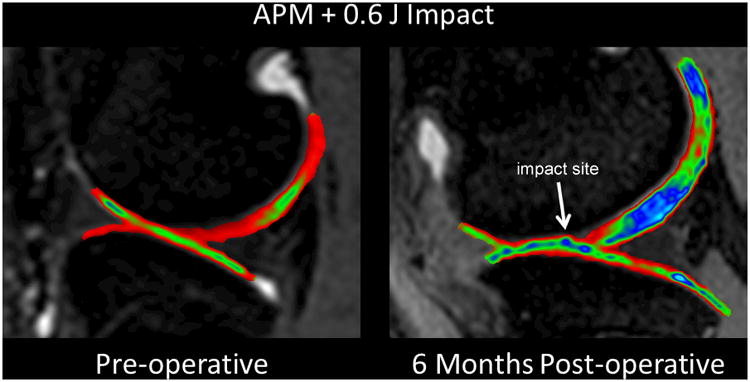
A limitation of the rabbit in vivo models is the inability to monitor the onset and progression of cartilage changes non-invasively; the thin articular cartilage and small size of the rabbit knee limits the ability to study changes in the joint without sacrificing the animals. For this reason we established a goat model of joint injury with progressive joint degeneration that can be monitored over time by MRI. The adult goat stifle joint anatomy is similar to that of the human knee, and the goat has relatively long legs with a knee that fits in the same MRI coil used for humans. To simulate human joint injuries that include acute cartilage impaction without fractures and mild residual instability, we performed anterior partial meniscectomy (APM) and impacted the goat medial femoral condyle through an anterior arthrotomy. In addition to introducing mild instability, the APM opening provided direct access for impaction of the load-bearing surface with a metal platen.
We document changes in the injured joints at multiple time points with MRI sequences routinely used to follow human ACL-rupture patients. The MRIs shown in Figure 2 are from an ongoing study of PTOA pathogenesis. Longitudinal T2-weighted fat-saturated fast spin echo MRI sequences were acquired to look at bone marrow edema, articular cartilage, and synovial fluid. Initial cartilage swelling and bone contusion progression over time are consistent with that documented in humans with ACL injuries during the acute time period of injury (3-day) to surgery (2-3 weeks).34-36
Figure 2. MRI images of goat knee before and six months after impaction and partial meniscectomy.
Yucatan Mini-Pig Intra-articular Fracture
Intra-articular fractures, even when anatomically reduced and stabilized, dramatically increase the risk of PTOA. The viable whole human joint model of intra-articular fractures showed that few chondrocytes are killed by the initial impact,26 raising the possibility that post-injury treatment of these severe injuries might decrease the risk or delay the onset of PTOA. However, small animal intra-articular fracture models have limitations including the difficulty in reducing and stabilizing the fractures and thin articular cartilage. For these reasons we designed a closed reproducible intra-articular fracture in Yucatan mini-pigs (Figure 3) that made it possible to perform fracture reductions and stabilizations similar to the current treatment of human intra-articular fractures, even using the same implants.37
Figure 3. Intraoperative fluoroscopic images of the creation and surgical repair of an intra-articular fracture of the distal tibia in the Yucatan mini-pig model.
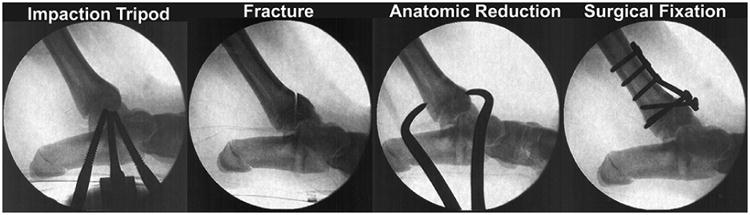
Intra-articular fractures were created in 22 Yucatan mini-pigs using identical fracture energy and treated either with anatomic reduction and surgical fixation or with a 2 mm articular surface step off and surgical fixation. Three months postoperatively, nearly all the animals with articular surface step offs, and many with anatomic reductions, developed significant cartilage degeneration, replicating the human problem where even anatomic reductions do not always prevent PTOA.
