

Abstract
Focal cerebral ischemia results in an ischemic core surrounded by the peri‐infarct region (penumbra). Most research attention has been focused on penumbra while the pattern of cell fates inside the ischemic core is poorly defined. In the present investigation, we tested the hypothesis that, inside the ischemic core, some neuronal and vascular cells could survive the initial ischemic insult while regenerative niches might exist many days after stroke in the adult brain. Adult mice were subjected to focal cerebral ischemia induced by permanent occlusion of distal branches of the middle cerebral artery (MCA) plus transient ligations of bilateral common carotid artery (CCA). The ischemic insult uniformly reduced the local cerebral blood flow (LCBF) by 90%. Massive cell death occurred due to multiple mechanisms and a significant infarction was cultivated in the ischemic cortex 24 h later. Nevertheless, normal or even higher levels of brain‐derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF) persistently remained in the core tissue, some NeuN‐positive and Glut‐1/College IV‐positive cells with intact ultrastructural features resided in the core 7‐14 days post stroke. BrdU‐positive but TUNEL‐negative neuronal and endothelial cells were detected in the core where extensive extracellular matrix infrastructure developed. Meanwhile, GFAP‐positive astrocytes accumulated in the penumbra and Iba‐1‐positive microglial/macrophages invaded the core several days after stroke. The long term survival of neuronal and vascular cells inside the ischemic core was also seen after a severe ischemic stroke induced by permanent embolic occlusion of the MCA. We demonstrate that a therapeutic intervention of pharmacological hypothermia could save neurons/endothelial cells inside the core. These data suggest that the ischemic core is an actively regulated brain region with residual and newly formed viable neuronal and vascular cells acutely and chronically after at least some types of ischemic strokes.
Keywords: cell survival, ischemic core, neurotrophic factors, neurovasculature, regeneration
Introduction
Stroke is a serious threat to human life and health with very few effective treatments available to stroke patients. Extensive research has improved our understanding on the mechanisms of ischemia‐induced cell death and developed many experimental treatments. Unfortunately, all neuroprotective treatments thus far have failed clinical trials. Facing the challenge of developing clinically effective therapies for ischemic stroke, it appears necessary to reevaluate some conventional perspectives in stroke pathology, pathophysiology, and current therapeutic strategies.
Cerebral ischemia typically induces an ischemic core in the supplying territory of the occluded artery. Massive excitotoxic cell death or pannecrosis occurs several hours later and continues for a few days after the insult 13, 24, 54. The classical and typical definition of the ischemic core is that of “a volume of tissue with which all cells (neuronal and glial), blood vessels (arteries, veins and capillaries) and nerve fibers (myelinated and non‐myelinated) have undergone necrosis” 58. Contiguous to core region, there is a transitional area where an intermediate severity of local cerebral blood flow (LCBF) reduction results in selective neuronal cell death 4, 37, 56, 65. This relatively minor injury occurs slower (days and weeks) and cells are more likely to undergo programmed cell death 5, 9, 20, 43, 73.
During many years of research on ischemic strokes, only scattered information on cell death in the ischemic core is available. An early pathological review in 1996 described “incomplete infarction”, referring to partial cell death in the ischemic area 23. Human and animal data supported an early assumption that “tissue necrosis would affect only a portion of the cells within the ischemic area if the arterial occlusion were of short duration or if ischemia were of moderate severity” 71. The incomplete infarction resulted in selective neuronal necrosis with preservation of some neurons, glia, and microvessels. Consistently, it was reported that “islands” of surviving neurons were observed in the ischemic core 2 days after stroke 53. However, ischemic core has been ignored for the last 20 years because attention in the stroke field has been drawn to the attractive concept of salvaging the penumbra, mostly caused by the greater possibility of saving neurons in this area where cell death progresses relatively slow and the increased understanding of the mechanisms of programmed cell death 1, 21, 28, 32, 38. In addition to the information from the few early investigations, the mechanisms of death and survival of neuronal and vascular cells inside the core are largely unknown.
The lack of specific focus on the ischemic core and the resultant knowledge gap have likely impaired the development of stroke therapy. For instance, it is not clear whether “incomplete infarction” occurs in common clinical cases or what types of ischemic stroke may result in the incomplete infarction. If we can delineate that under what condition/s cells inside the core might be savable, the instructive information or a game changing concept may improve the development of clinical stroke therapies. Moreover, many stem cell transplantation therapies after ischemic stroke are designed to avoid placing graft cells into the core region. Common justification for this practice is that there is no supporting infrastructure for grafted cells and nothing worthy of rescue inside the core region. More accurate information about the ischemic core will likely change and improve the stem cell transplantation therapy for ischemic stroke.
In contrast to the research focus on the peri‐infarct region, human stroke data suggests that the cortical ischemic core but not the penumbra is a determinant of clinical outcomes after acute ischemic stroke 36, 39. The percentage of penumbra tissue remains relatively constant from individual to individual, and that the percent core but not percent penumbra was significantly associated with outcomes. This clinical information reinforces that a more accurate understanding of cellular, molecular, and pathological events in the ischemic core is critically important for developing clinical stroke treatments. The present investigation examined cell fates and regenerative niche inside the core in two different ischemic stroke models of adult mice.
Methods and Materials
Animals
C57/BL adult mice (male, 26–28 g) were used in this investigation. In the experiment of collecting cortical cells for neuronal cultures, Nestin‐GFP transgenic adult mice were used to track neural progenitors. The animal protocol (DAR 2003027) was approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University School of Medicine. Animal procedures followed institutional guidelines that meet NIH standards. Principles of laboratory animal care” (NIH publication No. 86‐23, revised 1985) were followed, as well as specific national laws where applicable.
Focal cortical ischemic stroke in mice
A focal cerebral ischemic stroke targeting the right sensorimotor cortex was induced as previously described 14, 43. Mice were anesthetized with 3% isofluorane and maintained using 1.5% isoflurane supplemented with regular air during surgery. The cortex ischemia was achieved by permanent occlusion of the distal branches of the right middle cerebral artery (MCA) supplying the sensorimotor cortex. The MCA occlusion was paired with 10‐min ligation of both common carotid arteries (CCAs). Local cerebral blood flow (LCBF) was applied to verify the reduction of flow to the targeted brain region (see below).
