

Abstract
Background
Sturge-Weber syndrome (SWS) is a rare congenital neurocutaneous disorder characterized by facial and extracraniofacial capillary malformations and capillary-venule malformations in the leptomeninges. A somatic mosaic mutation in GNAQ (c.548G>A; p.R183Q) was found in SWS brain and skin capillary malformations. Our laboratory showed endothelial cells (ECs) in skin capillary malformations are enriched for the GNAQ mutation. The purpose of this study is to determine whether the GNAQ mutation is also enriched in ECs in affected SWS brain.
Methods
Two human SWS brain specimens were fractionated by fluorescence-activated cell sorting into hematopoietic (CD45), endothelial (CD31, VE-Cadherin and VEGFR2), perivascular (PDGFRβ) cells and cells negative for all markers. The sorted cell populations were analyzed for GNAQ p.R183Q mutation by droplet digital PCR. SWS patient-derived brain ECs were selected by anti-CD31-coated magnetic beads and cultured in endothelial growth medium in vitro.
Results
The GNAQ p.R183Q mutation was present in brain ECs in two SWS specimens, with mutant allelic frequencies of 34.7% and 24.0%. Cells negative for all markers also harbored the GNAQ mutation. The mutant allelic frequencies in these unidentified cells were 9.2% and 8.4%. SWS patient-derived brain ECs with mutant allelic frequencies of 14.7% and 21% survived and proliferated in vitro.
Conclusions
Our study provides evidence that GNAQ p.R183Q mutation is enriched in ECs in SWS brain lesions and thereby reveals ECs as a source of aberrant Gαq signaling. This will help to understand the pathophysiology of SWS, to discover biomarkers for predicting cerebral involvement and to develop therapeutic targets to prevent neurologic impairments in SWS.
Keywords: Sturge-Weber syndrome, capillary malformation, GNAQ, endothelial cell, ddPCR
Introduction
Sturge-Weber syndrome (SWS) is a sporadic, congenital neurocutaneous disorder that affects 1/20,000 to 1/50,000 newborns1, 2. It is characterized by cutaneous capillary malformation with ocular and/or brain abnormalities. The clinical features are largely variable and they can be progressive. Glaucoma is the most common ophthalmology complication; and typically the neurological problems include often medically resistant epilepsy, hemiparesis and neurocognitive impairment.
Facial capillary malformations (also known as port-wine stain) with specific distribution patterns are associated with an increased risk of SWS3, 4. If the birthmark involves the forehead and upper eyelids, the risk of having SWS is approximately 26%5. The strong association of facial capillary malformation and SWS suggests that they are caused by the same somatic mutation and that the timing of when the mutation occurs during the development likely impacts the clinical sequela and disease severity.
Recent genetic studies discovered a somatic mosaic mutation in GNAQ (c.548G>A; p.R183Q) in SWS brain and skin lesions and non-syndromic capillary malformation lesions6–8. GNAQ encodes Gαq, an alpha subunit of heterotrimeric G proteins. G proteins are a family of membrane bound guanosine triphosphatases (GTPase) that transmit signaling from transmembrane G-protein coupled receptors. The p.R183Q mutation impairs GTPase activity and maintains Gαq in its GTP–bound, active state9. Our laboratory recently showed that the GNAQ p.R183Q mutation is enriched in the endothelial cell population in SWS skin lesions and sporadic capillary malformations10. Another group reported GNAQ p.R183Q mutant cells located around blood vessels in port-wine stains11. In this study we set out to determine whether the GNAQ p.R183Q mutation is also enriched in endothelial cell population in SWS brain lesions.