The results of the explant and the rabbit femoral condyle impact studies discussed above indicate that inhibiting ROS production or scavenging ROS could potentially preserve chondrocyte viability and metabolism following joint injury. The Yucatan mini-pig model provides a vehicle for investigation of the effects of inhibiting ROS, other biologic interventions, and mechanical interventions on joint health in a model that simulates closed human intra-articular fractures and their surgical treatment. The model can also be used to study treatments designed to improve outcomes in joints with articular step offs and gaps similar to those found in severe intra-articular fractures in humans. However, compared to small animal models this model is expensive, and the creation and operative treatment of the intra-articular fractures requires greater surgical and anesthetic skills.
Class III: Patient-specific computational models (Table S-3)
Results from the in vitro and in vivo models suggest that it would be reasonable to initiate clinical studies to assess the effect of biologic or improved surgical treatments of injured or overloaded joints. However, a challenge in clinical studies of PTOA has been that it is not known what mechanical factors most strongly influence its development and what levels of increased contact stress predictably cause OA. For instance, the severity of an intra-articular fracture and the elevated contact stress exposure after treatment (due to residual surface incongruity) are two influential but uncontrolled and previously un-measurable mechanical factors. This has largely precluded comparative multi-center studies on intra-articular fractures, because to rigorously assess the effect of treatment of any condition, an investigator must be able to measure risk factors. To address this need, and to define the tolerance of human articular cartilage for overloading, we developed computational tools for quantitative patient-specific assessment of mechanical factors involved in PTOA development following intra-articular fracture.38-44 To assess the effect of joint overloading in un-injured joints we studied patients with hip dysplasia.
Intra-articular Fracture Severity
Clinical experience supports the contention that the intensity of the original joint trauma (fracture severity) is one of the most important factors contributing to PTOA. Based on fracture mechanics principles, we developed objective techniques to measure the fracture severity from CT scan data and validated them in surrogate bone specimens,38 bovine bone specimens,39 and in an initial prospective single-center patient series of tibial plafond intra-articular fractures.42,43 The fracture severity metric correlated strongly with PTOA development; in particular, fracture energy exceeding a specific threshold led to PTOA in every patient within two years. The PTOA risk in this instance was regardless of reduction quality, just as in the pig PTOA model. Recently we have expedited the analysis procedures so that extending our original small series to a larger cohort is now feasible.45 This approach has promise for identifying at-risk joints so that they can be stratified by their PTOA risk based on the energy of injury.
Joint Incongruity
Elevated contact stress due to residual articular incongruity after intra-articular fracture is another mechanical variable implicated in PTOA. Similar to fracture severity, the degree of mal-reduction is difficult to objectively measure in patients. Furthermore, it provides but a surrogate for the actual contact stress experienced by the joint. We developed computational techniques to directly measure the post-reduction contact stress exposure after intra-articular fracture treatment40,41,46 and have shown that areas of high contact stress correlate with progressive cartilage thinning. Our results have further suggested a cumulative contact stress exposure threshold above which cartilage degeneration is likely (Figure 4).44 More recently, we have developed expedited computational stress analysis techniques47 making larger multi-center studies now possible. Similar to fracture energy calculation this modeling allows objective stratification of an important risk factor for PTOA. These observations are now being explored further in the animal models of overload.
Figure 4.
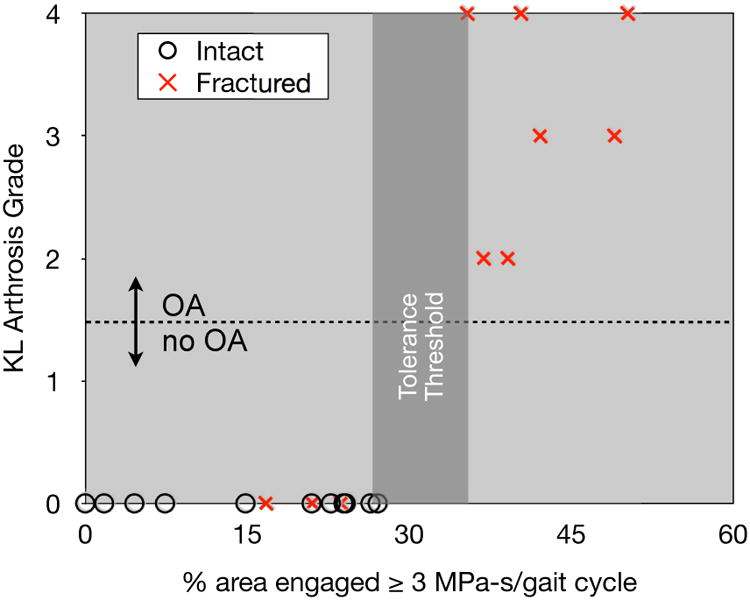
The relative proportion of articular surface area experiencing supra-threshold contact stress was perfectly predictive of OA status two years following surgical reduction of displaced intra-articular fractures of the distal tibia.