Body temperature was monitored during surgery and recovery period using a rectal probe and maintained at 37°C on a homoeothermic blanket in a ventilated incubator (Havard Apparatus, Holliston, MA, USA). The body temperature for mice received pharmacological hypothermia treatment was not intervened. Different time points after stroke, mice were anesthetized with 4% chloral hydrate and sacrificed by decapitation. Before and after surgery, the animals were housed at 4‐5 animals per cage, with ad libitum access to food and water.
Permanent embolic ischemic stroke in mice
A severe stroke model of permanent embolic MCA occlusion that damaged cortical and subcortical structures was also tested. Clot preparation followed earlier reports with a few modifications 7. Briefly, the blood collected by cardiac puncture was supplemented with human fibrinogen (10 mg/ml), and immediately clotted in PE‐50 tubing for 6 h at room temperature followed by storage at 4°C. Before use, the clot (2.5 cm) was transferred into a PE‐10 tube filled with sterile saline and retracted. A single clot was transferred to PE‐10 catheter for embolization. Mice were anesthetized with 3% isofluorane and maintained using 1.5% isoflurane during surgery. The right CCA, the right external carotid artery (ECA) and the internal carotid artery (ICA) were exposed via a ventral midline neck incision. The PE‐10 catheter containing a clot was introduced into the CCA lumen through a small hole, advanced into the ICA, and the clot was gently injected with saline. The catheter was removed immediately after thromboembolization. Animal temperature control and animal care during and after surgery were the same as in the focal cortical stroke.
Local cerebral blood flow (LCBF) measurement
We used two different methods of LCBF measurement: laser Doppler perfusion imaging using the PeriScan PIM II scanner system (Perimed AB, Stockholm, Sweden) and autoradiography of 14C‐iodoantipyrine.
Laser Doppler scan imaging
This measurement was performed before and during surgery, 5, 10 minutes, and 24 h after reperfusion of CCAs as previously described 70. Briefly, under anesthesia, a crossing skin incision was made on the head to expose the whole skull. Laser scanning imaging measurements and analysis were performed using the PeriScans system and LDPIwin 2s (Perimed AB, Stockholm, Sweden) on the intact skull. The scanning region had a center point of ML+ 4.1 mm, and the four edges of the infarct area were ML+ 2.9 mm, ML+ 5.3 mm, AP − 1.5 mm, and AP+ 2.0 mm, respectively. In laser scanning imaging, the “single mode” with medium resolution was used to scan the photo image of LCBF. The laser beam was pointed to the center of the ischemic core (ML + 4.1 mm, AP 0 mm), the scan range parameter was set up as 5 × 5 and the intensity was adjusted to 7.5–8.0. The conventional “duplex mode” was used to record the Doppler image with the laser beam pointed to exact the same point on the border of the stroke core (ML—0, 5 mm, AP 0 mm). Corresponding areas in the contralateral hemisphere were similarly surveyed as internal controls. This scanning measurement largely avoids inaccurate or bias results caused by inconsistent locations of the traditional single point measurement.
[14C]iodoantipyrine autoradiography
Regional LCBF was measured according to the established method of iodoantipyrine autoradiography 7, 48. Mice were anesthetized with a mixture of 1.5% halothane, 69% nitrous oxide, and 29.5% oxygen. Under the operating microscope, the femoral artery and femoral vein were catheterized on both sides of the animal with polyethylene tubing (PE‐10; 3.0 cm long). The wound was infiltrated with lidocaine‐HCl and closed with sutures. Body temperature was monitored and maintained at 37.0°C to 37.5°C with a heat lamp. Arterial blood pressure was continuously recorded, and arterial blood samples were taken for blood gas assays before the start of the actual measurement of flow. The measurement of cortical cerebral blood flow followed the procedures described by Jay et al 34 and Wei et al, 71 with some modifications. In brief, 5 to 10 μCi of [14C]iodoantipyrine (American Radiochemical) was infused into 1 femoral vein for 20 s; 6 well‐timed blood samples were collected on preweighed pieces of filter paper over this period. At 20 s, the mice were decapitated. The brains were removed from the severed heads and frozen in 2‐methylbutane cooled to −45°C within 30 s of decapitation. Frozen brains were stored at −80°C until the time of sectioning. 14C radioactivity was determined in the reweighed samples of blood by liquid scintillation counting. Tissue radioactivity was assayed by quantitative autoradiography in the same part of the cortical field of the MCA in all three strains. Coronal sections (20 μm thick) were serially cut in a cryostat set at −17°C, starting at the level of the area postrema and ending at the rostral end of the caudate putamen. These sections were placed in x‐ray cassettes along with an appropriate set of standards and a sheet of x‐ray film (BRS Kodak, New York, NY). Commercial standards were used for 14C quantification (American Radiolabeled Chemicals, St. Louis, MO). The exposure period of the sections and standards was 7–9 days. The optical densities of the brain images and of the standards were measured on the autoradiograms with an MCID image analysis system (Imaging Research Inc., Ontario, Canada)). Cortical cerebral blood flow was determined from the blood and tissue radioactivities and the equation of the method 34, 71.
Infarct volume measurement
Twenty‐four hours or 4–5 days after the onset of MCAO, animals were sacrificed for assessment of brain infarct formation. 2,3,5‐triphenyltetrazolium chloride (TTC; Sigma‐Aldrich) staining was used to reveal damaged/dead brain tissue as previously described 70. Brains were removed and placed in a brain matrix then sliced into 1‐mm coronal sections. Slices were incubated in 2% TTC solution at 37°C for 5 minutes, then stored in 10% buffered formalin for 24 h. Digital images of the caudal aspect of each slice were obtained by a flatbed scanner. Infarct, ipsilateral hemisphere, and contralateral hemisphere areas at different levels were measured using ImageJ software (NIH, Bethesda, MD, USA). The indirect method (subtraction of residual right hemisphere cortical volume from cortical volume of the intact left hemisphere) was used for total infarct volume calculation. Infarct measurements were performed under double‐blind conditions.