Materials and Methods
SWS brain specimens and cell isolation
Two individuals with SWS who underwent hemispherectomy as part of their clinical care were enrolled in the study. The Committee on Clinical Investigation at Boston Children’s Hospital approved this study. SWS brain lesions were collected during the neurosurgical procedure. The brain tissue of patient 1 (male, 11-month old) was from left lateral temporal lobe and of patient 2 (male, 12-year old) was from the occipital lobe. Both were about 0.5 cm3, including leptomeninges, cortex and superficial white matter. Single cell suspensions were prepared by digestion with LiberaseTM (Roche, Indianapolis, IN). Cells were fractionated into distinct populations by fluorescence-activated cell sorting (FACS) using the following antibodies: anti-human CD45 and anti-human Glycophorin A, both conjugated to allophycocyanin (APC) (eBioscience; San Diego, CA), anti-human platelet-derived growth factor receptor beta (PDGFRβ) conjugated to fluorescein isothiocyanate (FITC) (R&D Systems, Minneapolis, MN), and a mixture of anti-human CD31 (BD Biosciences Pharmingen; Bedford, MA), anti-human VE-Cadherin (R&D Systems) and anti-human vascular endothelial growth factor receptor 2 (VEGFR2, R&D Systems), each conjugated to phycoerythrin (PE). Genomic DNA was extracted from the sorted cells using the QIAamp DNA Mini kit (Qiagen, Germantown, MD).
Non-sorted brain cells from the single cell suspension were seeded on fibronectin-coated (1μg/cm2) tissue culture plates in EGM-2 complete medium (Lonza, Walkersville, MD) supplemented with 10% fetal bovine serum. After 10 days of culture, endothelial cells were selected using anti-human CD31 DynaBeads™ (Thermo Fisher Scientific, Cambridge, MA). Genomic DNA was extracted from CD31-positive and CD31-negative cell populations using the QIAamp DNA Mini kit.
Droplet digital PCR (ddPCR)-based analysis for GNAQ p.R183Q
Primers and probes were designed to target GNAQ p.R183Q as described previously10. Sequences used were: forward primer (5′-CCTGCCTACGCAACAAGAT-3′); reverse primer (5′-GTAAGTCAAAGGGGTATTCGAT-3′); reference probe (5′-TGCTTAGAGTTCGAGTCCCCACC-3′); mutant probe (5′-TGCTTAGAGTTCAAGTCCCCACC-3′). Briefly, the 20 μL ddPCR reaction mixture was composed of 1X SuperMix for Probes (Bio-Rad, Hercules, CA), mutant and reference probes (0.25 μM each), forward and reverse primer (0.9 μM each) and template DNA (30ng). The PCR reaction was partitioned into approximately 20,000 droplets using the QX100 Droplet Generator (Bio-Rad) and subjected to the following PCR cycle profile: 95°C for 10 min; 40 cycles of 94°C for 30 sec, 60°C for 60 sec, and 72°C for 30 sec; and final 98°C for 10 min. Droplets were measured in the QX100 Droplet Reader (Bio-Rad) and results were analyzed with the QuantaSoft software (Bio-Rad).
Immunostaining and confocal microscopy
Formalin-fixed, paraffin-embedded SWS brain sections were deparaffinized and immersed in antigen retrieval solution (Citrate-EDTA buffer: 10 mM citric acid, 2 mM EDTA, 0.05% Tween 20, pH 6.2 or 1mM EDTA pH 8.0) for 20 minutes. Sections were blocked for 30 minutes and stained with anti–human VE-Cadherin antibody (Sigma, HPA030562), anti-human CD31 antibody (Santa Cruz, C-20) and anti-human VEGFR2 antibody (55B11, Cell Signaling Technology, Danvers, MA) overnight. Purified class- and species-matched IgGs (Vector Lab) were the controls. Afterwards, sections were incubated with appropriate biotinylated secondary antibodies followed by DyLight 488 Streptavidin (Vector Lab). All slides were mounted using DAPI (Molecular Probe, Eugene, OR) to visualize nuclei. Images were acquired using a Leica TCS sp2 Acousto-Optical Beam Splitter confocal system equipped with DMIRE2 inverted microscope camera (Leica Microsystems, Wetzlar, Germany).
Flow cytometry
Cultured cells (1 × 105) were incubated at 4°C for 30 minutes in 100 μl of PBS plus 1% bovine serum albumin (BSA) with PE-conjugated anti-human CD31 antibody, washed three times, and analyzed by flow cytometry (Becton Dickinson, San Diego, CA). PE-conjugated mouse IgG1,k (R&D Systems) was used as isotype control. Data were analyzed using Flowjo_10v software (Tree Star, Inc. Ashland, OR).
