

Abstract
Background
The aberrant expression of CD40, a co-stimulatory receptor found on the antigen-presenting cells, is involved in the pathogenesis of various degenerative diseases. Our previous study demonstrated that the reduction of cytosolic phospholipase A2 alpha (cPLA2α) protein overexpression and activation in the spinal cord of a mouse model of ALS, hmSOD1 G93A, inhibited CD40 upregulation in microglia. The present study was designed to determine whether cPLA2α has a direct, participatory role in the molecular events leading to CD40 induction.
Methods
Cultures of primary mouse microglia or BV-2 microglia cell line exposed to lipopolysaccharide (LPS) or interferon gamma (IFNγ) for different periods of time, in order to study the role of cPLA2α in the events leading to CD40 protein induction.
Results
Addition of LPS or IFNγ caused a significant upregulation of cPLA2α and of CD40, while prevention of cPLA2α upregulation by a specific oligonucleotide antisense (AS) prevented the induction of CD40, suggesting a role of cPLA2α in the induction of CD40. Addition of LPS to microglia caused an immediate activation of cPLA2α detected by its phosphorylated form, while addition of IFNγ induced cPLA2α activation at a later time scale (4 h). The activation of cPLA2α is mediated by ERK activity. Suppression of cPLA2α activity inhibited superoxide production by NOX2-NADPH oxidase and activation of NF-κB detected by the phosphorylation of p65 on serine 536 at 15 min by LPS and at 4 h by IFNγ. Inhibition of NOX2 prevented NF-κB activation and CD40 induction but did not affect cPLA2α activation, suggesting cPLA2α is located upstream to NOX2 and NF-κB. The activation of cPLA2 by LPS was mediated by both adaptor proteins downstream to LPS receptor; TRIF and MyD88, while the activation of cPLA2α by IFNγ was mediated by the secreted TNF-α at 4 h. The early activation of STAT1α (detected by phospho-serine727 and phoshpo-tyrosine701) by IFNγ and the late activation of STAT1α by LPS were not affected in the presence of cPLA2α inhibitors, indicating that STAT1α is not under cPLA2α regulation.
Conclusions
Our results show for the first time that cPLA2 upregulates CD40 protein expression induced by either LPS or IFNγ, and this regulatory effect is mediated via the activation of NOX2-NADPH oxidase and NF-κB. Cumulatively, our results indicate that cPLA2α may serve as a pivotal amplifier of the inflammatory response in the CNS.
Keywords: Cytosolic phospholipase A2α, CD40, Microglia, Lipopolysaccharide, Interferon gamma, Nuclear factor-κB
Background
The co-stimulatory receptor, CD40 molecule, is a 50-kDa type I member of the tumor necrosis factor receptor superfamily that is widely expressed by the various immune and non-immune cells [1–7]. The interaction between CD40 and its ligand, CD40L (CD154), is one of multiple signals necessary for a productive immune response [8–10]. The CD40-CD154 interaction promotes a wide spectrum of molecular and cellular processes including, immunoglobulin class switching, cell differentiation and maturation, B-cell growth, and expression of other co-stimulatory molecules such as MHC class II, ICAM-1, VCAM-1, E-selectin, LFA-3, B7.1, and B7.2) [11, 12]. In addition, CD40-CD154 interaction induces the production of cytotoxic radicals and of various pro-inflammatory cytokines (TNF-α, IL6, IL-8, and IL-12) and chemokines (CCL-2) [13, 14].
In the central nervous system (CNS), the microglial cells are constantly in motion, surveying their environment to protect the nervous system acting as debris scavengers, killers of pathogens, and regulators of innate and adaptive immune responses. The microglia cells express the key surface molecules for antigen presentation (CD40, MHC-II, and B7); therefore, they are considered the most potent endogenous antigen-presenting cells in the CNS [15]. In a healthy nervous system, microglia constitutively expresses CD40 at a low level, which is enhanced under inflammatory conditions. Several studies show that the aberrant expression of CD40 is involved in the initiation and maintenance of various neurodegenerative diseases including multiple sclerosis, Alzheimer’s disease, HIV-1-associated dementia and cerebral ischemia [16–20], and other diseases as rheumatoid arthritis and atherosclerosis [18, 21, 22]. Blockade of CD40-CD40L signaling has been shown to provide a significant beneficial effect in a number of animal models of neurological human diseases [1, 18, 23–28].
Previous findings suggested that cPLA2α plays an important role in inflammation. cPLA2α specifically hydrolyzes phospholipids containing arachidonic acid at the sn-2 position [29, 30] and is generally thought to be the rate-limiting step in the generation of eicosanoids and platelet activating factor. These lipid mediators play critical roles in the initiation and modulation of inflammation and oxidative stress. cPLA2α is ubiquitous in the brain cells and is essential for their physiological regulation. However, elevated cPLA2α expression and activity were detected in the inflammatory sites in a vast array of inflammatory diseases [31], including neurodegenerative diseases such as Alzheimer’s disease, multiple sclerosis, and amyotrophic lateral sclerosis (ALS) [32–35]. Our previous study [36] in a mouse model of ALS, hmSOD1 G93A, demonstrated that the blunting cPLA2α protein expression and inhibition of its activity inhibited microglial-CD40 upregulation. This inhibitory effect could be a result of a direct regulatory role of cPLA2α on CD40 inductive process or an indirect effect due to damping of inflammation. The present study was designed to determine whether cPLA2α has a direct role in the events leading to CD40 protein induction. To this aim, we used mouse microglia cultures and two different stimuli, LPS and IFNγ that have been reported to induce CD40 upregulation. The signal transduction events leading to CD40 upregulation by both stimuli have been studied, and it was reported that they include two transcription factors NF-κB and STAT1α that are activated in different rank order and time scale by the two stimuli [37–39].
Methods
Materials
Glutamine, penicillin-streptomycin-nystatin, phosphate buffered saline (PBS) Dulbecco’s Modified Eagle’s Medium (DMEM), Hanks’ Balanced Salts Solution (HBSS), fetal bovine serum (FBS), HEPES, sodium pyruvate, Dulbecco’s Modified Eagle’s/F12 (HAM) medium (DMEM/F12) were from Beth Ha-Emek, Biological Industries, Israel.
Sodium azide, trypan blue, p-nitrophenylphosphate, phenylmethylsulfonyl fluoride, leupeptin, benzamidine, aprotinin, DMSO, Tween 20, Tris, 4,6-diamidino-2-phenylindole (DAPI), bovine serum albumin (BSA), Trypsin-EDTA, dihyroethidium (DHE), lipopolysaccharide (LPS), Skim Milk Powder, Poly-L-lysine, horseradish peroxidase (HRP), 1,2-Dioleoyl-sn-glycerol, Triton X-100, β-mercaptoethanol, Percoll, non-essential amino-acids, Diphenyliodonium chloride (DPI) were from Sigma Israel, Rehovot, Israel. Fetal calf serum was from GE Healthcare Life Sciences HyClone Laboratories, Inc., Logan Utah, USA. ECL detection kit for the immunoblot analysis was from PerkinElmer, MA, USA. Pyrrophenone was from Cayman Chemical, Michigan, USA. TNF-α-neutralizing antibody and U0126 (MEK1/2 inhibitor) were from Cell Signaling Technology, Danvers, MA, USA. Interleukin (IL)-4, IL-10, TNF-α, IFN-γ were from PeproTech Asia, NJ, USA.
Primary microglial cell culture
Microglia were isolated from the brains of mice C57BL 1-day-old pups as previously described [40] with minor modifications. Briefly, the pups were decapitated and the brains were taken out. The tissues were digested by incubation with an enzymatic solution containing papain (116 mM NaCl, 5.4 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, 1.5 mM CaCl2, 1 mM MgSO4, 0.5 mM EDTA, 25 mM glucose, 1 mM cysteine, and 20 U/ml papain) for 60 min at 37 °C, 5% CO2. The enzymatic solution was quenched with 20% FBS in HBSS and centrifuged for 4 min at ×200g. A second digestion procedure was performed by treating the brain tissues with 0.5 mg/ml DNase-I (Worthington Biochemical Corp., NJ, USA) for 5 min and gently passing it through a fire-polished Pasteur pipettes several times. Then, the digested tissues were filtered through a 70 micron cell strainer (Corning, NY, USA) and centrifuged at 200g for 4 min. The pellet was resuspended in 20% isotonic percoll in HBSS. Fresh HBSS was carefully added and then the tubes were centrifuged at ×200g for 20 min with slow acceleration and no brakes. The pellet containing the mixed glial cells were washed with HBSS, centrifuged at ×200g for 4 min and then suspended in DMEM-F12 medium (10% FCS, 1% non-essential amino-acids, 11.4 μm β-mercaptoethanol, 10 mM HEPES, 1 mM sodium pyruvate 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 12.5 U/ml nystatin). The cells were seeded into Poly-L-lysine coated flasks and kept at 37 °C in a humidified atmosphere of 5% CO2. The growth medium was replaced with a fresh after 4 days. After two weeks, the microglial cells were separated from the astroglial cell monolayer by shaking the flasks for 1 h at 120 rpm on a rotator shaker and subjected to mild trypsinization with DMEM containing 0.25% Trypsin-EDTA (1:3) for about 90 min at 37 °C and then exchange with fresh DMEM. Then, the isolated microglial cultures were treated with 0.25% Trypsin for approximately 15 min at 37 °C and carefully detached. The cells were suspended with DMEM-F12 (containing 2% FBS 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 12.5 U/ml Nystatin) and cultured (6 × 105 cells/ml) in 24 wells on cover-slips coated with Poly-L-lysine at 37 °C in a humidified atmosphere of 5% CO2 for a week before the experiment. The purity of microglial cell preparations was confirmed by testing their immunoreactivity to the Iba-1 (Wako Chemicals, Richmond, VA, USA) marker.