TUNEL staining and cell death assessments
A terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit was used to examine cell death by detecting fragmented DNA in 10‐µm‐thick coronal fresh frozen sections as described previously (n = 8 per group) 40. After fixation (10% buffered formalin for 10 minutes and then ethanol:acetic acid (2:1) solution for 5 minutes) and permeabilization (0.2% Triton X‐100 solution), brain sections were incubated in equilibration buffer for 10 minutes. Recombinant terminal deoxynucleotidyl transferase (rTdT) and nucleotide mixture were then added on the slide at 37°C for 60 minutes in the dark. Reactions were terminated by 2x SSC solution for 15 minutes. Nuclei were counterstained with Hoechst 33342 (1:20,000; Molecular Probes) for 5 minutes. Cell counting was performed as described previously 40. Cell counting was performed following the principles of design based stereology. Systematic random sampling was used to ensure accurate and nonredundant cell counting. Eight brain sections per animal were collected at 90 µm distance between sections for nonoverlapping multistage random sampling. For each animal, 8 “area of interest” regions per slide were selected. Each field was scanned at 200× magnifications for cell counting. ImageJ (NIH) was used to analyze each picture. All analysis was performed in a double‐blinded fashion.
Western blot analysis
Western blot analysis was used to detect the expression of trophic factors brain‐derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF). Brain cortical tissue was lysed in a lysis buffer containing 0.02 M Na4P2O7, 10 mM Tris‐HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA (pH 8.0), 1% Triton, 1 mM EGTA, 2 mM Na3VO4, and a protease inhibitor cocktail (Sigma‐Aldrich). The supernatant was collected after centrifugation at 15,000 g for 10 minutes at 4°C. Protein concentration was determined with a bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL, USA). Equivalent amounts of total protein were separated by molecular weight on an SDS‐polyacrylamide gradient gel, and then transferred to a polyvinyl difluoride (PVDF) membrane. The blot was incubated in 5% bovine serum albumin (BSA) for at least 1 h and then reacted with primary antibodies at 4°C for overnight. The primary antibodies used in this investigation included: rabbit anti‐cleaved caspase‐3 (1:500; Cell Signaling, Danvers, MA, USA), anti‐BDNF antibody (1:2000; Cell Signaling) and anti‐VEGF antibody (1:5000; Sigma). After washing with Tris‐buffered saline with Tween (TBST), membranes were incubated with AP‐conjugated or HRP‐conjugated secondary antibodies (GE Healthcare, Piscataway, NJ, USA) for 1–2 h at room temperature. After final washing with TBST, the signals were detected with bromochloroidolylphosphate/nitroblue tetrazolium (BCIP/NBP) solution (Sigma‐Aldrich) or film. Signal intensity was measured by ImageJ and normalized to the actin signal intensity.
Immunohistochemical staining and cell counting
Frozen brain tissues were sliced into 10 µm‐thick coronal sections using a cryostat vibratome (Leica CM 1950; Leica Microsystems, Buffalo Grove, IL, USA). Sections were dehydrated on a slide warmer for 30 minutes, fixed with 10% formalin buffer, washed with −20°C precooled ethanol: acetic acid (2:1) solution for 10 minutes, and finally permeabilized with 0.2% Triton‐X 100 solution for 5 minutes. All slides were washed 3 times with PBS (5 minutes each) after each step. Then, tissue sections were blocked with 1% fish gelatin (Sigma‐Aldrich) in PBS for 1 h at room temperature, and subsequently incubated with the primary antibody: mouse anti‐NeuN (1:400; Millipore, Billerica, MA, USA), rabbit anti‐Glut‐1 (1:400, AB1341, Chemicon, Temecula, USA), and rabbit anti‐Beclin (1:5000; Abcam, Cambridge, MA, USA), goat anti‐collagen type IV (1:400; Millipore, Billerica, MA, USA) overnight at 4°C. Next day, the slides were washed three times with PBS for 5 minutes, then reacted with the secondary antibodies Alexa Fluor®488 goat anti‐mouse or rabbit (1:300; Life Technologies, Grand Island, NY, USA) and Cy3‐conjugated donkey anti‐rabbit (1:300; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) or Cy5‐conjugated donkey anti‐mouse or rabbit (1:400; Jackson ImmunoResearch Laboratories) for 80 minutes at room temperature. After 3 washes with PBS, nuclei were stained with Hoechst 33342 (1:20,000; Molecular Probes, Eugene, OR, USA) for 5 minutes as a counterstain. And then the brain sections were mounted, coverslipped, imaged, and photographed under a fluorescent microscope (BX51, Olympus, Japan) and laser scanning confocal microscope (Carl Zeiss Microimaging, Inc., Thornwood, NY, USA).
Primary cell cultures and immunocytochemistry
Wild type and Nestin‐GFP adult mice were subjected to the sensorimotor focal ischemia. The Nestin‐GFP mouse permanently expresses GFP under the Nestin promoter, consequently labeling all newly generated neurons green. This works as a reporter for neurogenesis in vitro assays. Primary cortical cells were isolated from the ischemic core region following the dissection procedure described previously 77. For labeling of all newly divided cells, 5‐bromo‐2′‐deoxyuridine (BrdU, 50 mg/kg, i.p.; Sigma) was administered daily starting from 1 day before stroke to 6 days after stroke. Seven days after stroke, the mice were anesthetized with isoflurane. The cortical tissue of core region and the corresponding area of the contralateral cortex tissue were isolated by an ophthalmic scissor and a forceps under a dissection microscope. Dissociated cells from the ipsilateral and contralateral cortex were plated separately under the same culture condition. Trypsin‐treated dissociated cells were plated on poly‐D‐lysine and laminin‐coated glass (VWR, West Chester, PA, USA) at the density of 8 × 104 cells/mL in MEM containing 10% horse serum (Invitrogen, Carlsbad, CA, USA). One hour later, medium was changed to Neurobasal‐medium (Invitrogen) supplemented with B‐27 (Invitrogen), L‐glutamine (0.5 mM, Invitrogen) and 20 ng/mL NGF (Sigma‐Aldrich). Cells were cultured at 37°C and 5% CO2 for 3–7 days. No inhibitor of glial cell proliferation was added in order to allow growth of all cell types. Half amount of the medium was changed with fresh medium every 2 days.