Hip Dysplasia
The in vivo rabbit meniscal destabilization model and patient-specific models of articular surface incongruity showed that excessive contact stresses lead to cartilage loss and OA. These observations raise the question: could decreasing excessive chronic contact stresses decrease the risk of OA, and perhaps more importantly relieve symptoms? Dysplastic human hips provide an opportunity to investigate this question. Studies of 84 patients at our institution with unilateral hip dysplasia treated without surgery and followed for an average of 29.2 years showed that increasing cumulative contact stress (a combination of elevated contact stress acting over time) predicted joint degeneration with 90% relability.48,49
In contemporary practice, peri-acetabular osteotomy (PAO) is a surgical treatment performed in young adult patients with symptomatic dysplastic hips with the intent of relieving excessive joint overloading and thereby decreasing joint pain and the likelihood of progressive OA. Utilizing retrospective patient-specific computational stress analyses performed in a series of consecutive patients treated with PAO, we found that decreased peak contact stress during normal walking gait correlated significantly with improved clinical outcome (pain/function) scores.50 Changes in visual analog scale (VAS) pain scores (post-op – pre-op) correlated well with changes in maximum contact stress (n = 18, R2 = 0.6, p = 0.001 – Figure 5).
Figure 5.
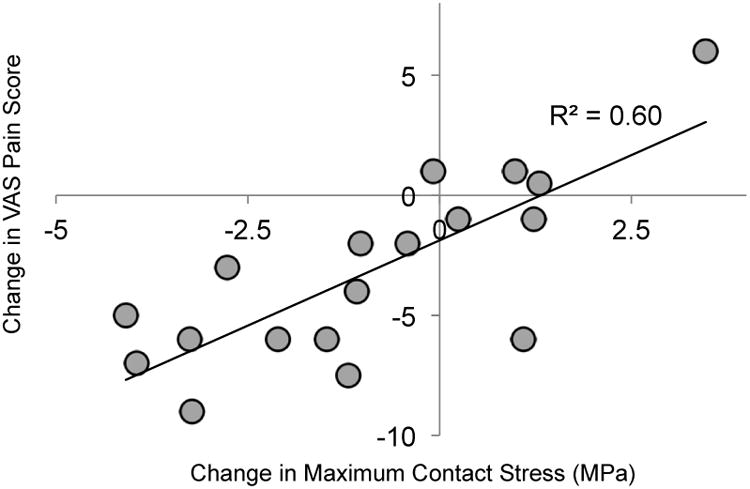
Plot of changes in visual analog scale (VAS) pain score (post-op – pre-op) vs. change in maximum contact stress. VAS pain scores range from 0 to 10, with higher scores indicating more pain. Lower contact stress values were computed in patients who experienced less pain.
The principal advantage of patient-specific computational models is that they enable quantitative assessment of the severity of injury and degree of chronic increased contact stresses using measures that reliably predict which patients will later develop OA and stratifies them for the degree of mechanical overload seen by the joint. Thus far, only patient-specific models of intra-articular fractures and joint dysplasia have been developed and tested. Patient-specific computational models of other joint injuries, for example meniscal or ligamentous tears, would help advance understanding of the relationships between these injuries and OA.