Cell cultures
BV2 immortalized murine microglial cell line was a kind gift from Prof. Rosario Donato (Department of Biochemical Sciences, University of Perugia, Italy). The cells were maintained in DMEM containing 5% FBS 2 mM L-glutamine, 100u/ml penicillin, 100 μg/ml streptomycin, and 12.5 U/ml Nystatin at 37 °C and 5% CO2 until they reached confluence. The cells (3.5 × 105 cells/ml) were suspended in DMEM containing 2% FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 12.5 U/ml Nystatin and seeded in plates of 24 or 6 wells at 37 °C in a humidified atmosphere of 5% CO2.
Flow cytometry
The microglial cells were suspended in PBS and counted by Trypan Blue. The cells were pre-incubated with rat anti-mouse Fc Blocker (BD Pharmingen, San Jose, CA) at 4 °C for 10 min. For detection of CD40, the cells were incubated with PE anti-mouse CD40 (BioLegend, San Diego, CA) for 2 h on ice in the presence of Fc Blocker. Next, the cells were washed three times with PBS and subjected to fluorescence-activated cell sorter (FACS FC 500, Switzerland, Beckman Coulter) analysis. The median (median of fluorescence intensity) was calculated by subtracting the non-specific fluorescence.
Immunofluorescence analysis
Microglia were suspended in DMEM (2% FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 12.5 U/ml Nystatin) and seeded on cover slips. The cells were fixed with ice-cold methanol for 3 min and then washed with HBSS. For immunofluorescence detection, the fixed microglial cells were incubated with the first antibody 1:50 in 5% BSA/PBS (anti cPLA2α (Santa Cruz Biotechnology, CA, USA), anti CD40 (Serotec, Cambridge, UK), anti CD206 (R&D Systems, Minneapolis, USA) Serotec, Oxfordshire, UK) for 90 min at room temperature. The cells were washed three times in HBSS and incubated with Cy3 anti-rabbit, DyLight anti-rabbit, and Cy3 anti-goat (1:50 in 5% BSA/PBS; Jackson ImmunoResearch Laboratories, Inc., PA, USA) for 60 min at room temperature. The cells were washed three times in HBSS, and the nuclei were stained with DAPI. Then, final wash was performed and the cells were taken to fluorescence microscope analysis (Olympus, BX60, Hamburg, Germany).
Intracellular superoxide anion assay
O2 − production was measured using dihyroethidium (DHE). The cells were incubated in a 24-well plate on cover slips for 24 h at 37 °C. The next day the medium was replaced with heated HBSS containing 10 μm DHE, and the cells were incubated for 45 min at 37 °C. Then, the cells were stimulated with IFN-γ or LPS for 15 min. Then, the cells were stained with DAPI, washed, and fixed with ice-cold methanol for 3 min. the fluorescence intensity was measured by fluorescence microscope (Olympus, BX60, Hamburg, Germany).
Inhibition of cPLA2α expression using antisense oligonucleotides
An oligodeoxy-nucleotide antisense (tcaaaggtctcattccaca) and its corresponding sense with phosphorothioate modifications on the last three bases at both 5′ and 3′ ends were used as described in our previous article [35]. The specificity to cPLA2α was analyzed by blast search program and was demonstrated in our previous study [31].
Immunoblot analysis
Microglial cell lysates were prepared using lysis buffer containing: 2% Triton X-100, 50 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 10 μm MgCl2, 10 μg/ml leupeptin, 1 mM phenylmethylsulphonylfluoride, 10 μg/ml aprotonin, 1 mM benzamidine, 20 mM para-nitrophenyl phosphate, 5 mM sodium orthovanadate, 10 mM sodium fluoride, and 50 mM β-glycerophosphate). Cell lysates were analyzed by SDS-PAGE on 9% gels. The amount of protein in each sample was quantified with the Pierce BCA Proteins Assay using BSA standards. The resolved proteins were transferred to nitrocellulose and blocked in 5% BSA in TBS-T (10 mM Tris, 135 mM NaCl, pH 7.4, 0.1% Tween 20). The blots were incubated overnight at 4 °C with primary antibodies (anti-cPLA2α and anti-phospho-(serine-505)-cPLA2α from Sigma, anti-NF-κB p65, anti-phospho-(serine-536)-NF-κB p65, anti phopho-p44/42 ERK1/2 (Thr202/Tyr204), anti-p44/42 ERK1/2, anti-STAT1α, anti-phospho-(serine-727)-STAT1α, anti-phospho-(Thr-701)-STAT1α from Cell Signaling, MA, USA; washed and incubated with peroxidase-conjugated secondary antibodies (Amersham Pharmacia Biotech, NJ, USA) for 1.5 h at room temperature. Detection of immunoreactive bands was carried out using enhanced chemiluminescence. Changes in protein expression or phosphorylation were quantified by densitometry using ImageJ program. The intensity of each band was divided by the intensity of each total protein band and expressed as arbitrary units. The quantitative measurements are adequate to determine the changes of each protein in the same immunoblot.
Separation of plasma membranes and immunoprecipitation
Plasma membranes were separated as described before ([41]). Cell 108/ml suspended in relaxation buffer (100 mM KCl, 3 mM NaCl, 3.5 mM MgCl2, 1.25 mM EGTA, 1 mM ATP, 10 mM PIPES, pH7.4) containing 1 mM PMSF and 100 μm leupeptin at 4 °C and sonicated , resulting in 95% cell breakage. After centrifugation (5 min; ×15,600g) to remove the granules, nuclei, and unbroken cells, the supernatant was centrifuge in a Beckman Airfuge (Beckman Instrument, Fulletron, CA) 30 min; ×134,000g to obtain cell membrane pellet and cytosol supernatant. The membranes were suspended at 109 cell equivalent/ml in 0.34 sucrose/half-strength relaxation buffer. The microglial cell membranes subjected to immunoprecipitation with goat anti-serum raised against recombinant p47phox (gift from Dr. T Leto, NIAID, NIH, Bethesda, USA). Immunoprecipitation was at a final volume of 0.5 ml at 4 °C. Recombinant protein A–Sepharose beads (Zymed Laboratories Inc., CA, USA) were added to each sample, and the samples were tumbled end-over-end for 1 h. The beads were then washed six times with lysis buffer boiled in lamely buffer and subjected to SDS-PAGE analysis.
TNF-a detection–using mouse TNF-α high sensitivity ELISA, eBioscience, Vienna, Austria.
Statistical analysis
Significant differences between the parameters evaluated were determined by ANOVA using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA) followed by multiple comparisons Bonferroni post hoc correction. p value less than 0.05 were considered statistically significant.
Results
cPLA2α upregulation regulates the overexpression of CD40 in microglia
Addition of 50 ng/ml LPS or 10 ng/ml IFNγ to BV-2 microglia cell line for 24 h caused a significant (p < 0.0001) elevation of and cPLA2α of CD40 protein expression, as shown in the double-immunofluorescence staining analysis (Fig. 1a, b). To determine whether cPLA2α upregulation is involved in the induction of CD40 by either LPS or IFNγ, cPLA2α upregulation was prevented by a specific antisense oligo-deoxy-nucleotide against cPLA2α (AS). As shown in Fig. 1a, b, impeding cPLA2α upregulation by addition of 4 μm AS 24 h prior to addition of LPS or of IFNγ prevented CD40 protein induction. Incubation with the corresponding sense that had no effect on the elevation of cPLA2α protein expression by either of the inducers did not affect the elevation of CD40 protein expression. To further support these results, the reduction of both cPLA2α and CD40 in the presence of AS was validated by western blot analysis (Fig. 1c, d). Similar results were obtained in primary mouse microglia cultures. As shown in the immunofluorescence staining (Fig. 1e, f) and western blot analysis (Fig. 1g, h), preventing cPLA2α upregulation prevented the elevated CD40 protein expression induced by LPS or IFNγ. BV-2 is an immortalized mouse microglia cell line that is reported to share many characteristics with primary microglia [42]. Since the BV-2 cells act similarly to primary microglia cultures, they were used to study the role of cPLA2α in the regulation of CD40 upregulation by either LPS or IFNγ.
Fig. 1.