Transmission electron microscopy
The cellular and sub‐cellular structures of the brain cells was examined by electron microscopy. Seven days after stroke, brains were perfusion fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) and then stored in the same fixative overnight at 4 C. Brains were then sliced into 100 µm sections using a vibrating microtome. After washes with 0.1 M cacodylate buffer (pH 7.4), sections were post‐fixed with 1% osmium tetroxide and 1.5% potassium ferrocyanide in the same buffer for 1 h, dehydrated in an ethanol series up to 100%, infiltrated with Epoxy resin, and then flat‐embedded in the pure resin. Ultrathin sections of stroke affected cortical area were cut at 70 nanometers on a Leica Ultracut S ultramicrotome (Buffalo Grove, IL) and counterstained with 5% aqueous uranyl acetate followed by 2% lead citrate. Ultrathin sections were examined on a JEOL JEM‐1400 transmission electron microscope (Tokyo, Japan) equipped with a Gatan US1000 CCD camera (Pleasanton, CA).
Statistics
All data were analyzed for normality using the D'Agostino and Pearson omnibus normality test (where N > 7) and Kolmogorov‐Smirnov normality test (where N < 7). Unpaired Student's t‐test or Fisher's test were used for pairwise comparisons. Comparisons of multiple groups were analyzed using one‐ or two‐way ANOVA followed by post hoc Tukey's test. All results are expressed as mean ± S.E.M. Statistical comparisons were generated using Graph Pad Prism 6 (Graph Pad Software, Inc., San Diego, CA). P < 0.05 was considered significant for all comparisons.
Results
Ischemic core and infarction formation after focal cerebral ischemic stroke in mice
A focal cortical ischemic stroke model of adult mice was tested first in this investigation. The ischemic stroke was induced by permanent occlusion of distal branches of the right MCA and transient ligations (10 minutes) of both CCAs 14, 43. The MCA/CCA blockade markedly decreased the local cerebral blood flow (LCBF) to the lethal level in the sensorimotor cortex involving the barrel cortex as well as the surrounding motor cortex. In Laser Doppler scanning imaging, the LCBF in the targeted cortical region was 11.5 ± 1.8% of the basal level. Release of CCA ligations returned the flow to 44.0 ± 1.0% of the original LCBF (Figure 1A). Using 14C‐IAP autoradiography, we verified that there was a marked and homogenous reduction in LCBF within the ischemic core area (Figure 1B). Fourteen days later, the LCBF in this ischemic region was 60.0 ± 5.5% of the basal flow. It is known that the mouse brain is more sensitive/vulnerable to ischemic damage compared to the rat's brain, therefore shorter ischemic insult can cause comparable brain damage as in rats 10. The ischemic insult in the mouse model induced significant and consistent infarct formation revealed by the TTC staining negative area shown in white color (Figure 1C). The cortical infarct volume progressed from 24 mm3 to 33 mm3 at 1–3 days after stroke and the infarct ratio was around 10% of the hemispheric volume (Figure 1C). These data indicated that the permanent MCA occlusion paired with transient CCA ligations was severe enough to decrease LCBF in the targeted region, resulting in an unambiguous infarction in the mouse brain.
Figure 1.

Focal ischemic stroke in the mouse and ischemia‐induced cell death. Focal cerebral ischemia targeting the right sensorimotor cortex was induced by permanent occlusion of the distal braches of the right MCA and transient (10 minutes) ligation of both CCAs. A. The Laser Doppler scanner was used to measure the local cerebral blood flow (LCBF) in the ischemic region shown on the skull sketch (squire box) on the left. LCBF was reduced to about 10% of basal LCBF during the MCA/CCA blockage. B. 14C‐iodoantipyrine autoradiography was applied to assess regional flow in the brain after MCA/CCA occlusions. The blue area in the right cortex shows a marked and uniformed reduction in LCBF, surrounded by less reduced penumbra. C. One and 3 days after the ischemic insult, TTC staining revealed a well defined ischemic core region in the senserimotor cortex. N = 12 animals for 24 h and 15 for 72 h after stroke. D. DNA damage representing neuronal cell death was identified using TUNEL staining (green) at different times after stroke. In the ischemic core, Hoechst 33342 (blue) was applied to label nuclei of all cells, NeuN (red) was used as a mature neuronal marker. As late as 7 days after stroke, there were still some NeuN+ staining that did not overlay with TUNEL (arrows). E. NeuN+/TUNEL+ cells were counted in the ischemic core region. Quantified cell numbers were from six random survey fields and six brain sections. Neuronal cell death increased from 6 h after stroke and reached to more than 90% at 7 days after stroke. F. Non‐neuronal cells were identified as Hoechst 33342 labeled NeuN negative cells. The number of these cells was relatively stable during the first day after stroke, but showed a reduction at 2 days after stroke. A huge increase of non‐neuronal cells was seen at 7 days after stroke when massive invasion of inflammatory immune cells into the ischemic core. N = 10, Six brain sections were obtained from each animal and six random fields on the core of each section were counted.
In experimental assays, the center area within the TTC staining negative white area was identified as the ischemic core (Figure 1C). The peri‐infarct region or penumbra was defined as previously described by a 500 μm boundary extending from the edge of the infarct core, medial, and lateral to the infarct 61. In TTC staining, this is an area often appears in pink or light red color.