Results
Two SWS patients underwent hemispherectomy due to extensive brain parenchyma abnormalities that causing drug-resistant epilepsy. Magnetic resonance imaging (MRI) in Figure 1A–B demonstrated marked abnormalities in the left hemisphere including the extensive leptomeningeal enhancement, cerebral atrophy, choroid plexus hypertrophy and tortuous and enlarged veins. Hematoxylin and eosin (H&E) staining highlighted the malformed vessels in the leptomeninges (Figure 1C–D). Anti-human VE-Cadherin (Figure 1E–F), anti-human CD31 (Figure 1G–H) and anti-human VEGFR2 (Figure 1I–J) staining shows strong staining along vessel lumens in the leptomeninges and cortex, consistent with endothelial cells, that validated the use of these antibodies for brain endothelial cell sorting. Sparse anti-VEGFR2 staining of non-vascular cells was also observed (Figure 1I–J). Red blood cell autofluorescence was evident in sections stained with anti-VEGFR2, which may be due to the antigen retrieval method that was different from anti-VE-Cadherin and anti-CD31 or to the weak but specific anti-VEGFR2 signal wherein red blood cell autofluorescence was similar in intensity.
Figure 1. Brain specimens from two SWS patients.
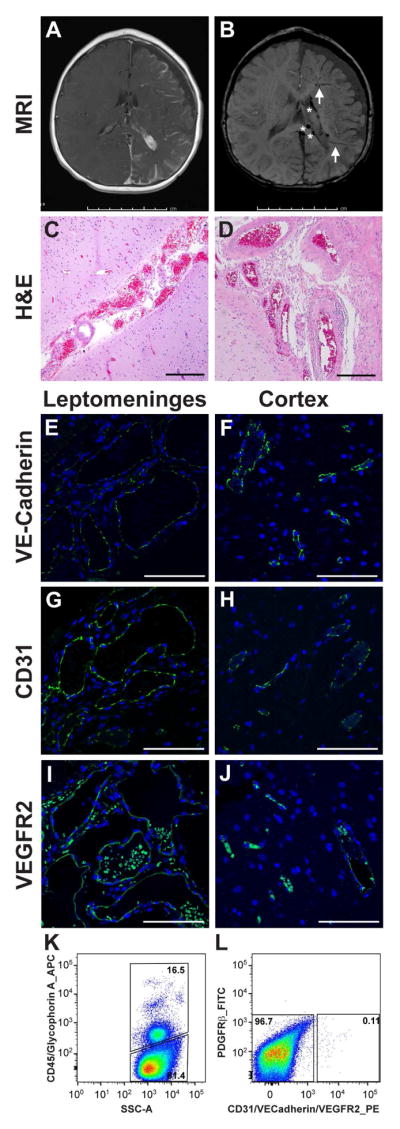
(A) Post-contrast axial T1-weighted MRI in patient 1 shows extensive left hemisphere leptomeningeal enhancement, left choroid plexus hypertrophy and marked left cerebral atrophy. (B) Axial susceptibility-weighted MRI in patient 1 shows enlarged medullary veins (arrows) and dilated deep veins (*). (C) Hematoxylin and eosin (H&E) staining of patient 1 brain tissue section shows abnormal clusters of leptomeningeal vascular channels of variable calibers. The underlying cortex demonstrates focal loss of neurons in layer 2 and subpial gliosis. Scale bar, 100μm. (D) H&E staining, viewed at higher magnification, shows some of these vascular walls are thick and rigid. Scale bar, 200μm. The histologic findings are consistent with leptomeningeal angiomatosis. (E–J) Anti human VE-cadherin, anti-human CD31 and anti-human VEGFR2 staining highlights enlarged vessels in leptomeningeal angiomas ( E,G and I, green) and shows positive staining predominately in the endothelium in the cortex (F,H and J, green) in patient 1. Nuclei counterstained with DAPI (blue). Scale bar, 100μm. (G–H) Fluorescence-activated cell sorting of freshly isolated brain cells from patient 2. Cells were sorted into hematopoietic and non-hematopoietic cells based on CD45/Glycophorin A expression (G). Non-hematopoietic cells were further fractioned into endothelial cells (using a set of anti-endothelial antibodies against CD31, VE-Cadherin and VEGFR2), PDGFRβ-positive cells and cells negative for all markers (H). Percentages of cells in each population are shown.