In silico biomathematical models: integrating knowledge for translation
Explant, human whole joint, and animal models of joint injuries and chronic joint overloading provide crucial insight into the cellular processes that lead to PTOA. We have focused these studies on a unique chondrocyte mechanotransduction pathway that regulates critical aspects of energy metabolism. Mitochondria play a central role in the pathway by producing free radicals in quantities proportional to load magnitude. Over-activation of the pathway by acute and chronic overloading leads to oxidative stress, metabolic derangement, and cell death. Drugs targeting the pathway have shown strong cytoprotective and chondroprotective effects in ex vivo and in vivo models of PTOA.17,51 The patient-specific computational models, and future refinements of these methods, can define the relationships between acute and chronic joint overloading and the onset and progression of PTOA. The remaining challenge is to integrate the knowledge gained about cellular responses to joint overloading into the treatment of specific patients or to predict the effects of therapeutic interventions at different points in time after joint injury.
We have created in silico mathematical models of cellular and biochemical processes in injured joints to meet this challenge. We refer to these models as “biomathematical” models, and they offer a powerful method of clarifying complex interactions between multiple variables. We first established a framework for the complex relationship between chondrocyte behavior and the chemical signaling within cartilage after a blunt impact that determines whether trauma leads to PTOA.52 Next, we modeled the effect of repetitive joint overloading, and we introduced a mathematical structure into the general modeling framework to gain flexibility in modeling delayed cellular responses to cytokines and increase the speed of computation.53 Most recently, we have explicitly introduced cartilage biomechanics into our biomathematical models.
The value of biomathematical models is in their potential to help predict specific patient outcomes across a wide range of joint injuries. By distilling the data from explant and animal assays into parameters for biomathematical models, mathematics can translate experimental data to clinically relevant knowledge. As one example, an appropriate biomathematical model could be combined with a model of the severity of intra-articular fractures, such as described above, to create a virtual joint. Such a tool would allow a clinician to assess the severity of the trauma in a given patient, determine the probability and time frame for PTOA pathogenesis, and help prescribe appropriate treatments at different time points following injury.
Summary and Conclusions
The observations presented above show that OA is not the result of straightforward mechanical wearing or acute high energy structural disruption of joint surfaces; instead it is a multifaceted complex process that involves manifold interactions of contact stresses and cellular responses. The daunting challenge of defining these interactions and the interplay between joint loading and cell responses that cause OA, or maintain joint health, is one of the reasons it has been difficult to find therapies that prevent or slow the progression of OA.
Study of PTOA can reveal the pathways likely to have a role in all forms of OA. Study of in vitro explant and whole joint cartilage injury models showed that cartilage injury causes chondrocytes to overproduce ROS, which leads to progressive cell death and long-lasting metabolic dysfunction.54 Interruption of the ROS pathway following joint injury with multiple agents decreases cell death and maintains healthy chondrocyte metabolism.54 The results of the models of joint overloading in the absence of joint injury suggest that deleterious levels of ROS release have a role in all forms of OA – a suggestion that will require further study.
Computational patient-specific models show that acute joint loading and cumulative joint loading exceeding defined thresholds lead to the rapid loss of articular cartilage. These results create the potential for identifying populations of patients who will certainly develop OA and therefore would be candidates for experimental therapies. Evidence that decreasing joint contact stresses in patients with hip pain and dysplasia relieves symptoms spurs pursuit of new treatments of people at risk of OA caused or exacerbated by joint overloading. Results from the in vitro and in vivo models reveal a cellular mechano-transduction process that underlies the joint overloading-progressive cartilage loss phenomenon, thereby establishing targets for drug interventions for these patients.
In conclusion, we have determined thresholds of acute and repetitive contact stress that cause cartilage loss, identified cellular responses to acute and chronic mechanical overload of articular surfaces that damage cartilage, and have discovered methods of interrupting those pathways. Somewhat surprisingly, remarkably few chondrocytes are killed by even extreme impact injuries, and acute and chronic contact stress induced chondrocyte dysfunction may play a more important role in OA caused by joint injury. Preliminary short term in vivo experiments show that several of these methods maintain chondrocyte viability and metabolic function following joint injury. We are currently engaged in longer-term studies to determine if strategies to safely disrupt the overload-ROS pathway restore joint health following injury or chronic joint overloading and to see if they can be integrated into clinical practice.