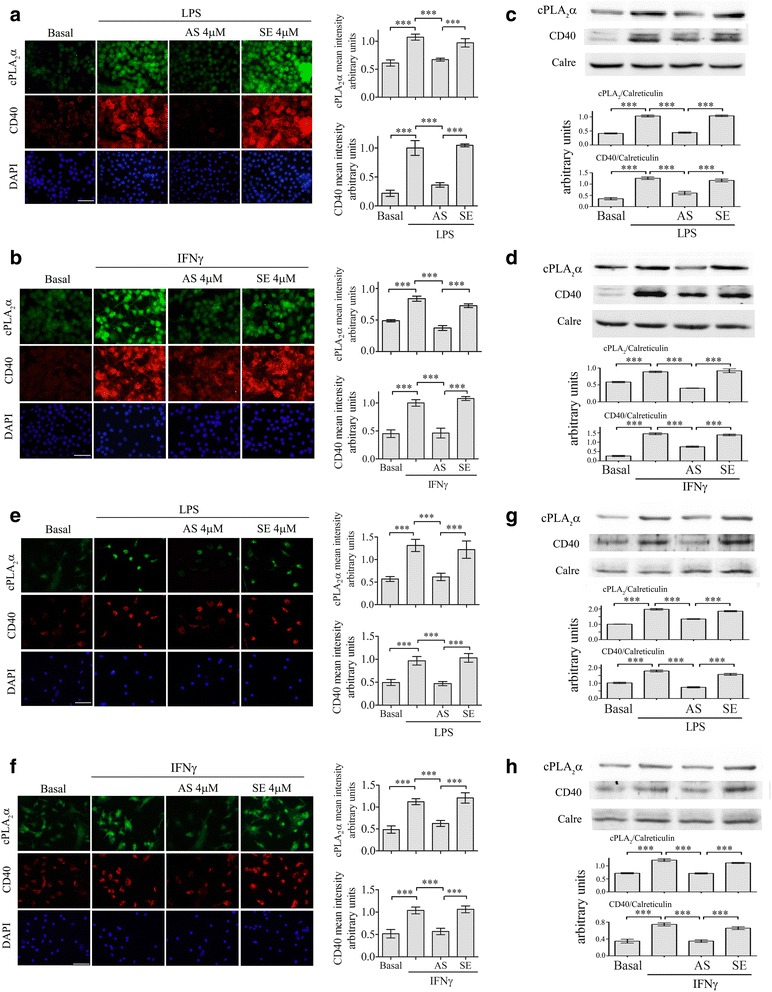
Elevated CD40 expression by LPS or IFNγ in the BV-2 and primary microglia cells is regulated by cPLA2α. A representative double-immunofluorescence staining of cPLA2α (green) and CD40 (red) in unstimulated or stimulated microglia by LPS or IFNγ in the absence or presence of AS or sense (SE). DAPI staining shows cell nuclei. The BV-2 cells were treated with (a) 50 ng/ml LPS or (b) 10 ng/ml IFNγ for 24 h. The mouse primary microglia cells were treated with (e) 50 ng/ml LPS or (f) 25 ng/ml IFNγ for 48 h. Scale bars = 50 μm. Four micrometer AS or the corresponding sense (SE) were added to the cultures 24 h before addition of the stimuli. The intensity of CD40 or cPLA2α were quantitated for the cell and expressed in the bar graph as arbitrary units. A representaive immunoblot analysis of cPLA2α and CD40 for the BV-2 microglia cells (c, d) and primary mouse microglia (g, h) treated as (a), (b), (e), and (f). The intensity of each cPLA2α or CD40 band after quantification by densitometry was divided by the intensity of each calreticulin (Calre) band and expressed as arbitrary units. The bar graphs are the mean ± SE from three independent experiments. (***p < 0.0001)
To study whether cPLA2α affect microglia induction towards M2 phenotype, the BV-2 cells were cultured with 20 ng/ml IL4 + 20 ng/ml IL10 for 24 h. IL4 + IL10 caused a significant (p< 0.0001) elevation of CD206 (Fig. 2a). Pre-incubated of the BV-2 cells with AS or the corresponding sense for 24 h prior to addition of IL4 + IL10 for 24 h did not affect the elevated expression of CD206. Similar results were obtained with respect to the induction of arginase 1, another marker of M2 microglia; the presence of AS or sense did not affect the elevated expression of arginase 1 induced by IL4 + IL10 detected by immunoblot analysis (Fig. 2b).
Fig. 2.
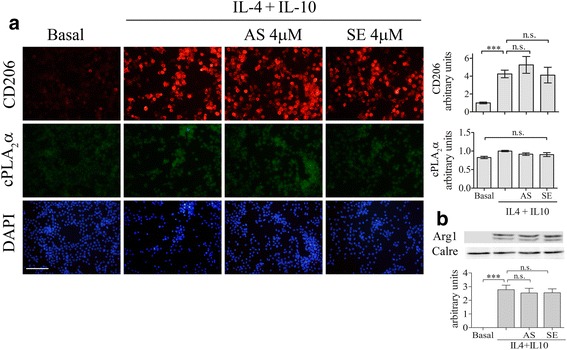
cPLA2α is not involved in the induction of the BV-2 cells towards M2 phenotype induced by IL-4 + IL-10. A representative double-immunofluorescence staining of cPLA2α (green) and CD206 (red) in the unstimulated or stimulated BV-2 cells with IL-4 (20 ng/ml) + IL-10 (20 ng/ml) for 24 h in the absence or presence of AS or SE. DAPI staining shows cell nuclei. Scale bars = 100 μm. a The intensity of CD206 or cPLA2α were quantitated and expressed in the bar graph as arbitrary units. The bar graphs are the mean ± SE from three independent experiments. b A representative immunoblot analysis of arginase 1 protein expression in the cells treated as in a. The intensity of each Arg1 band after quantification by densitometry was divided by the intensity of each calreticulin band and expressed as arbitrary units. The bar graphs are the mean ± SE from three independent experiments. (***p < 0.0001, n.s. not significant)
The location of cPLA2α in the signal transduction leading to CD40 upregulation by LPS
Next, we aimed to determine the location of cPLA2α in the suggested signal transduction pathways induced by either LPS or IFNγ [37–39]. We first focused on the signaling induced by LPS and analyzed the time dependent of cPLA2α activation determined by the appearance of its phosphorylated form on serine 505 in BV-2 cells lysates. As shown in Fig. 3a, a significant (p< 0.0001) transient activation was detected at 15 min after addition of 50 ng/ml LPS to the BV-2 cells. This phosphorylation was prevented in the presence of 2 μm of pyrrophenone added 60 min before activation (Fig. 3b). Since we have previously reported that cPLA2α activity regulates NOX2-NADPH oxidase activity in the phagocytic cells including primary rat microglia [40], we studied whether under LPS stimulation the oxidase is also regulated by cPLA2α. Addition of the oxidase inhibitor, 5 μm DPI, prior to LPS added for 15 min did not affect the activation of cPLA2α (Fig. 3b) while addition of 4 μm AS for 24 h or pyrrophenone for 60 min prior to addition of LPS for 15 min caused a significant (p < 0.0001) inhibition of superoxide production as measured by DHE reduction, similar to that caused by the presence of a specific inhibitor of the oxidase, 200 μm apocynin (Fig. 3c). Pre-incubation of the BV-2 cells with 4 μm of the corresponding sense had no effect on superoxide production. These results indicate that cPLA2α activity regulates NOX2-NADPH oxidase activity in BV-2 microglia stimulated by LPS. The immunoprecipitation of the cytosolic subunit of NOX2-NADPH oxidase with the phosphorylated form of cPLA2α in the membrane fraction of the activated microglia cells further support the role of cPLA2α in the regulation of the oxidase (3D). Our previous studies demonstrated that arachidonic acid restored the inhibited NOX2-NADPH oxidase activity in the absence of cPLA2α activity [43]. In line with our previous results, addition of arachidonic acid to the stimulated cells in the presence of antisense against cPLA2α restored the expression of CD40 (Fig. 3e). To further support that the NOX2-NADPH oxidase is located in the signal events leading to CD40 upregulation by LPS, the effect of its inhibition was studied on CD40 expression. The presence of a specific inhibitor of NOX2-NADPH oxidase activity (200 μm apocynin) prevented the elevation of CD40 protein expression induced by LPS, as determined by FACS analysis, (Fig. 3f) similar to the effect caused by the presence of the inhibitor of cPLA2α activity, pyrrophenone. The results indicate that the NOX2-NADPH oxidase activity participates in the signal transduction pathway leading to CD40 induction by LPS and is located downstream to cPLA2α. As shown in Fig. 3g, addition of LPS for 15 min caused activation of NFκB, detected by the phosphorylation of its p65 subunit on serine 536. This phosphorylation was similarly reduced by inhibition of cPLA2α or NOX2-NADPH oxidase activity by the presence of either pyrrophenone or DPI, respectively. The presence of a specific inhibitor of MEK1/2 activation, U0126, caused inhibition of ERK1/2, cPLA2α, and NFκB activation induced by LPS as detected by their phosphorylated forms (Fig. 3h). Taken together, these results suggest that early after the addition of LPS to the BV-2 cells, ERK activates cPLA2α that in turn activates the assembled NOX2-NADPH oxidase that mediates the activation of NF-κB.
Fig. 3.