Acute and chronic neuronal cell death in the ischemic core
The ischemic insult to the cortex triggered massive neuronal cell death revealed by the DNA fragmentation marker TUNEL staining. In the ischemic area, about 40–60% of NeuN‐positive (NeuN+) cells were marked with TUNEL staining from 6 to 48 h after the cerebral ischemia (Figure 1D, E). This data suggested that a large population of neuronal cells die within hours after the ischemic strike. Conversely, it also implied that another half population of neurons in the ischemic area survived the initial ischemic insult. Seven days after stroke, the majority (>90%) of neurons inside the core became TUNEL positive (Figure 1E). During the 7 day period after stroke, we also examined non‐neuronal cells by counting NeuN negative cells that were labeled with nuclei staining of Hoechst 33342. These cells showed a trend of gradual reduction and reached the lowest level at 48 h after stroke (Figure 1F). There was, however, a marked increase of non‐neuronal cells 7 days after stroke, resulting from a massive invasion of inflammatory cells into the ischemic core (Figure 1F and see below).
Several days after stroke, there were around 5% of NeuN+ cells that were TUNEL negative in the ischemic core. A close look revealed, however, that some of them exhibited their NeuN immunoreactivity inside of Iba‐1 positive (Iba‐1+) macrophages/microglial cells (Figure 2E). These cells appeared to be undergoing the cleaning up process by phagocytosis. Conversely, some NeuN+ cells were neither overlaid with Iba‐1 nor with TUNEL staining. As late as 7–14 days after stroke, we still detected these surviving neurons in the ischemic core region (Figures 1D and 2C).
Figure 2.

Neuronal cell fate in the ischemic core. Immunohistochemical staining was performed in brain sections using NeuN antibody to label neurons and Iba‐1 antibody to label microglial cells in the ischemic cortex. A to D. Neuronal and microglial cells at 7, 14, and 21 days after stroke. NeuN+ cells can be seen inside the ischemic core (*). The enlarged images in C illustrate intact NeuN+ staining (red, arrows) in the core region 14 days after stroke. Meanwhile, there were significant numbers of Iba −1+ migcroglial and macrophages (green) located into the core. Hoechst 33342 staining (blue) marks the nuclei of all cells. E. A magnified image showing Hoechst 33342, NeuN and Iba‐1 labeling in a core area. The NeuN immunoreactivity appears inside the Iba‐1+ cells (arrows), suggesting that neuronal cells and/or debris were cleaned up by microglial/macrophages in the process of phagocytosis. This appears the case for most NeuN positive staining in the core region. F. As a negative control, the image was taken from the core region of a brain section showing the absence of neuron staining with the secondary Cy3 antibody but without adding the primary antibody NeuN.
Cell survival inside the core after a severe ischemic stroke in the mouse
To verify the conclusion obtained from the cortical ischemic stroke model of partial reperfusion, some examinations were repeated in a permanent ischemic stroke model that had enlarged damage to subcortical structures (Figure 3). The right MCA was permanently occluded using an autologous blood clot; the embolic ischemia lead to an infarction that occupied most of the right hemisphere in TTC staining (Figure 3A). Like other permanent ischemia models, the infarct formation reached to a maximal volume around 2–3 days after the onset of ischemia 27. At 7 days after stroke, we observed surviving neurons and vascular cells in the center of the core region (Figure 3B–D). Because of the higher mortality rate of the severe stroke model, we continued the investigation using the partial reperfusion stroke model.
Figure 3.

Cell death and survival in the ischemic core after embolic stroke of sever ischemia. A severe ischemic stroke was induced in adult mice by permanent occlusion of the MCA using an autologous blood clot. A. TTC staining of brain sections at 1 and 4 days after the ischemic insult. B. Immunohistochemical staining of NeuN (red), TUNEL (green) and Hoechst 33342 (blue) in the core region for the inspection of neuronal cell death at 7 days after stroke. NeuN positive but TUNEL negative cells can be seen in the region. The frames show the enlarged areas in C and D. C. Three dimensional images show a NeuN positive but TUNEL negative neurons (arrow). D. Glut‐1 staining was used for vascular endothelial cells. The 3‐D images show endothelial cells that were TUNEL negative. Representative of three animals.
Mechanism of neuronal cell death in the ischemic core
It was shown that acute cell death after ischemic stroke is caused by excitotoxicity involving excessive glutamate release, intracellular Ca2+ accumulation, production of reactive oxygen species, and a number of other injurious factors 13, 51, 60. This acute cell death in the core was widely believed to be necrotic in nature. However, we detected significant activation of caspase‐3 in the ischemic core at 1 day after stroke, similar to the caspase‐3 activity in penumbra (Figure 4A,B). Caspase‐3 activities were also detected in the core tissue 7 days after stroke (data not shown). These results suggested that apoptotic events played a noticeable part in the ischemic core as a mechanism of cell death.
Figure 4.
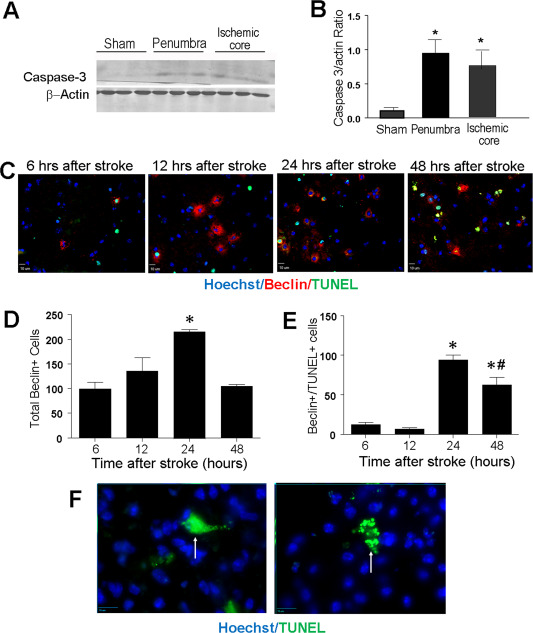
Apoptosis and autophagy contributed to cell death in the ischemic core. Signals in the apoptotic and autophagic cascades were examined in the ischemic cortex. A and B. Western blotting shows activation of caspase‐3 in the penumbra and core regions 1 days after stroke. N = 7. *. P < 0.05 vs. sham controls. C. The autophagy marker beclin 1 was detected in immunostaining of the ischemic core area (arrows). D. Quantification of total beclin 1+ cells per animal (six brain sections and six random fields on each section) in the ischemic core different time points after stroke. E. Double labeling of beclin 1 and TUNEL revealed a relationship between the beclin 1 immunoreactivity and cell death, especially 24 to 48 h after stroke. Comparing to the bar graph in D, approximately 50% of beclin cells were also TUNEL+ at these time points. N = 3 animals per time point. *. P < 0.05 vs. 6 h, #. P < 0.05 vs. 24 h. F. Differential morphology of TUNEL+ neurons. The image on the left shows smear TUNEL staining without noticeable nucleus fragmentation (Type 1 TUNEL+ cells; arrow). The image on the right shows fragmented DNA of TUNEL staining (Type 2 TUNEL+ cells; arrow).