Clinical Significance.
Considered collectively, studies extending from explants to humans show that thresholds of joint overloading that cause cartilage loss can be defined, that to at least some extent both forms of joint overloading act through the same biologic pathways, and interventions that interrupt these pathways prevent cartilage damage. These observations suggest that treatments that decrease the risk of all forms of OA progression can be discovered.
Acknowledgments
This research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award numbers P50 AR048939 and P50 AR055533. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This research was also supported by the Department of Defense, the Veterans Administration and the University of Iowa Department of Orthopaedics and Rehabilitation. The critical contributions of Thomas D. Brown, Todd O. McKinley and Yuki Tochigi to the investigations and concepts presented in this manuscript are gratefully acknowledged.
Footnotes
Author Contributions Statement: All authors made substantial contributions to the research design, data acquisition, and analysis/interpretation of data. DDA, JAM, and JAB were involved in the drafting of the paper, and all authors provided subsequent critical review. All authors have read and approved the final submitted manuscript.
References
- 1.Buckwalter JA, Mankin HJ, Grodzinsky AJ. Articular cartilage and osteoarthritis. Instr Course Lect. 2005;54:465–480. [PubMed] [Google Scholar]
- 2.Cross JD, Ficke JR, Hsu JR, Masini BD, Wenke JC. Battlefield orthopaedic injuries cause the majority of long-term disabilities. J Am Acad Orthop Surg. 2011;19(1):S1–7. doi: 10.5435/00124635-201102001-00002. [DOI] [PubMed] [Google Scholar]
- 3.Rivera JC, Wenke JC, Buckwalter JA, Ficke JR, Johnson AE. Post-Traumatic Osteoarthritis Caused by Battlefield Injuries is the Primary Source of Disability in Warriors. J Am Acad Orthop Surg. 2012;20:S64–S69. doi: 10.5435/JAAOS-20-08-S64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson DD, Chubinskaya S, Guilak F, Martin JA, Oegema TR, Olson SA, Buckwalter JA. Post-traumatic osteoarthritis: Improved understanding and opportunities for early intervention. J Orthop Res. 2011;29:802–809. doi: 10.1002/jor.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buckwalter JA, Brown TD. Joint injury, repair, and remodeling: roles in post-traumatic osteoarthritis. Clin Orthop Relat Res. 2004;423:7–16. [PubMed] [Google Scholar]
- 6.Buckwalter JA, Felson DT. Post-Traumatic Arthritis: Definitions and Burden of Disease. In: Olson SA, Guilak F, editors. Post-Traumatic Arthritis. New York: Springer; 2015. pp. 7–15. [Google Scholar]
- 7.Gillquist J, Messner K. Anterior cruciate ligament reconstruction and the long-term incidence of gonarthrosis. Sports Med. 1999;27:143–156. doi: 10.2165/00007256-199927030-00001. [DOI] [PubMed] [Google Scholar]
- 8.Roos H, Adalberth T, Dahlberg L, Lohmander LS. Osteoarthritis of the knee after injury to the anterior cruciate ligament or meniscus: the influence of time and age. Osteoarthritis Cartilage. 1995;3:261–267. doi: 10.1016/s1063-4584(05)80017-2. [DOI] [PubMed] [Google Scholar]
- 9.Roos H, Lauren M, Adalberth T, Roos EM, Jonsson K, Lohmander LS. Knee osteoarthritis after meniscectomy: prevalence of radiographic changes after twenty-one years, compared with matched controls. Arthritis Rheum. 1998;41:687–693. doi: 10.1002/1529-0131(199804)41:4<687::AID-ART16>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 10.von Porat A, Roos EM, Roos H. High prevalence of osteoarthritis 14 years after an anterior cruciate ligament tear in male soccer players: a study of radiographic and patient relevant outcomes. Ann Rheum Dis. 2004;63:269–273. doi: 10.1136/ard.2003.008136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furman BD, Mangiapani DS, Zeitler E, Bailey KN, Horne PH, Huebner JL, Kraus VB, Guilak F, Olson SA. Targeting pro-inflammatory cytokines following joint injury: acute intra-articular inhibition of interleukin-1 following knee injury prevents post-traumatic arthritis. Arthritis Res Ther. 2014;16:R134. doi: 10.1186/ar4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takebe K, Rai MF, Schmidt EJ, Sandell LJ. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthritis Cartilage. 2015;23:454–461. doi: 10.1016/j.joca.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Brouillette MJ, Ayati BP, Martin JA. A validated model of the pro- and anti-inflammatory cytokine balancing act in articular cartilage lesion formation. Front Bioeng Biotechnol. 2015;3:25. doi: 10.3389/fbioe.2015.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beecher BR, Martin JA, Pedersen DR, Heiner AD, Buckwalter JA. Antioxidants block cyclic loading induced chondrocyte death. Iowa Orthop J. 2007;27:1–8. [PMC free article] [PubMed] [Google Scholar]
- 15.Goodwin W, McCabe D, Sauter E, Reese E, Walter M, Buckwalter JA, Martin JA. Rotenone prevents impact-induced chondrocyte death. J Orthop Res. 2010;28:1057–1063. doi: 10.1002/jor.21091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jang KW, Buckwalter JA, Martin JA. Inhibition of cell-matrix adhesions prevents cartilage chondrocyte death following impact injury. J Orthop Res. 2014;32:448–454. doi: 10.1002/jor.22523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin JA, McCabe D, Walter M, Buckwalter JA, McKinley TO. N-acetylcysteine inhibits post-impact chondrocyte death in osteochondral explants. J Bone Joint Surg Am. 2009;91:1890–1897. doi: 10.2106/JBJS.H.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sauter E, Buckwalter JA, McKinley TO, Martin JA. Cytoskeletal dissolution blocks oxidant release and cell death in injured cartilage. J Orthop Res. 2012;30:593–598. doi: 10.1002/jor.21552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seol D, McCabe DJ, Choe H, Zheng H, Yu Y, Jang K, Walter MW, Lehman AD, Ding L, Buckwalter JA, Martin JA. Chondrogenic progenitor cells respond to cartilage injury. Arthritis Rheum. 2012;64:3626–3637. doi: 10.1002/art.34613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu Y, Zheng H, Buckwalter JA, Martin JA. Single cell sorting identifies progenitor cell population from full thickness bovine articular cartilage. Osteoarthritis Cartilage. 2014;22:1318–1326. doi: 10.1016/j.joca.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckwalter JA, Anderson DD, Brown TD, Tochigi Y, Martin JA. The roles of mechanical stresses in the pathogenesis of osteoarthritis: Implications for treatment of joint injuries. Cartilage. 2013;4:286–294. doi: 10.1177/1947603513495889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolff KJ, Ramakrishnan PS, Brouillette MJ, Journot BJ, McKinley TO, Buckwalter JA, Martin JA. Mechanical stress and ATP synthesis are coupled by mitochondrial oxidants in articular cartilage. J Orthop Res. 2013;31:191–196. doi: 10.1002/jor.22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin JA, Martini A, Molinari A, Morgan W, Ramalingam W, Buckwalter JA, McKinley TO. Mitochondrial electron transport and glycolysis are coupled in articular cartilage. Osteoarthritis Cartilage. 2012;20:323–329. doi: 10.1016/j.joca.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramakrishnan P, Hecht BA, Pedersen DR, Lavery MR, Maynard J, Buckwalter JA, Martin JA. Oxidant conditioning protects cartilage from mechanically induced damage. J Orthop Res. 2010;28:914–920. doi: 10.1002/jor.21072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sillat T, Barreto G, Clarijs P, Soininen A, Ainola M, Pajarinen J, Korhonen M, Konttinen YT, Sakalyte R, Hukkanen M, Ylinen P, Nordstrom DC. Toll-like receptors in human chondrocytes and osteoarthritic cartilage. Acta Orthop. 2013;84:585–592. doi: 10.3109/17453674.2013.854666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tochigi Y, Buckwalter JA, Martin JA, Hillis SL, Zhang P, Vaseenon T, Lehman AD, Brown TD. Distribution and progression of chondrocyte damage in a whole-organ model of human ankle intra-articular fracture. J Bone Joint Surg Am. 2011;93:533–539. doi: 10.2106/JBJS.I.01777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKinley TO, Rudert MJ, Koos DC, Brown TD. Incongruity versus instability in the etiology of posttraumatic arthritis. Clin Orthop Relat Res. 2004;423:44–51. doi: 10.1097/01.blo.0000131639.89143.26. [DOI] [PubMed] [Google Scholar]