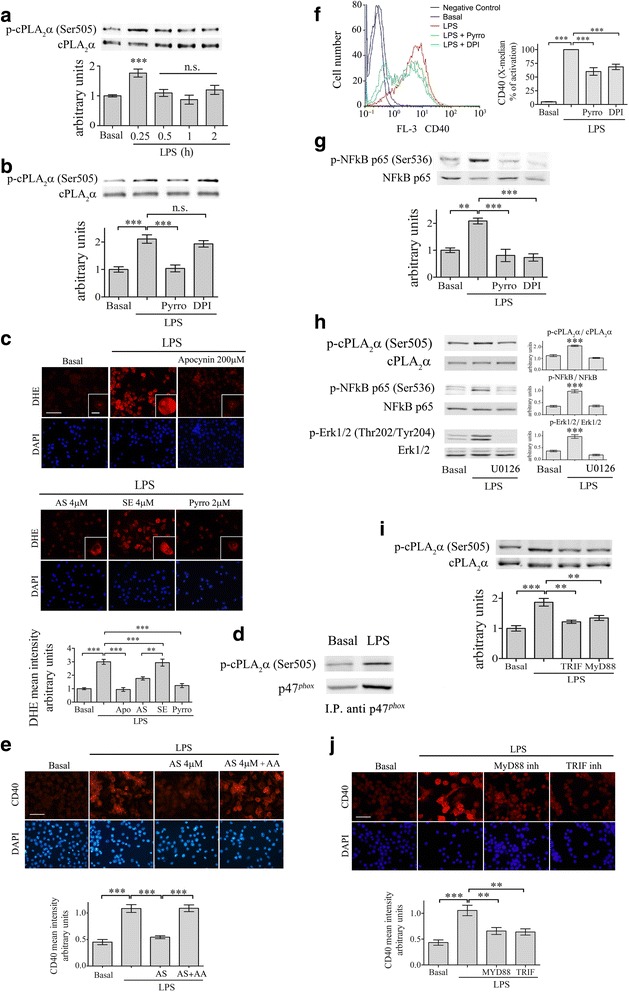
cPLA2α activates NFκB through activation of NOX2-NADPH oxidase in the BV-2 microglia cells under LPS stimulation. a A representative immunoblot analysis of the kinetics of cPLA2α phosphorylation induced by 50 ng/ml LPS, out of three independent experiments. The intensity of each phosphorylated cPLA2α (p-cPLA2α Ser-505) band after quantification by densitometry was divided by the intensity of each cPLA2α band and expressed as arbitrary units. The bar graphs are the mean ± SE from three independent experiments. b The BV-2 cells were treated with 2 μm pyrrophenone (Pyrro) or 5 μm DPI for 60 min before stimulation with 50 ng/ml LPS for 15 min. The intensity of phosphorylated cPLA2α was quantitated by densitometry as described in (a). The bar graphs are the mean ± SE from three independent experiments. c The effect of cPLA2α inhibition on superoxide production in the unstimulated or stimulated BV-2 cells with 50 ng/ml LPS for 15 min was detected by DHE reduction. Two micrometer pyropheonoe (Pyrro) or 200 μm apocynin (used as a positive control) were added to the cells 60 min before stimulation with LPS. AS or sense were added 24 h prior to addition of LPS. DAPI staining shows cell nuclei. Scale bars large = 50 μm, insert = 20 μm. The intensity of reduced DHE was quantitated and expressed in the bar graph as arbitrary units. The bar graphs are the mean ± SE from three independent experiments. d Immunoprecipitation of p47phox and phoshpo cPLA2α (pcPLA2α) in the membrane fraction of unstimulated microglia and stimulated with LPS for 15 min. Shown a representative immunoblot of three experiments. e Addition of 10 μM arachidonic acid together with LPS to cells pretreated for 24 h with antisense against cPLA2α restored the expression of CD40 protein. Shown a representative immunofluorescence staining of CD40. DAPI staining shows cell nuclei. The intensity of CD40 was quantitated for the cell and expressed in the bar graph as arbitrary units. Scale bars = 50 μm. The bar graph is the mean ± SE from three independent experiments. (***p < 0.0001). f FACS analysis of CD40 protein expression in the unstimulated or stimulated BV-2 cells with 50 ng/ml LPS for 24 h in the absence or presence of 2 μm pyrrophenone or 5 μm DPI (added to the cells 60 min before stimulation with LPS). The bar graphs are the X-median ± SE from five independent experiments. g A representative immunoblot analysis of NF-κB p-65 phosphorylation (p-NFκB p-65 Ser-536) in unstimulated or stimulated BV-2 microglia by 50 ng/ml LPS for 15 min in the absence or presence of 2 μm pyrrophenone or 5 μm DPI. The intensity of each phosphorylated NF-κB p-65 band after quantification by densitometry was divided by the intensity of each NFκB p-65 band and expressed as arbitrary units. The bar graphs are the mean ± SE from three independent experiments. h A representative immunoblot analysis of phoshpo cPLA2α and phospho NF-κB p-65 subunit and phosspho ERK1/2 in unstimulated or stimulated BV-2 microglia by 50 ng/ml LPS for 15 min in the absence or presence of 5 μM OU126. The bar graphs are the mean ± SE of the intensity of the quantitated phosphorylated forms divided by the non-phophorylated forms of three independent experiments. i The involvement of TRIF and MyD88 pathways in activation of cPLA2α in the signaling leading to CD40 upregualtion. The BV-2 cells were incubated with TRIF or MyD88 peptide inhibitors for 60 min before stimulation with 50 ng/ml LPS for 15 min. A representative immunoblot analysis of cPLA2α activity, out of three independent experiments is presented. The intensity of phosphorylated cPLA2α (p-cPLA2α Ser-505) was quantitated by densitometry as described in A. j Shown is a representative immunofluorescence analysis of CD40 protein expression in the cells treated as in i. DAPI staining shows cell nuclei. Scale bars = 50 μm. The intensity of cPLA2α and of CD40 was quantitated and expressed in the bar graph as arbitrary units. The bar graph is the mean ± SE from three independent experiments. (***p < 0.0001,**p < 0.001, n.s. not significant)
Next, we determined whether MyD88- or TRIF- pathways in the signal events induced by LPS are capable of activating cPLA2α. We used specific peptide inhibitors for either MyD88 signaling by inhibiting its homodimerization or TRIF signaling by interfering with TLR-TRIF interaction. As shown in the immunoblot, each peptide inhibitor inhibited the activation of cPLA2α detected by its phosphorylated form induced by LPS, to the levels detected in the unstimulated cells (Fig. 3i). Moreover, either of the inhibitors totally prevented the induction of CD40 induced by LPS, suggesting that both pathways participate in cPLA2α activation and in CD40 upregulation (Fig. 3j).
Since activation of STAT1α was reported to signal the induction of CD40 by LPS [39], we studied whether STAT1α activation is dependent on cPLA2α activity. Kinetics analysis of STAT1α activation detected by its phosphorylation revealed that significant phosphorylation of STAT1α on tyrosine 701 (Fig. 4a) and on serine 727 (Fig. 4b) was detected at 4 h of stimulation with LPS. Neither tyrosine 701 nor serine 727 phosphorylation induced by LPS for 4 h were affected by the presence of cPLA2α inhibitor, pyrrophenone, or the presence of the NOX2-NADPH oxidase inhibitor, DPI (Fig. 4c, d), suggesting that STAT1α is not under cPLA2α regulation in the signal cascade leading to CD40 induction by LPS in the BV-2 cells.
Fig. 4.
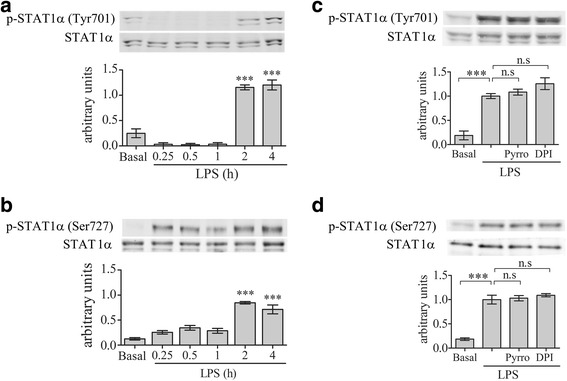
STAT1α activation is not under regulation of cPLA2α or NOX2-NADPH oxidase in the BV-2 microglia cells under LPS stimulation. A representative immunoblot analysis of the kinetics of STAT1α phosphorylation on (a) tyrosine 701 or (b) serine 727 induced with 50 ng/ml LPS. A representative immunoblot analysis of STAT1α phosphorylation on (c) tyrosine 701 or on (d) serine 727 in unstimulated or stimulated with 50 ng/ml LPS for 4 h in the absence or presence of 2 μm pyropheonoe (Pyrro) or 5 μm DPI added to the cells 1 h before stimulation. The intensity of each phosphorylated STAT1α (p-STAT1α) band after quantification by densitometry was divided by the intensity of each STAT1α band and expressed as arbitrary units. The Bar graphs are the mean ± SE from three experiments (***p < 0.0001, n.s. not significant)
The location of cPLA2α in the signal transduction leading to CD40 upregulation by IFNγ
To determine the location of cPLA2α in the signal transduction events leading to CD40 protein induction by IFNγ, we first studied the time-dependent activation of cPLA2α. As shown in Fig. 5a, a significant (p < 0.0001) cPLA2α activation detected by its phosphorylation on serine 505 appeared at 240 min of stimulation with 10 ng/ml IFNγ in BV-2 cell lysates. This phosphorylation was prevented in the presence of 2 μm pyrrophenone but not by the presence of 5 μm DPI (Fig. 5b), suggesting that the NOX2-NADPH oxidase is downstream to cPLA2α. Inhibition of cPLA2α activity by addition of either AS for 24 h or pyrrophenone for 60 min prior to stimulation by IFNγ for 4 h caused inhibition of NOX2-NADPH oxidase activity detected by DHE reduction that was similar to that achieved in the presence of 200 μm apocynin (Fig. 5c), while the presence of sense had no effect. The immunoprecipitation of NOX2-NADPH oxidase cytosolic subunit p47phox with phopho-cPLA2α in the membrane fraction of the activated mircroglia cells at 4 h further support the role of cPLA2α in regulating the oxidase activity (Fig. 5d). Addition of arachidonic acid to the IFNγ stimulated cells in the presence of antisense against cPLA2α restored the expression of CD40 (Fig. 5e). The presence of DPI or pyrrophenone significantly inhibited the induction of CD40 as shown by FACS analysis (Fig. 5f). These results suggest that NOX2-NADPH oxidase is downstream to cPLA2α in the signal transduction pathway leading to CD40 upregulation induced by IFNγ. Inhibition of cPLA2α or NOX2-NADPH oxidase by the presence of either pyrrophenone or DPI inhibited NF-κB activity detected by the phosphorylation of its p65 subunit on serine 536 at 4 h of stimulation with IFNγ (Fig. 5g), suggesting that NF-κB is downstream to cPLA2α and NOX2-NADPH oxidase. The presence of a specific inhibitor of MEK1/2 activation, U0126, caused inhibition of ERK1/2, cPLA2α, and NF-κB activation at 4 h of induction by IFNγ as detected by their phosphorylated forms (Fig. 5h).
Fig. 5.