Meanwhile, immunohistochemical examination in the ischemic core region revealed beclin‐1 activity as early as 6 h after stroke, suggesting that autophagy took place at this time (Figure 4C,D). Most beclin‐1 reactive cells at 6 and 12 h after stroke were not TUNEL positive (Figure 4D,E). Thus, this early autophagic activity was likely a physiological event of clearing away dead cells. The beclin‐1 activation reached to its peak at 24 h after stroke, while many beclin‐1+ cells now became TUNEL positive, indicating that, one day after stroke, excessive autophagy contributed to cell death in the ischemic core (Figure 4D,E). We also noticed different morphological features of TUNEL+ cells. Some TUNEL+ cells showed smear fluorescence of TUNEL staining in cell nuclei, which was previously named Type I TUNEL+ cells mainly injured by necrosis 73. Conversely, Type II TUNEL+ cells had fragmented TUNEL staining, consistent to an apoptotic feature (Figure 4F) 73. Among the cells examined, there were approximately 70% of Type I TUNEL+ cells and 30% Type II TUNEL+ cells. These cellular and subcellular changes indicate that a mixed form of cell death takes place in the ischemic core, which may include necrosis, apoptosis as well as autophagy.
Endothelial cell and vasculature fate in the ischemic core
Glut‐1 is a uniporter protein expressed in the endothelial cell membrane. The Glut‐1 55‐kDa isoform is specifically expressed in brain microvascular endothelium and has been used as a specific marker of microvessel/capillary cells in the brain 75. We noted that Glut‐1+ vessels remained at about the same level for up to 2 days after stroke and then started to decrease and reached a lower level by day 7 after stroke (Figure 5A,B). Interestingly, Glut‐1+ microvessel structures increased significantly 14 days after stroke and the majority of Glut‐1+ cells were not labeled by TUNEL (Figure 5C). Furthermore, Collagen IV staining revealed that extensive extra‐matrix networks formed at this time throughout the ischemic core (Figure 5C). This extra‐matrix infrastructure forms the foundation of a favorable microenvironment for angiogenesis.
Figure 5.
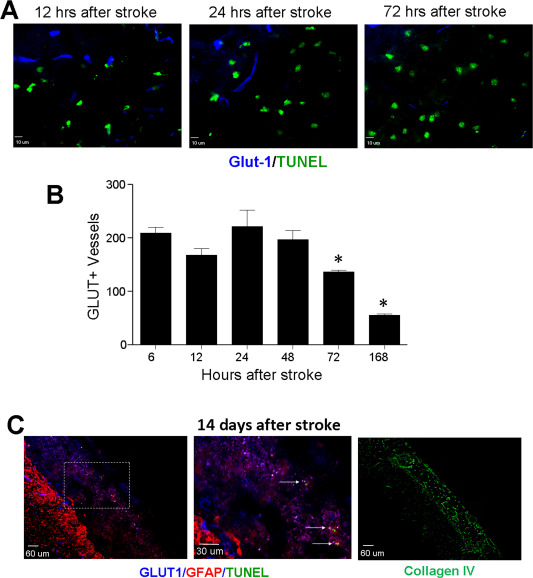
Vascular endothelial cells and microvessel structures in the ischemic core. Vascular endothelial cells were examined using the glucose transporter 1 antibody in immunohistochemical examination. A. Immunoflourenscent images of Glut‐1 (blue) and TUNEL (green) staining in the ischemic core at different days after stroke. B. Quantified data of the experiment in A. The number of Glut‐1+ endothelial cells/vessels remained relatively constant for up to 2 days after ischemia. Glut‐1+ cells decreased gradually from 3 to 7 days after stroke. N = 7 per time point, *. P < 0.05 vs. 6 h data). C. At 14 days after stroke, Glut‐1 (blue) markers was still widely detectable inside the ischemic core, while GFAP labeled glial cells (astrocytes) were mostly located in the peri‐infarct region. In the enlarged image from the frame shown in the left, TUNEL staining revealed some cell death process at this very delayed time point. The Collagen IV staining verified the extramatrix networks developed in the ischemic core 14 days after stroke.
Invasion of microglial/microphage into the ischemic core after stroke
One of the main cellular events after stroke is the invasion of activated microglia and macrophages into the ischemic core. Using the anti‐Iba1 antibody, we detected a gradual migration and accumulation of microglial cells and macrophages in the core region from 2 to 21 days after stroke (Figure 6A,B, also see Figure 2A–D). By day 14, it was estimated that more than 80% of cells resident in the core region were microglia or macrophages and the rest of cells were some NeuN+ cells and non‐neuronal cells such as vascular endothelial cells and a few astrocytes (Figures 5C, 6B and 6C).
Figure 6.