- 28.McKinley TO, Tochigi Y, Rudert MJ, Brown TD. The effect of incongruity and instability on contact stress directional gradients in human cadaveric ankles. Osteoarthritis Cartilage. 2008;16:1363–1369. doi: 10.1016/j.joca.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKinley TO, Tochigi Y, Rudert MJ, Brown TD. Instability-associated changes in contact stress and contact stress rates near a step-off incongruity. J Bone Joint Surg Am. 2008;90:375–383. doi: 10.2106/JBJS.G.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tochigi Y, Rudert MJ, McKinley TO, Pedersen DR, Brown TD. Correlation of dynamic cartilage contact stress aberrations with severity of instability in ankle incongruity. J Orthop Res. 2008;26:1186–1193. doi: 10.1002/jor.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tochigi Y, Buckwalter JA, Brown TD. Toward improved clinical relevance of cartilage insult models in the rabbit knee: surgical access to the habitual weight-bearing region. Iowa Orthop J. 2013;33:196–201. [PMC free article] [PubMed] [Google Scholar]
- 32.Arunakul M, Tochigi Y, Goetz JE, Diestelmeier BW, Heiner AD, Rudert J, Fredericks DC, Brown TD, McKinley TO. Replication of chronic abnormal cartilage loading by medial meniscus destabilization for modeling osteoarthritis in the rabbit knee in vivo. J Orthop Res. 2013;31:1555–1560. doi: 10.1002/jor.22393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tochigi Y, Vaseenon T, Heiner AD, Fredericks DC, Martin JA, Rudert MJ, Hillis SL, Brown TD, McKinley TO. Instability dependency of osteoarthritis development in a rabbit model of graded anterior cruciate ligament transection. J Bone Joint Surg Am. 2011;93:640–647. doi: 10.2106/JBJS.J.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Potter HG, Jain SK, Ma Y, Black BR, Fung S, Lyman S. Cartilage injury after acute, isolated anterior cruciate ligament tear: immediate and longitudinal effect with clinical/MRI follow-up. Am J Sports Med. 2012;40:276–285. doi: 10.1177/0363546511423380. [DOI] [PubMed] [Google Scholar]
- 35.Pedersen DR, Martin JA, Thedens DR, Klocke NF, Roberts NH, Goetz JE, Amendola A. Imaging biopsy composition at ACL reconstruction. Orthop Res Rev. 2013;5:35–41. doi: 10.2147/ORR.S43973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klocke NF, Amendola A, Thedens DR, Williams GN, Luty CM, Martin JA, Pedersen DR. Comparison of T1rho, dGEMRIC, and quantitative T2 MRI in preoperative ACL rupture patients. Acad Radiol. 2013;20:99–107. doi: 10.1016/j.acra.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goetz JE, Fredericks D, Petersen E, Rudert MJ, Baer T, Swanson E, Roberts N, Martin J, Tochigi Y. A clinically realistic large animal model of intra-articular fracture that progresses to post-traumatic osteoarthritis. Osteoarthritis Cartilage. 2015;23:1797–1805. doi: 10.1016/j.joca.2015.05.022. [DOI] [PubMed] [Google Scholar]
- 38.Beardsley CL, Bertsch CR, Marsh JL, Brown TD. Interfragmentary surface area as an index of comminution energy: proof of concept in a bone fracture surrogate. J Biomech. 2002;35:331–338. doi: 10.1016/s0021-9290(01)00214-7. [DOI] [PubMed] [Google Scholar]