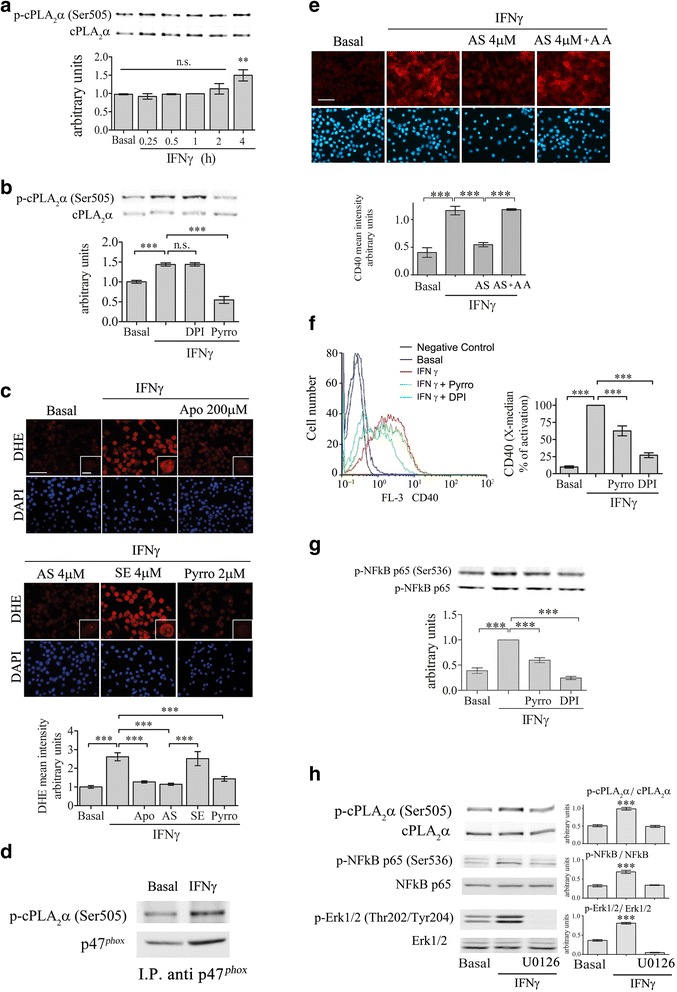
cPLA2α activates NF-κB through activation of NOX2-NADPH oxidase in the BV-2 microglia cells under IFNγ stimulation. a A representative immunoblot analysis of the kinetics of cPLA2α phosphorylation induced by 10 ng/ml IFNγ, out of three independent experiments. The intensity of phosphorylated cPLA2α (p-cPLA2α Ser-505) was quantitated by densitometry as described in Fig. 3a. b The BV-2 microglia cells were treated with 2 μm pyrrophenone (Pyrro) or 5 μm DPI for 1 h before stimulation with 10 ng/ml IFNγ for 4 h. Phosphorylated cPLA2α (p-cPLA2α Ser-505) intensity was quantitated by densitometry as described in Fig. 3a. The bar graphs are the mean ± SE from three independent experiments. c The effect of cPLA2α inhibition on superoxide production in the unstimulated or stimulated BV-2 cells with 10 ng/ml IFNγ for 4 h detected by DHE reduction. Two micrometer pyropheonoe (Pyrro) or 200 μm apocynin (used as a positive control) were added to the cells 60 min before stimulation with IFNγ for 4 h. AS or sense (SE) were added 24 h prior to addition of IFNγ. DAPI staining shows cell nuclei. The intensity of reduced DHE was quantitated and expressed in the bar graph as arbitrary units. Scale bars large = 50 μm, insert = 20 μm. The bar graphs are the mean ± SE from three independent experiments. d Immunoprecipitation of p47phox and phoshpo cPLA2α (pcPLA2α) in the membrane fraction of unstimulated microglia and stimulated with IFNγ for 4 h. Shown a representative immunoblot of three experiments. e Addition of 10 μM arachidonic acid togeher with IFNγ to the cells pre-treated for 24 h with antisense against cPLA2α restored the expression of CD40 protein. Shown a representative immunofluorescence staining of CD40. DAPI staining shows cell nuclei. The intensity of CD40 was quantitated for the cell and expressed in the bar graph as arbitrary units. Scale bars = 50 μm. f FACS analysis of CD40 protein expression in the unstimulated or stimulated BV-2 cells with10 ng/ml IFNγ for 24 h in the absence or presence of 2 μm pyrrophenone (Pyrro) or 5 μm DPI added to the cells 1 h before stimulation. The bar graphs are the X-median ± SE from five independent experiments. g A representative immunoblot analysis of phosphorylated NF-κB p65 (p-NFκB p-p65(Ser-536) in unstimulated or stimulated BV-2 microglia by 10 ng/ml IFNγ for 4 h in the absence or presence of 2 μm pyrrophenone (Pyrro) or 5 μm DPI. The intensity of each p-NFκB p-p65(Ser-536) band after quantification as described in Fig. 3g. The bar graphs are the mean ± SE from three independent experiments. h A representative immunoblot analysis of phoshpo cPLA2α and phospho NF-κB p-65 subunit and phosspho ERK1/2 in unstimulated or stimulated BV-2 microglia by 10 ng/ml IFNγ for 4 h in the absence or presence of 5 μM OU126. The bar graphs are the mean ± SE of the intensity of the quantitated phosphorylated forms divided by the non-phophorylated forms of three independent experiments. (***p < 0.0001)
It was reported that TNF-α is secreted from macrophages and microglia after 4 h of stimulation [37] and has an autocrine effect on the cells. To determine whether the activation cPLA2α is mediated by endogenous release of TNF-α, we first studied the time-dependent activation of cPLA2α by TNF-α. As shown in Fig. 6a, TNF-α caused a rapid and transient activation of cPLA2α that was similar to that induced by IFNγ for 4 h. We then measured the release of TNF-α form microglia stimulated by IFNγ (Table 1). TNF-α could be significantly detected in the supernatant of microglia cultures for 4 h with IFNγ. The dose-dependent activation of cPLA2α by TNF-α showed that cPLA2α was significantly (p < 0.01) activated by 0.5 ng/ml TNF-α, while 2 and 10 ng/ml were yet more significant (p < 0.0001) with a similar effect (Fig. 6b). A similar dose-dependent effect was detected for NF-kB activated by TNF-α (Fig. 6c). To determine the role of the released TNF-α in activation of cPLA2α and of NF-κB by IFNγ, its autocrine effect was prevented by pre-incubation of the cells with anti-TNF-α-neutralizing antibody before stimulation. The presence of anti-TNF-α prevented the activation of cPLA2α as detected by its phosphorylated forms on serine 505 (Fig. 6d) and prevented the activation of NF-κB p-65 as detected by it phosphorylation on serine 536 (Fig. 6e) at 4 h of IFNγ stimulation. As shown in Fig. 6f, the presence of anti-TNF-α prevented the induction of CD40 by IFNγ in the BV-2 microglia cells. Addition of TNF-α alone to the cells did not induce CD40 protein expression, as expected since both NF-kB and STAT1 transcription factors are required for the induction of CD40 by IFNγ, while TNF-α was shown to activate only NF-κB that is located downstream to STAT1α in the signal events [38].
Fig. 6.
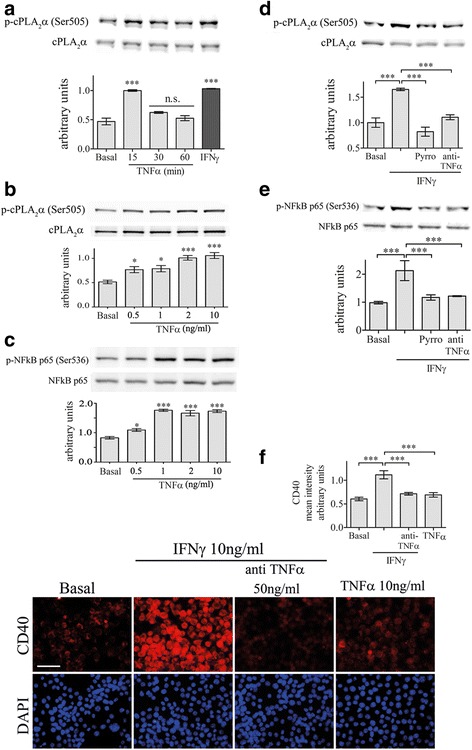
Endogenously produced TNF-α regulates cPLA2α activation in the BV2 cells under IFNγ stimulation. a A representative immunoblot analysis of the kinetics of cPLA2α activation detected by its phosphorylated form induced by 10 ng/ml TNF-α in BV-2 microglia lysates. A representative immunoblot analysis of a dose-dependent activation of the cPLA2α (b) and of NF-kB (c) detected by their phosphorylated induced by TNF-α in BV-2 microglia lysates. Representative immunoblot analysis of (d) cPLA2α phosphorylation on Ser-505 and (e) NF-kB p-65 phosphorylation on Ser-536 in unstimulated or stimulated BV-2 microglia by 10 ng/ml IFNγ for 4 h in the absence or presence of 2 μm pyrrophenone (Pyrro) or 50 ng/ml TNF-α-neutralizing antibody (anti-TNF-α) added to the cells 60 min before stimulation with IFNγ stimulation. The intensity of each p-cPLA2α(Ser-505) or p-NFκB p65(Ser-536) band after quantification by densitometry was divided by the intensity of each cPLA2α or NFκB p65 band, respectively, and expressed as arbitrary units. The results are the mean ± SE from four experiments. f Immunofluorescence analysis of CD40 protein expression in unstimulated or stimulated BV-2 microglia with 10 ng/ml IFNγ for 24 h in the absence or presence of 50 ng/ml TNF-α-neutralizing antibody or stimulated by 10 ng/ml TNF-α. Scale bars = 50 μm. The intensity of CD40 was quantitated and expressed in the bar graph as arbitrary units. The bar graph is the mean ± SE from four independent experiments. DAPI staining shows cell nuclei. (***p < 0.0001, n.s. not significant)
Table 1.