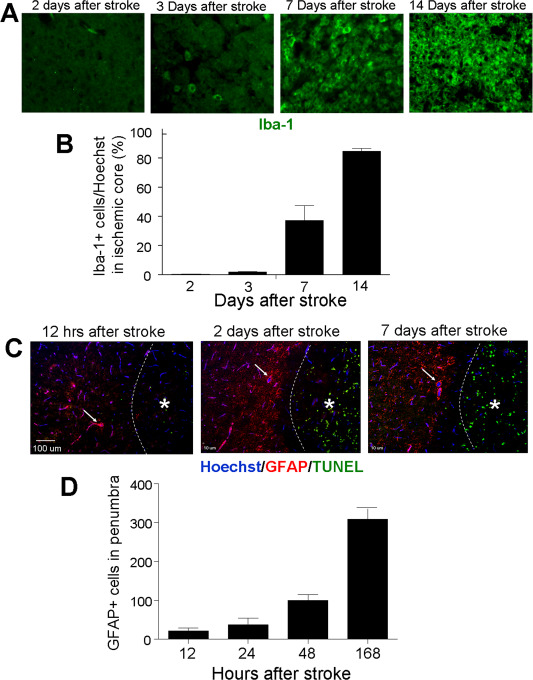
Distribution of microglia/macrophages and astrocytes in the ischemic and penumbra regions after stroke. Immunohistochemical staining was performed to track the invasion and localization of Iba‐1+ and GRAP+ cells in the ischemic cortex. A and B. Iba‐1 staining (green) in the ischemic core at different days after stroke. Iba‐1+ microglial/macrophages were rare in the core during the first few days after stroke. These cells started to occupy the ischemic core tissue several days after stroke, by day 14 about 90% of cells in the core were Iba‐1+ cells. N = 10 animals. Six brain sections were obtained from each animal and six random fields on the core of each section were counted. C and D. GFAP staining (red) was applied to identify activated astrocytes and TUNEL staining (green) was used to detect dead cells. Hoechst 33342 (blue) labeled nuclei of all cells. GFAP+ astrocytes started to accumulate in the peri‐infarct area 12 h after stroke, and the number increased gradually as long as 7 days after stroke. Arrows point to some GFAP/Hoechst positive cells in the peri‐infarct area. TUNEL staining was concentrated in the ischemic core (*), demonstrating massive cell death in the core. The bar graph shows the average total counted numbers of cells per animal. N = 8–12 animals. Six brain sections were obtained from each animal and six random fields on the core of each section were counted.
Astrocytes fate in the ischemic core and penumbra
As shown in previous investigations, activated astrocytes accumulated in the peri‐infarct region surrounding the ischemic core but not inside the core. GFAP staining illustrated that at 12 to 48 h after stroke, an increasing number of astrocytes started to locate in the peri‐infarct area (Figure 6C). The number of astrocytes steadily increased and formed an astrocytic scar around the core 7–14 days after stroke (Figure 6D).
Ultrastructure of surviving cells in the ischemic core
To further understand the structural integrity of the residual cells in the ischemic core, surviving cells in this region were examined using electron microscopy at 7 days after stroke. Under an electron microscope, viable looking cells were spread throughout survey fields. Some of them showed neuronal features including a large nucleus with intact plasma membrane and numerous cellular organelles in the cytoplasm (Figure 7B,C). Moreover, there were synaptic contacts between cells, composed of intact synapses with pre‐ and post‐synaptic features such as presynaptic vesicles and a postsynaptic density (PSD) (Figure 7D). Some glial‐like cells were observed and most of them appeared to be microglia or microphages (Figure 7E). Interestingly, we also identified myelinated axons (Figure 7F) and many blood vessel like structures composed endothelial cells and astrocytes/pericytes that resembled the neurovascular unit (Figure 7H,I). Consistent to the earlier observation of the autophagy marker, typical autophagosomes were observed in microglial cells (Figure 7G).
Figure 7.

Ultrastructural evidence of surviving cells in the ischemic core 7 days after stroke. A transmission electron microscope was used to examine the control brain section and the ischemic core of brain sections 7 days after ischemia. A. A control EM image of normal cortical neurons in the contra lateral cortex. The neurons had clear boundaries and a large nuclear (*), chromatin was uniformly distributed, the plasma membrane was continuous and clear, and many organelles including mitochondria and endoplasmic reticulum (ER) were visible. B. A representative surviving neuronal cells in the ischemic core. The cell contains a relatively large and universal nucleus, although the total cell size appeared smaller than the control neurons. The cell was surrounded by an intact plasma membrane and abundant cellular organelles existed in the cytoplasm. C. Another example of surviving neuronal cells in the ischemic core. In addition to the near normal nucleus (*) and chromatin, there were numerous intracellular organelles including mitochondria and ER (arrows) contained in the axonal shape of the cell and the intact membrane. Some lysosomes existed in the cytoplasm, suggesting the progress of degeneration. D. Remaining synaptic structures in the ischemic core region. Several presynaptic terminals and transmitter vesicles (V) inside the terminal can be seen in this image. The dark post‐synaptic density (PSD) was located opposite to the pre‐synaptic membrane. E. A microglial or microphagic cell showing two processes containing many lysosomes in the cytosol. Note the small size of this type of cell. F. Myelinated axon as shown in this image can be seen in the ischemic core. The image shows a cross section of the myelin (M) surrounding nerve fibers. G. Some autophagosomes (A) were identified, suggesting the process of autophagy. H and I. Surviving microvessels and endothelial cells (E) as well as the infrastructure of the neurovascular unit were easily detectable in the ischemic core. Astrocytes and/or pericytes (#) and basement membrane were surrounding endothelial cells. The lumen space was clearly formed inside the neurovascular unit.
Expression of regenerative factors in the ischemic core
In line with the existence of different cell populations in the ischemic core, Western blotting detected significant expression of trophic/growth factors including brain derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF). At 3 days after stroke, BDNF expression in the core tissue showed a two fold increase compared to the expression in the corresponding cortex of the sham control mice (Figure 8A). BDNF in the penumbra increased to a less extent (Figure 8A). The VEGF expression was not significantly altered in the core but increased in the penumbra (Figure 8A). Seven days after stroke, the BDNF level returned to about the sham control levels while the VEGF level significantly increased in both the core and penumbra tissues, which was consistent with the extensive vasculature formation in the ischemic region around this time (Figures 8B and 5C). These different regulations of BDNF and VEGF also verified that the tissues examined were distinctively from the core and peri‐infarct region, respectively.
Figure 8.