- 39.Beardsley CL, Anderson DD, Marsh JL, Brown TD. Interfragmentary surface area as an index of comminution severity in cortical bone impact. J Orthop Res. 2005;23:686–690. doi: 10.1016/j.orthres.2004.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson DD, Goldsworthy JK, Shivanna K, Grosland NM, Pedersen DR, Thomas TP, Tochigi Y, Marsh JL, Brown TD. Intra-articular contact stress distributions at the ankle throughout stance phase-patient-specific finite element analysis as a metric of degeneration propensity. Biomech Model Mechanobiol. 2006;5:82–89. doi: 10.1007/s10237-006-0025-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li W, Anderson DD, Goldsworthy JK, Marsh JL, Brown TD. Patient-specific finite element analysis of chronic contact stress exposure after intraarticular fracture of the tibial plafond. J Orthop Res. 2008;26:1039–1045. doi: 10.1002/jor.20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson DD, Mosqueda T, Thomas T, Hermanson EL, Brown TD, Marsh JL. Quantifying tibial plafond fracture severity: absorbed energy and fragment displacement agree with clinical rank ordering. J Orthop Res. 2008;26:1046–1052. doi: 10.1002/jor.20550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas TP, Anderson DD, Mosqueda TV, Van Hofwegen CJ, Hillis SL, Marsh JL, Brown TD. Objective CT-based metrics of articular fracture severity to assess risk for posttraumatic osteoarthritis. J Orthop Trauma. 2010;24:764–769. doi: 10.1097/BOT.0b013e3181d7a0aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson DD, Van Hofwegen C, Marsh JL, Brown TD. Is elevated contact stress predictive of post-traumatic osteoarthritis for imprecisely reduced tibial plafond fractures? J Orthop Res. 2011;29:33–39. doi: 10.1002/jor.21202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson DD, Kilburg AT, Thomas TP, Marsh JL. Expedited CT-based methods for evaluating fracture severity to assess risk of post-traumatic OA after articular fractures. Iowa Orthop J. 2016;36:46–52. [PMC free article] [PubMed] [Google Scholar]
- 46.Anderson DD, Goldsworthy JK, Li W, James Rudert M, Tochigi Y, Brown TD. Physical validation of a patient-specific contact finite element model of the ankle. J Biomech. 2007;40:1662–1669. doi: 10.1016/j.jbiomech.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kern AM, Anderson DD. Expedited patient-specific assessment of contact stress exposure in the ankle joint following definitive articular fracture reduction. J Biomech. 2015;48:3427–3432. doi: 10.1016/j.jbiomech.2015.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hadley NA, Brown TD, Weinstein SL. The effects of contact pressure elevations and aseptic necrosis on the long-term outcome of congenital hip dislocation. J Orthop Res. 1990;8:504–513. doi: 10.1002/jor.1100080406. [DOI] [PubMed] [Google Scholar]
- 49.Maxian TA, Brown TD, Weinstein SL. Chronic stress tolerance levels for human articular cartilage: two nonuniform contact models applied to long-term follow-up of CDH. J Biomech. 1995;28:159–166. doi: 10.1016/0021-9290(94)00054-8. [DOI] [PubMed] [Google Scholar]
- 50.Townsend KC. Master of Science. University of Iowa; Iowa City, IA, U.S.A: 2015. Validation and applications of discrete element analysis in the hip joint. [Google Scholar]
- 51.Coleman MC, Ramakrishnan PS, Brouillette MJ, Martin JA. Injurious Loading of Articular Cartilage Compromises Chondrocyte Respiratory Function. Arthritis Rheumatol. 2016;68:662–671. doi: 10.1002/art.39460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Graham JM, Ayati BP, Ding L, Ramakrishnan PS, Martin JA. Reaction-diffusion-delay model for EPO/TNF-alpha interaction in articular cartilage lesion abatement. Biol Direct. 2012;7:9. doi: 10.1186/1745-6150-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Ayati BP, Brouillete MJ, Graham JM, Ramakrishnan PS, Martin JA. Modeling and simulation of the effects of cyclic loading on articular cartilage lesion formation. Int J Numer Method Biomed Eng. 2014;30:927–941. doi: 10.1002/cnm.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coleman MC, Buckwalter JA, Martin JA. Potential Mechanisms of PTA: Oxidative Stress. In: Olson SA, Guilak F, editors. Post-Traumatic Arthritis. New York: Springer; 2015. pp. 211–219. [Google Scholar]