TNF-α secretion induced by INFγ
| Medium treated | IFNγ treated (4 h) |
|---|---|
| 0.06 ± 0.004 | 2.7 ± 0.05 |
| Cells were treated with 10 ng/ml IFNγ | |
The results are means ± SED from three independent experiments
There is a significant difference between the treated and non-treated cells (p < 0.0001)
Addition of IFNγ caused a rapid and significant (p < 0.0001) activation of STAT1α on either serine 727 or tyrosine 701, detected at 15 min of activation (Fig. 7a, b). Both phosphorylation were not affected by the presence of cPLA2α inhibitor, pyrrophenone, or the presence of NOX2-NADPH oxidase inhibitor, DPI (Fig. 7c, d), suggesting that STAT1α is not under cPLA2α regulation in the signal cascade leading to CD40 induction by IFNγ in the BV-2 cells.
Fig. 7.
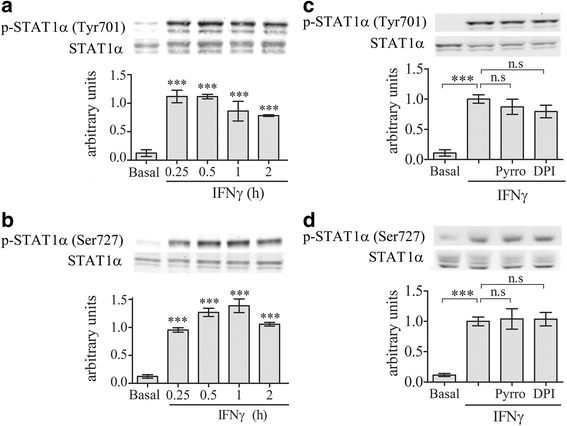
cPLA2α or NADPH oxidase did not affect STAT1α activation under IFNγ stimulation. A representative immunoblot analysis of the kinetics of STAT1α phosphorylation on (a) tyrosine 701 or (b) serine 727 induced by 10 ng/ml IFNγ. A representative immunoblot analysis of STAT1α phosphorylation on (c) tyrosine 701 or on (d) serine 727 in unstimulated or stimulated BV-2 microglia by 10 ng/ml IFNγ for 15 min in the absence or presence of 2 μm pyrrophenone (Pyrro) or 5 μm DPI (added to the cells 60 min before stimulation). The intensity of each phosphorylated STAT1α (p-STAT1α) band was quantitated as described in Fig. 4. The Bar graphs are the mean ± SE from three experiments (***p < 0.0001, n.s. not significant)
Discussion
The present study shows that cPLA2α is involved in the induction of CD40 by either LPS or IFNγ. Reduction of cPLA2α upregulation by a specific antisense or inhibition of cPLA2α activity by a specific inhibitor prevented the induction of CD40 protein expression by either LPS or IFNγ. The results suggest that cPLA2α has a direct role in CD40 upregulation, a feature of the pro-inflammatory M1-phenotype. In accordance with this view, the regulatory role of cPLA2α in the induction of several characters of M1 phenotype in microglia and macrophages, such as iNOS, COX2, NOX2-NADPH oxidase as well as production of eicosanoids and pro-inflammatory mediators, was reported by us and others [40, 44]. cPLA2α, however, is not involved in the transformation to M2-phenotype, as its protein level was not elevated by addition of IL4 + IL10, and the presence of AS did not affect the significant induction of CD206 or of arginase 1 in microglia. In accordance with our results, it was reported that the antiinflammatory cytokines IL4 or IL10 by themselves did not affect cPLA2α activation or biosynthesis [45, 46], further supporting the role of cPLA2α in inflammatory processes.
The results of the present study show that superoxides generated by NOX2-NADPH oxidase participate in upregulation of CD40 expression induced by LPS or IFNγ in microglia, since inhibition of NOX-2 NADPH oxidase prevented the induction of CD40. We show here that in BV-2 microglia cell line, inhibition of the activation of cPLA2α induced by either LPS or IFNγ, as demonstrated by the use of antisense against cPLA2α or the specific inhibitor of cPLA2α activity, pyrrophenone, inhibited the production of superoxides by the NOX2-NADPH oxidase. Inhibition of the oxidase activity did not affect cPLA2α activation detected by its phosphorylated form. These results suggest that the NOX2-NADPH oxidase is regulated by cPLA2α in microglia stimulated with either LPS or IFNγ, that is similar to ours and other studies related to the various phagocytic cells stimulated with a variety of agonists [31, 40, 44, 47–50]. We show here that phoshpo-cPLA2α translocated to the cell membranes of activated microglia, where it binds p47phox subunit of NOX2-NADPH oxidase, in accordance with our previous studies in other phagocytic cells as well as in primary rat microglia [40, 43, 48, 50]. The binding between p-cPLA2α and 47phox was detected at 15 min when the microglia cells were stimulated with LPS and at 4 h when stimulated with IFNγ in correlation with the detection of superoxide production and the kinetic of cPLA2α phosphorylation by the two stimuli. Our previous study [43] demonstrated that arachidonic acid activated the assembled oxidase in activated cPLA2α-deficient cells, although the precise mechanism is not known. The restoration of CD40 upregulation in the activated cells that were pretreated with AS against cPLA2α by addition of arachidonic acid is probably due to the activation of the NOX2-NADPH oxidase. The activation of cPLA2α at 15 min by LPS and at 4 h by IFNγ was mediated by ERK activation since the presence of MEK inhibitor inhibited cPLA2α activation in accordance with ours and other earlier studies [44, 51].
The involvement of two transcription factors, NF-κB and STAT1α, was reported in the signal transduction pathways leading to induction of CD40 by either LPS or IFNγ [37–39]. While NF-κB was shown to be rapidly activated by LPS, it was activated only at 4 h following exposure to IFNγ. In contrast, STAT1α was rapidly activated by IFNγ and only at 4 h by LPS. Time-dependent activation of cPLA2α detected by its phosphorylation on serine 505 revealed that cPLA2α is rapidly activated by LPS and only considerably later (4 h) by IFNγ, that is in accordance with a previous report [44]. We show in the present study that the kinetic of activation of cPLA2α coincided with the kinetic of NF-kB activation and that the activation of cPLA2α is required for the activation of NF-κB in BV-2 microglia cell line, a finding consonant with our earlier study in microglia activated with amyloid beta [40]. While superoxide production by NOX2-NADPH oxidase is extremely important for killing invading pathogens, it is also an important activator of diverse cell signaling pathways such as mitogen activated protein kinase and NF-κB to regulate the expression of genes encoding a variety of pro-inflammatory factors [40, 52, 53]. The activation of NF-κB by either LPS or IFNγ shown in the present study detected by the phosphorylation of its p-65 subunit on serine 536 is probably mediated by superoxides produced by the NOX2-NADPH oxidase since the inhibition of the oxidase activity prevented NF-kB action. In line with this suggestion, the phosphorylation of p65 NF-kB RelA on Ser-536 is known to be redox-sensitive [54]. The activation of NF-kB by NOX2-NADPH oxidase activity is consistent with our previous studies in microglia and macrophages [40, 55] and with other in various systems and by various agonist [56, 57].
It was reported that the activation of NF-kB under IFNγ stimulation is mediated by an autocrine effect of released TNF-α from the stimulated cells [37]. Consistent with this observation, we show here that the activation of cPLA2α and of NF-kB and the induction of CD40 by IFNγ are mediated by an autocrine effect of TNF-α, since TNF-α secretion from the activated cells was detected and the levels of secreted TNF-α activated cPLA2α and NF-kB. In addition, the presence of antibodies against TNF-α in microglia stimulated with IFNγ of all three processes were inhibited, suggesting that cPLA2α activation by TNF-α regulates the induction of CD40 via NF-kB activation. The activation of cPLA2α by TNF-α coincided with other reports in microglia and macrophages [46, 58]. However, addition TNF-α is not sufficient to induce CD40, although it activates cPLA2α, probably since it stimulates the activation of NF-κB but not the activation of STAT1α that is also required for CD40 induction.
The activation of cPLA2α and NF-κB in the signals leading to CD40 upregulation by LPS is mediated by both MyD88 and TRIF pathways, since inhibition of each pathway inhibited cPLA2α and NF-κB activation and abolished CD40 induction. In accordance with our results, the activation of cPLA2α by MyD88 and by TRIFF adaptive protein was shown in macrophages stimulated by LPS [59]. The activation of NF-κB leading to CD40 upregulation by LPS was suggested to be mediated only by MyD88 adaptive protein in macrophages [39]. However, several studies reported, similar to our results, that both pathways are mediating NF-κB by TLR4 receptor in macrophages and other cell types [59–61].
Conclusions
Our results show for the first time that cPLA2α regulates CD40 protein induction in microglia by either LPS or IFNγ, and this regulation is mediated via activation of NOX2-NADPH oxidase and NF-κB. STAT1α transcription factor, that was reported to participate in CD40 induction, was early activated by IFNγ and late activated by LPS as detected by the phosphorylation on either serine 727 or tyrosine 701, but this activation was not under cPLA2α regulation. As shown in Fig. 8, cPLA2α is located in the early event induced by LPS and its activation is mediated by both adaptor proteins, TRIF and MyD88. While, under IFNγ stimulation, cPLA2α is activated at a later time (4 h) by the autocrine effect of released TNF-α. Under both stimuli, cPLA2α activation is mediated by ERK activity. The role of cPLA2α in the induction of the CD40 suggests that cPLA2α may serve as an amplifier of the inflammatory response in the CNS and the reduction of its levels in the inflamed organ can lead to therapeutic effect.