Small pieces of brain tissue from patient 1 and patient 2 were digested to prepare single cell suspensions. The brain cell preparations from each SWS patient were fractioned into different cellular populations by FACS. Hematopoietic cells were detected and gated using anti-human CD45 and anti-human Glycophorin A (GlyA) antibodies (Figure 1G). Endothelial cells were labeled with a panel of PE-conjugated antibodies against endothelial markers CD31, VE-Cadherin, and VEGFR2 to ensure all endothelial cells were captured. FITC-conjugated anti-human PDGFRβ antibody was used to detect pericytes 12, 13. However, very few PDGFRβ-positive cells were detected in these two SWS brain specimens (Figure 1H). The hematopoietic cells, endothelial cells and cells negative for all markers from SWS brain lesions were sorted for ddPCR to determine the GNAQ p.R183Q allelic frequency.
The two SWS brain lesions were first confirmed to have the GNAQ p.R183Q mutation by performing ddPCR on unfractionated brain cells. Mutant allelic frequencies were 8.7% and 6.2% for patient 1 and patient 2, respectively (Table 1). Remarkably, the endothelial cell fraction from patient 1 showed a mutant allelic frequency of 34.7% and endothelial cells from patient 2 showed a mutant allelic frequency of 24.0%. This corresponds to 70% and 48% mutant cells, respectively, for each of the sorted endothelial populations. These results indicate that GNAQ p.R183Q mutation is enriched in endothelial cells in SWS brain lesions, similar to what we reported for skin capillary malformation lesions 14. Mutant cells were also detected in the cell population negative for all markers, which may include un-captured endothelial cells, neuronal cells, pericytes or other stromal cells. Additional specimens and studies are needed to determine the identity of the mutant cells in this population. Very few mutant cells were found in the hematopoietic cell population.
Table 1.
GNAQ p.R183Q Mutational Analysis in SWS Brain Cell Populations
| Cell Population | Mutant Allelic Frequency (%)
|
|
|---|---|---|
| Patient 1 | Patient 2 | |
| Unfractionated cells from brain lesion | 8.7 | 6.2 |
| CD45/Glycophorin A – positive cells | 0.2 | 0.5 |
| Endothelial cells | 34.7 | 24.0 |
| PDGFRβ – positive cells | na | na |
| All negative cells | 9.2 | 8.4 |
na: not available
An aliquot of unfractionated brain cells from each patient was plated on a fibronectin-coated dish and cultured in endothelial growth medium. A viable and robust primary culture was observed after 10 days of in vitro culture (Figure 2A). Endothelial cells and non-endothelial cells were isolated from the primary culture using anti-CD31-coated magnetic beads. CD31-positive and CD31-negative cells were analyzed by flow cytometry to verify purity (Figure 2B). The GNAQ mutation was detected in CD31-positive endothelial cells but not in CD31-negative cells (Table 2). These results demonstrate that we were able to isolate and culture SWS patient-derived GNAQ mutant brain endothelial cells for further characterization. However, the mutant allelic frequencies of 14.7% and 21% demonstrate that the cultures are a mix of mutant and non-mutant endothelial cells. For the patient-1-derived EC culture at passage 4, it is approximately 30% mutant: 70% non-mutant; the patient 2-derived endothelial cell culture at passage 4 is 42% mutant: 58% non-mutant.
Figure 2. Cells isolated from SWS brain lesion.

(A) Primary culture from patient 1 brain lesion. Dashed circle highlights cells with endothelial morphology. Scale bar, 50μm (B) Flow cytometric analysis of cells in the primary culture after selection using anti-CD31-coated magnetic beads: CD31-positive cells and CD31-negative cells. Black lines depict cells incubated with anti-CD31 antibody; shaded grey areas are cells incubated with isotype-matched control antibody.