Fig. 8.
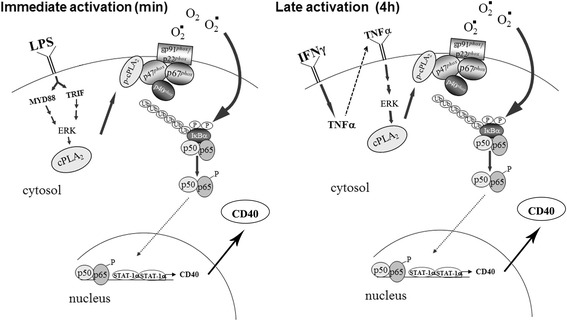
The involvement of cPLA2α in the proposed schematic NF-κB signaling pathways of CD40 protein expression induced by LPS or IFNγ (described in the “Discussion”section)
Acknowledgements
We thank Dr. Sergio Lamprecht for assistance in editing the English text. This research was supported by a grant from the Israel Sciences Foundation founded by the Israel Academy of Sciences and Humanities 1012/09.
Funding
This research was supported by a grant from the Israel Sciences Foundation founded by the Israel Academy of Sciences and Humanities 1012/09 for Prof. R. Levy. She designed the study, directed the study, and wrote the manuscript.
Availability of data and materials
Information about experimental methods and data described in this paper are available to scientific and medical communities for review, verification, and research studies.
Authors’ contributions
YPME designed and carried out all the experiments, researched the data, prepared the figures, and participated in writing the manuscript. NH participated in the design of the study and guided some methodologies. RL designed the study, directed the study, and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicative
Ethics approval
The study was approved by the Ben-Gurion University Institutional Animal Care and Use Committee (IL-37-05-2012).
Abbreviations
- AS
Antisense oligonucleotide against cPLA2α
- COX-2
Cyclooxygenase-2
- cPLA2α
Cytosolic phospholipase A2 alpha
- DHE
Dihyroethidium
- IFNγ
Interferon gamma
- iNOS
Inducible nitric oxide synthase
- LPS
Lipopolysaccharide
- LPS
Lipopolysaccharide
- NF-kB
Nuclear factor-kappaB
Contributor Information
Yafa Fetfet Malada-Edelstein, Email: ymalda@post.bgu.ac.il.
Nurit Hadad, Email: nurith@bgu.ac.il.
Rachel Levy, Phone: 972-8-6403186, Email: ral@bgu.ac.il.
References
- 1.Chen K, Huang J, Gong W, Zhang L, Yu P, Wang JM. CD40/CD40L dyad in the inflammatory and immune responses in the central nervous system. Cell Mol Immunol. 2006;3:163–169. [PubMed] [Google Scholar]
- 2.Banchereau J, Dubois B, Fayette J, Burdin N, Briere F, Miossec P, Rissoan MC, van Kooten C, Caux C. Functional CD40 antigen on B cells, dendritic cells and fibroblasts. Adv Exp Med Biol. 1995;378:79–83. doi: 10.1007/978-1-4615-1971-3_16. [DOI] [PubMed] [Google Scholar]
- 3.van Kooten C, Banchereau J. Functions of CD40 on B cells, dendritic cells and other cells. Curr Opin Immunol. 1997;9:330–337. doi: 10.1016/S0952-7915(97)80078-7. [DOI] [PubMed] [Google Scholar]
- 4.Alderson MR, Armitage RJ, Tough TW, Strockbine L, Fanslow WC, Spriggs MK. CD40 expression by human monocytes: regulation by cytokines and activation of monocytes by the ligand for CD40. J Exp Med. 1993;178:669–674. doi: 10.1084/jem.178.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I, Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J Exp Med. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollenbaugh D, Mischel-Petty N, Edwards CP, Simon JC, Denfeld RW, Kiener PA, Aruffo A. Expression of functional CD40 by vascular endothelial cells. J Exp Med. 1995;182:33–40. doi: 10.1084/jem.182.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galy AH, Spits H. CD40 is functionally expressed on human thymic epithelial cells. J Immunol. 1992;149:775–782. [PubMed] [Google Scholar]
- 8.Ponomarev ED, Shriver LP, Dittel BN. CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J Immunol. 2006;176:1402–1410. doi: 10.4049/jimmunol.176.3.1402. [DOI] [PubMed] [Google Scholar]
- 9.Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foy TM, Aruffo A, Bajorath J, Buhlmann JE, Noelle RJ. Immune regulation by CD40 and its ligand GP39. Annu Rev Immunol. 1996;14:591–617. doi: 10.1146/annurev.immunol.14.1.591. [DOI] [PubMed] [Google Scholar]
- 11.Fuleihan R, Ramesh N, Geha RS. Role of CD40-CD40-ligand interaction in Ig-isotype switching. Curr Opin Immunol. 1993;5:963–967. doi: 10.1016/0952-7915(93)90113-7. [DOI] [PubMed] [Google Scholar]
- 12.Zhang B, Wu T, Chen M, Zhou Y, Yi D, Guo R. The CD40/CD40L system: a new therapeutic target for disease. Immunol Lett. 2013;153:58–61. doi: 10.1016/j.imlet.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 13.D'Aversa TG, Weidenheim KM, Berman JW. CD40-CD40L interactions induce chemokine expression by human microglia: implications for human immunodeficiency virus encephalitis and multiple sclerosis. Am J Pathol. 2002;160:559–567. doi: 10.1016/S0002-9440(10)64875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Aversa TG, Eugenin EA, Berman JW. CD40-CD40 ligand interactions in human microglia induce CXCL8 (interleukin-8) secretion by a mechanism dependent on activation of ERK1/2 and nuclear translocation of nuclear factor-kappaB (NFkappaB) and activator protein-1 (AP-1) J Neurosci Res. 2008;86:630–639. doi: 10.1002/jnr.21525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aloisi F, Ria F, Penna G, Adorini L. Microglia are more efficient than astrocytes in antigen processing and in Th1 but not Th2 cell activation. J Immunol. 1998;160:4671–4680. [PubMed] [Google Scholar]
- 16.Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med. 2001;193:967–974. doi: 10.1084/jem.193.8.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salemi J, Obregon DF, Cobb A, Reed S, Sadic E, Jin J, Fernandez F, Tan J, Giunta B. Flipping the switches: CD40 and CD45 modulation of microglial activation states in HIV associated dementia (HAD) Mol Neurodegener. 2011;6:3. doi: 10.1186/1750-1326-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJ, Claassen E. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A. 1996;93:2499–2504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchhave P, Janciauskiene S, Zetterberg H, Blennow K, Minthon L, Hansson O. Elevated plasma levels of soluble CD40 in incipient Alzheimer’s disease. Neurosci Lett. 2009;450:56–59. doi: 10.1016/j.neulet.2008.10.091. [DOI] [PubMed] [Google Scholar]
- 20.Garlichs CD, Kozina S, Fateh-Moghadam S, Handschu R, Tomandl B, Stumpf C, Eskafi S, Raaz D, Schmeisser A, Yilmaz A, et al. Upregulation of CD40-CD40 ligand (CD154) in patients with acute cerebral ischemia. Stroke. 2003;34:1412–1418. doi: 10.1161/01.STR.0000074032.64049.47. [DOI] [PubMed] [Google Scholar]
- 21.MacDonald KP, Nishioka Y, Lipsky PE, Thomas R. Functional CD40 ligand is expressed by T cells in rheumatoid arthritis. J Clin Invest. 1997;100:2404–2414. doi: 10.1172/JCI119781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laman JD, de Smet BJ, Schoneveld A, van Meurs M. CD40-CD40L interactions in atherosclerosis. Immunol Today. 1997;18:272–277. doi: 10.1016/S0167-5699(97)80022-9. [DOI] [PubMed] [Google Scholar]
- 23.Lincecum JM, Vieira FG, Wang MZ, Thompson K, De Zutter GS, Kidd J, Moreno A, Sanchez R, Carrion IJ, Levine BA, et al. From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat Genet. 2010;42:392–399. doi: 10.1038/ng.557. [DOI] [PubMed] [Google Scholar]
- 24.Howard LM, Miga AJ, Vanderlugt CL, Dal Canto MC, Laman JD, Noelle RJ, Miller SD. Mechanisms of immunotherapeutic intervention by anti-CD40L (CD154) antibody in an animal model of multiple sclerosis. J Clin Invest. 1999;103:281–290. doi: 10.1172/JCI5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laman JD, t Hart BA, Brok H, Meurs M, Schellekens MM, Kasran A, Boon L, Bauer J, Boer M, Ceuppens J. Protection of marmoset monkeys against EAE by treatment with a murine antibody blocking CD40 (mu5D12) Eur J Immunol. 2002;32:2218–2228. doi: 10.1002/1521-4141(200208)32:8<2218::AID-IMMU2218>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 26.Samoilova EB, Horton JL, Zhang H, Chen Y. CD40L blockade prevents autoimmune encephalomyelitis and hampers TH1 but not TH2 pathway of T cell differentiation. J Mol Med (Berl) 1997;75:603–608. doi: 10.1007/s001090050145. [DOI] [PubMed] [Google Scholar]
- 27.Giunta B, Rezai-Zadeh K, Tan J. Impact of the CD40-CD40L dyad in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9:149–155. doi: 10.2174/187152710791012099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ. Role of CD40 ligand in amyloidosis in transgenic Alzheimer’s mice. Nat Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- 29.Kramer RM, Roberts EF, Manetta J, Putnam JE. The Ca2(+)-sensitive cytosolic phospholipase A2 is a 100-kDa protein in human monoblast U937 cells. J Biol Chem. 1991;266:5268–5272. [PubMed] [Google Scholar]