Table 2.
GNAQ p.R183Q Mutational Analysis in Cultured Cells Isolated from SWS Brain Lesion
| Sample | Mutant Allelic Frequency (%)
|
|
|---|---|---|
| Patient 1 | Patient 2 | |
| CD31–positive, p4 | 14.7 | 21.0 |
| CD31–negative, p4 | 0.1 | 2.6 |
Discussion
In this study, we demonstrated that the GNAQ p.R183Q mutation resides within endothelial cells in SWS brain lesions. This is consistent with our previous findings for the GNAQ p.R183Q mutation in skin capillary malformations10. Here we extend our studies to show the SWS brain endothelial cells can be isolated and expanded in culture and further confirm that the GNAQ p.R183Q mutation is present in brain endothelial cells.
The GNAQ mutant allelic frequency in the CD45 negative/endothelial marker negative/PDGFRβ negative cell population from the SWS brain specimens was 9.2 and 8.4%, higher than what was found in the same population in capillary malformation 14. These mutant cells may include endothelial cells that were not sorted due to insufficient antibody capture; addition of anti-glucose transporter 1 (GLUT1) may provide a more complete capture of all brain endothelial cells as GLUT1 is specifically expressed in brain endothelial cells 15, 16. We also observed GLUT1 expression in SWS brain endothelium by immnuostaining (data not shown). Alternatively, some of cells could be neuronal cells; sub-sorting of CD45 negative/endothelial marker negative/PDGFRβ negative cell population with antibodies against neuronal markers would be needed to address this. These refinements will be incorporated into future studies as additional SWS brain specimens become available.
We used anti-VE-Cadherin, anit-CD31 and anti-VEGFR2 antibodies to sort endothelial cells from the SWS brain specimens based on previous studies14, 17. Anti-VEGFR2 was included to capture cells that may be undergoing angiogenesis and thereby have increased VEGFR2 on the cell surface18, 19. A limitation of this strategy is that VEFGR2 expression is not strictly confined to endothelial cells and therefore the sorted endothelial population may contain non-endothelial cells. We can rule out that the mutant cells are composed entirely of non-endothelial VEGFR2-positive cells because the CD31-selected cells from a primary culture of total brain cells showed high mutant allelic frequency for GNAQ p. R183Q (Figure 2, Table 2). Together, our data demonstrate that the predominate mutant cells in SWS brain are endothelial cells
One feature of SWS is tortuous, malformed vessels in thickened leptomeninges (shown in Figure 1C–D). The aberrant pattern of arterial and venous cortical circulation underneath meningeal angiomas is also documented20, 21. In particular, venous congestion and stasis is recognized in SWS brain, which causes cerebral hypoperfusion and intraparenchymal dystrophic calcification; and potentially leads to frequent seizures, long epilepsy duration, severe cerebral atrophy, and brain functional deterioration. Given that the GNAQ mutation appears to reside primarily in endothelial cells in SWS brain, we speculate that GNAQ mutation causes some type of endothelial dysfunction, which in turn leads to the abnormal vascular structure and perhaps abnormal interaction with adjacent cells. The abnormal vessels in the leptomeninges and cortex may lead to faulty neuro-vascular interactions, which could in turn contribute to the cortical malformation, epileptogenesis and ultimately neurological deficits in SWS. Therapies directed at the impaired endothelial cell function and/or vascular-neuronal crosstalk at the early stage might prevent or alleviate some of the most devastating consequences of SWS.
The extent of leptomeningeal angiomas has been correlated with the severity of seizures, brain atrophy and cognitive loss 22. In our previous study, we noticed an association between capillary malformation severity and endothelial GNAQ mutant allelic frequency. That is, patients with more severe capillary malformations had higher endothelial GNAQ mutant allelic frequency 14. Therefore, it would be of interest to know whether endothelial GNAQ mutant allelic frequency in skin capillary malformation is correlated with mutant allelic frequency in the brain, and associated with the extent of leptomeningeal angiomas, and can further predict the severity of neurological impairment. If so, a biopsy of capillary malformation along with neuroimaging could potentially be performed in early childhood to predict the neurological problems. For example, if the endothelial mutant allelic frequency is high in skin capillary malformation, the patient may be expected to have a larger leptomeningeal angioma, and a higher risk of neurologic impairments. Future studies can test this hypothesis.