- 30.Clark JD, Milona N, Knopf JL. Purification of a 110-kilodalton cytosolic phospholipase A2 from the human monocytic cell line U937. Proc Natl Acad Sci U S A. 1990;87:7708–7712. doi: 10.1073/pnas.87.19.7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raichel L, Berger S, Hadad N, Kachko L, Karter M, Szaingurten-Solodkin I, Williams RO, Feldmann M, Levy R. Reduction of cPLA2alpha overexpression: an efficient anti-inflammatory therapy for collagen-induced arthritis. Eur J Immunol. 2008;38:2905–2915. doi: 10.1002/eji.200838545. [DOI] [PubMed] [Google Scholar]
- 32.Stephenson DT, Lemere CA, Selkoe DJ, Clemens JA. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in Alzheimer’s disease brain. Neurobiol Dis. 1996;3:51–63. doi: 10.1006/nbdi.1996.0005. [DOI] [PubMed] [Google Scholar]
- 33.Clemens JA, et al. Reactive glia express cytosolic phospholipase A2 after transient global forebrain ischemia in the rat. Stroke. 1996;27:527–535. doi: 10.1161/01.STR.27.3.527. [DOI] [PubMed] [Google Scholar]
- 34.Stephenson D, Rash K, Smalstig B, Roberts E, Johnstone E, Sharp J, Panetta J, Little S, Kramer R, Clemens J. Cytosolic phospholipase A2 is induced in reactive glia following different forms of neurodegeneration. Glia. 1999;27:110–128. doi: 10.1002/(SICI)1098-1136(199908)27:2<110::AID-GLIA2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 35.Sagy-Bross C, Kasianov K, Solomonov Y, Braiman A, Friedman A, Hadad N, Levy R. The role of cytosolic phospholipase A alpha in amyloid precursor protein induction by amyloid beta : implication for neurodegeneration. J Neurochem. 2014;132:559–571. doi: 10.1111/jnc.13012. [DOI] [PubMed] [Google Scholar]
- 36.Solomonov Y, Hadad N, Levy R. Reduction of cytosolic phospholipase A2alpha upregulation delays the onset of symptoms in SOD1G93A mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2016;13:134. doi: 10.1186/s12974-016-0602-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen VT, Benveniste EN. Critical role of tumor necrosis factor-alpha and NF-kappa B in interferon-gamma -induced CD40 expression in microglia/macrophages. J Biol Chem. 2002;277:13796–13803. doi: 10.1074/jbc.M111906200. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen VT, Benveniste EN. Involvement of STAT-1 and ets family members in interferon-gamma induction of CD40 transcription in microglia/macrophages. J Biol Chem. 2000;275:23674–23684. doi: 10.1074/jbc.M002482200. [DOI] [PubMed] [Google Scholar]
- 39.Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN. LPS induces CD40 gene expression through the activation of NF-kappaB and STAT-1alpha in macrophages and microglia. Blood. 2005;106:3114–3122. doi: 10.1182/blood-2005-02-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szaingurten-Solodkin I, Hadad N, Levy R. Regulatory role of cytosolic phospholipase A2alpha in NADPH oxidase activity and in inducible nitric oxide synthase induction by aggregated Abeta1-42 in microglia. Glia. 2009;57:1727–1740. doi: 10.1002/glia.20886. [DOI] [PubMed] [Google Scholar]
- 41.Levy R, Rotrosen D, Nagauker O, Leto TL, Malech HL. Induction of the respiratory burst in HL-60 cells. Correlation of function and protein expression. J Immunol. 1990;145:2595–2601. [PubMed] [Google Scholar]
- 42.Bocchini V, Mazzolla R, Barluzzi R, Blasi E, Sick P, Kettenmann H. An immortalized cell line expresses properties of activated microglial cells. J Neurosci Res. 1992;31:616–621. doi: 10.1002/jnr.490310405. [DOI] [PubMed] [Google Scholar]
- 43.Dana R, Leto TL, Malech HL, Levy R. Essential requirement of cytosolic phospholipase A2 for activation of the phagocyte NADPH oxidase. J Biol Chem. 1998;273:441–445. doi: 10.1074/jbc.273.1.441. [DOI] [PubMed] [Google Scholar]
- 44.Chuang DY, Simonyi A, Kotzbauer PT, Gu Z, Sun GY. Cytosolic phospholipase A2 plays a crucial role in ROS/NO signaling during microglial activation through the lipoxygenase pathway. J Neuroinflammation. 2015;12:199. doi: 10.1186/s12974-015-0419-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alaaeddine N, Di Battista JA, Pelletier JP, Kiansa K, Cloutier JM, Martel-Pelletier J. Inhibition of tumor necrosis factor alpha-induced prostaglandin E2 production by the antiinflammatory cytokines interleukin-4, interleukin-10, and interleukin-13 in osteoarthritic synovial fibroblasts: distinct targeting in the signaling pathways. Arthritis Rheum. 1999;42:710–718. doi: 10.1002/1529-0131(199904)42:4<710::AID-ANR14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 46.Kuroda A, Sugiyama E, Taki H, Mino T, Kobayashi M. Interleukin-4 inhibits the gene expression and biosyntheis of cytosolic phospholipase A2 in lipopolysaccharide stimulated U937 macrophage cell line and freshly prepared adherent rheumatoid synovial cells. Biochem Biophys Res Commun. 1997;230:40–43. doi: 10.1006/bbrc.1996.5885. [DOI] [PubMed] [Google Scholar]
- 47.Dana R, Malech HL, Levy R. The requirement for phospholipase A2 for activation of the assembled NADPH oxidase in human neutrophils. Biochem J. 1994;297:217–223. doi: 10.1042/bj2970217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shmelzer Z, Karter M, Eisenstein M, Leto TL, Hadad N, Ben-Menahem D, Gitler D, Banani S, Wolach B, Rotem M, Levy R. Cytosolic phospholipase A2alpha is targeted to the p47phox-PX domain of the assembled NADPH oxidase via a novel binding site in its C2 domain. J Biol Chem. 2008;283:31898–31908. doi: 10.1074/jbc.M804674200. [DOI] [PubMed] [Google Scholar]
- 49.Zhao X, Bey EA, Wientjes FB, Cathcart MK. Cytosolic phospholipase A2 (cPLA2) regulation of human monocyte NADPH oxidase activity. cPLA2 affects translocation but not phospho-rylation of p67phox AND p47phox. J Biol Chem. 2002;277:25385–25392. doi: 10.1074/jbc.M203630200. [DOI] [PubMed] [Google Scholar]
- 50.Shmelzer Z, Haddad N, Admon E, Pessach I, Leto TL, Eitan-Hazan Z, Hershfinkel M, Levy R. Unique targeting of cytosolic phospholipase A2 to plasma membranes mediated by the NADPH oxidase in phagocytes. J Cell Biol. 2003;162:683–692. doi: 10.1083/jcb.200211056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hazan I, Dana R, Granot Y, Levy R. Cytosolic phospholipase A2 and its mode of activation in human neutrophils by opsonized zymosan. Correlation between 42/44 kDa mitogen-activated protein kinase, cytosolic phospholipase A2 and NADPH oxidase. Biochem J. 1997;326:867–876. doi: 10.1042/bj3260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lal MA, Brismar H, Eklof AC, Aperia A. Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney Int. 2002;61:2006–2014. doi: 10.1046/j.1523-1755.2002.00367.x. [DOI] [PubMed] [Google Scholar]
- 53.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res. 1999;85:753–766. doi: 10.1161/01.RES.85.8.753. [DOI] [PubMed] [Google Scholar]
- 54.Pantano C, Reynaert NL, van der Vliet A, Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid Redox Signal. 2006;8:1791–1806. doi: 10.1089/ars.2006.8.1791. [DOI] [PubMed] [Google Scholar]
- 55.Hadad N, Levy R. The synergistic anti-inflammatory effects of lycopene, lutein, beta-carotene, and carnosic acid combinations via redox-based inhibition of NF-kappaB signaling. Free Radic Biol Med. 2012;53:1381–1391. doi: 10.1016/j.freeradbiomed.2012.07.078. [DOI] [PubMed] [Google Scholar]
- 56.Hoeck WG, Ramesha CS, Chang DJ, Fan N, Heller RA. Cytoplasmic phospholipase A2 activity and gene expression are stimulated by tumor necrosis factor: dexamethasone blocks the induced synthesis. Proc Natl Acad Sci U S A. 1993;90:4475–4479. doi: 10.1073/pnas.90.10.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heller RA, Kronke M. Tumor necrosis factor receptor-mediated signaling pathways. J Cell Biol. 1994;126:5–9. doi: 10.1083/jcb.126.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang S, Leonard SS, Castranova V, Vallyathan V, Shi X. The role of superoxide radical in TNF-alpha induced NF-kappaB activation. Ann Clin Lab Sci. 1999;29:192–199. [PubMed] [Google Scholar]
- 59.Qi HY, Shelhamer JH. Toll-like receptor 4 signaling regulates cytosolic phospholipase A2 activation and lipid generation in lipopolysaccharide-stimulated macrophages. J Biol Chem. 2005;280:38969–38975. doi: 10.1074/jbc.M509352200. [DOI] [PubMed] [Google Scholar]
- 60.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 61.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Information about experimental methods and data described in this paper are available to scientific and medical communities for review, verification, and research studies.