Recently, somatic mutations in GNAQ and GNA11 had been identified in congenital hemangioma 23, indicating GNAQ plays essential roles in vascular growth and vascular development. In addition, somatic mutations in GNA14, another Gαq family member, were recently reported in tufted angiomas, kaposiform hemangioendotheliomas and pyogenic granulomas 24. In these studies, the cellular residence of the mutations in the vascular tumors was not determined although Lim and colleagues expressed the GNA14 p.Q205L mutation in human umbilical vein endothelial cells. Further studies aimed to understand the mechanism by which altered Gαq signaling in endothelial cells contributes to pathophysiology of SWS and capillary malformation, as well as congenital hemangiomas and vascular tumors, may help to determine early biomarkers for predicting brain involvement and provide therapeutic targets to prevent skin and brain lesion progression.
Acknowledgments
Funding: Translational Neuroscience Center Pilot Study Award, Boston Children’s Hospital (J.B., A.K.G.), National Institutes of Health R01 HL127030-01A1 (J.B. A.K.G.) and Lisa’s Sturge-Weber Foundation Research Fellowship Award (L.H).
We thank the Dana-Farber/Harvard Cancer Center for use of their Flow Cytometry Core, Boston Children’s Hospital Department of Pathology-Histology for sectioning service and Kristin Johnson of the Vascular Biology Program at Boston Children’s Hospital for preparing the figures. This research was supported by a Pilot Study Grant from the Translational Neuroscience Center, Boston Children’s Hospital, Boston, MA and National Institutes of Health R01 HL127030-01A1 (J.B. and A.K.G.) and Lisa’s Sturge-Weber Foundation Research Fellowship Award (L.H.).
Footnotes
Disclosure of potential conflicts of interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5:257–264. doi: 10.1089/lrb.2007.1016. [DOI] [PubMed] [Google Scholar]
- 2.Sudarsanam A, Ardern-Holmes SL. Sturge-Weber syndrome: from the past to the present. Eur J Paediatr Neurol. 2014;18:257–266. doi: 10.1016/j.ejpn.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Waelchli R, Aylett SE, Robinson K, Chong WK, Martinez AE, Kinsler VA. New vascular classification of port-wine stains: improving prediction of Sturge-Weber risk. Br J Dermatol. 2014;171:861–867. doi: 10.1111/bjd.13203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dutkiewicz AS, Ezzedine K, Mazereeuw-Hautier J, Lacour JP, Barbarot S, Vabres P, Miquel J, Balguerie X, Martin L, Boralevi F, Bessou P, Chateil JF, Leaute-Labreze C. A prospective study of risk for Sturge-Weber syndrome in children with upper facial port-wine stain. J Am Acad Dermatol. 2015;72:473–480. doi: 10.1016/j.jaad.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Ch’ng S, Tan ST. Facial port-wine stains - clinical stratification and risks of neuro-ocular involvement. J Plast Reconstr Aesthet Surg. 2008;61:889–893. doi: 10.1016/j.bjps.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 6.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971–1979. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakashima M, Miyajima M, Sugano H, Iimura Y, Kato M, Tsurusaki Y, Miyake N, Saitsu H, Arai H, Matsumoto N. The somatic GNAQ mutation c. 548G>A (p R183Q) is consistently found in Sturge-Weber syndrome. J Hum Genet. 2014;59:691–693. doi: 10.1038/jhg.2014.95. [DOI] [PubMed] [Google Scholar]
- 8.Lian CG, Sholl LM, Zakka LR, OTM, Liu C, Xu S, Stanek E, Garcia E, Jia Y, MacConaill LE, Murphy GF, Waner M, Mihm MC., Jr Novel genetic mutations in a sporadic port-wine stain. JAMA Dermatol. 2014;150:1336–1340. doi: 10.1001/jamadermatol.2014.1244. [DOI] [PubMed] [Google Scholar]
- 9.O’Hayre M, Vazquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, Gutkind JS. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer. 2013;13:412–424. doi: 10.1038/nrc3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Couto JA, Huang L, Vivero MP, Kamitaki N, Maclellan RA, Mulliken JB, Bischoff J, Warman ML, Greene AK. Endothelial Cells from Capillary Malformations Are Enriched for Somatic GNAQ Mutations. Plast Reconstr Surg. 2016;137:77e–82e. doi: 10.1097/PRS.0000000000001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan W, Nadora DM, Gao L, Wang G, Mihm MC, Jr, Nelson JS. The somatic GNAQ mutation (R183Q) is primarily located within the blood vessels of port wine stains. J Am Acad Dermatol. 2016;74:380–383. doi: 10.1016/j.jaad.2015.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Crisan M, Corselli M, Chen WC, Peault B. Perivascular cells for regenerative medicine. J Cell Mol Med. 2012;16:2851–2860. doi: 10.1111/j.1582-4934.2012.01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Couto JA, Huang L, Vivero MP, Kamitaki N, Maclellan RA, Mulliken JB, Bischoff J, Warman ML, Greene AK. Endothelial Cells from Capillary Malformations are Enriched for Somatic GNAQ Mutations. Plast Reconstr Surg. 2015 doi: 10.1097/PRS.0000000000001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farrell CL, Yang J, Pardridge WM. GLUT-1 glucose transporter is present within apical and basolateral membranes of brain epithelial interfaces and in microvascular endothelia with and without tight junctions. J Histochem Cytochem. 1992;40:193–199. doi: 10.1177/40.2.1552163. [DOI] [PubMed] [Google Scholar]
- 16.Froehner SC, Davies A, Baldwin SA, Lienhard GE. The blood-nerve barrier is rich in glucose transporter. J Neurocytol. 1988;17:173–178. doi: 10.1007/BF01674204. [DOI] [PubMed] [Google Scholar]
- 17.Huang L, Nakayama H, Klagsbrun M, Mulliken JB, Bischoff J. Glucose transporter 1-positive endothelial cells in infantile hemangioma exhibit features of facultative stem cells. Stem Cells. 2015;33:133–145. doi: 10.1002/stem.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016;17:611–625. doi: 10.1038/nrm.2016.87. [DOI] [PubMed] [Google Scholar]
- 20.Comi AM. Presentation, diagnosis, pathophysiology, and treatment of the neurological features of Sturge-Weber syndrome. Neurologist. 2011;17:179–184. doi: 10.1097/NRL.0b013e318220c5b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caraballo R, Bartuluchi M, Cersosimo R, Soraru A, Pomata H. Hemispherectomy in pediatric patients with epilepsy: a study of 45 cases with special emphasis on epileptic syndromes. Childs Nerv Syst. 2011;27:2131–2136. doi: 10.1007/s00381-011-1596-5. [DOI] [PubMed] [Google Scholar]
- 22.Pinto AL, Chen L, Friedman R, Grant PE, Poduri A, Takeoka M, Prabhu SP, Sahin M. Sturge-Weber Syndrome: Brain Magnetic Resonance Imaging and Neuropathology Findings. Pediatr Neurol. 2016;58:25–30. doi: 10.1016/j.pediatrneurol.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Ayturk UM, Couto JA, Hann S, Mulliken JB, Williams KL, Huang AY, Fishman SJ, Boyd TK, Kozakewich HP, Bischoff J, Greene AK, Warman ML. Somatic Activating Mutations in GNAQ and GNA11 Are Associated with Congenital Hemangioma. Am J Hum Genet. 2016;98:1271. doi: 10.1016/j.ajhg.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim YH, Bacchiocchi A, Qiu J, Straub R, Bruckner A, Bercovitch L, Narayan D, McNiff J, Ko C, Robinson-Bostom L, Antaya R, Halaban R, Choate KA. GNA14 Somatic Mutation Causes Congenital and Sporadic Vascular Tumors by MAPK Activation. Am J Hum Genet. 2016 doi: 10.1016/j.ajhg.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]