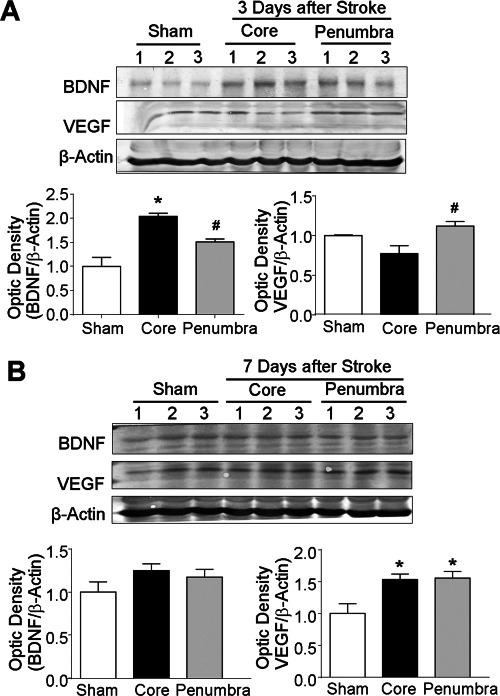
Expression of trophic factors in the ischemic core and penumbra. Western blotting was applied to assay the expression levels of BDNF and VEGF in the ischemic core and penumbra regions 3 and 7 days after stroke. A. BDNF and VEGF expression levels 3 days after stroke in sham control, core and penumbra brain tissues. B. BDNF and VEGF levels at 7 days after stroke in the three brain regions. The bar graph illustrates the expression ratios normalized to sham control after correction with loading controls. *. P < 0.05 vs. sham, #. P < 0.05 vs. core; n = 6 animals per group.
Regenerative activities in the ischemic core
Since we observed normal or even increased levels of BNDF and VEGF as well as expending vasculature structures inside the ischemic core many days after stroke, we speculated that regenerative niches might exist in this region. To label newly formed cells, BrdU (50 mg/kg, i.p.) was injected daily from day 1 after stroke until the day before sacrifice. In immunohistochemical assays of the ischemic cortex 7–14 days after stroke, BrdU/NeuN positive but TUNEL negative cells were detected in the core region, indicating newly generated neuronal cells (Figure 9A). Some Glut‐1+ vasculatures in the ischemic core were also BrdU positive, suggesting newly formed microvessels (Figure 9B). This was consistent with the abundant collagen IV expression on extra‐matrix basal lamina in the core (see Figure 5C) and partially recovered blood flow in the core (see Figure 1).
Figure 9.
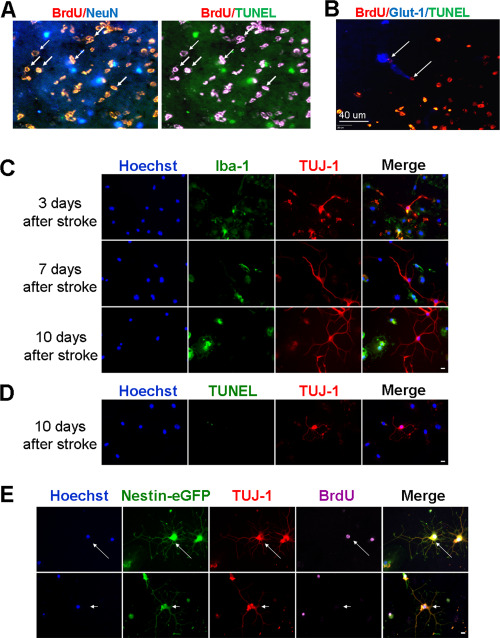
Regenerative niche exited in the ischemic tissue. Regenerative activities in the ischemic core were inspected in the brain and under cultured conditions. A and B. Stroke animals received BrdU injections daily to label proliferating cells. Seven days after stroke, immunostaining of the core region revealed the existence of BrdU/NeuN double positive cells that were TUNEL negative (A). BrdU/Glut1 positive but TUNEL negative endothelial cells were also observed in the core (B). Arrows point to BrdU/NeuN or BrdU/Glut1 double positive cells. To detect regenerative niche that might reside in the ischemic core tissue, we dissected the core tissue at 7 days after stroke and plated the dissociated cells in PDL/Laminin coated dishes. C. Different days after in vitro, cells were fixed and stained with immature neuronal marker TUJ‐1 (red) and microglia cell marker Iba‐1 (green). There were increasing numbers of TUJ‐1+ cells from 3 to 10 days after stroke. Many of them did not overlay with Iba1. D. In these images taken 10 days after stroke, TUJ‐1+ cells (red) were negative for TUNEL (green) staining suggesting they were viable cells. Scale bar = 10 μm. E. Nestin‐eGFP transgenic mice were subjected to the focal ischemic stroke. BrdU (50 mg/kg) was injected daily from 1 day after stroke. Cells from the ischemic core of 7 days after stroke were cultured for 7 days. Nestin‐eGFP (green) was easily detectable in this culture and it overlaid with the immature neuronal marker TUJ‐1 (red) as well as with the proliferation marker BrdU (purple) (arrow). There were a few cells that were eGFP and TUJ‐1 positive but BrdU negative (arrowhead), implying that they might be surviving original cells. Scale bar = 10 μm.
To verify the regenerative niche in the core region, we dissected the ischemic core tissue at 7 days after stroke and cultured the cells in a neuronal culture media for 3–10 days. As a control, the corresponding cortical region in the contralateral hemisphere was dissected and similarly cultured. It is commonly known that neurons acutely dissected from the adult brain cannot survive long‐term cell cultures. All neuronal cells from the contralateral side die by 7‐10 days in culture (data not shown). In the cultures from the ischemic core region, the majority of cells were Iba1‐positive microglia and macrophage cells, which agreed with the cellular component in this region. We noticed some NeuN immunoreactivity in day 1 cultures. The NeuN staining, however, mostly appeared inside the cytoplasm of Iba1‐positive cells, similarly as seen in the brain section of the core region where neuronal debris were phagocytized by immunoinflammatory cells (see Figure 2E). In day 3 cultures, labeling by the immature neuronal cell marker TUJ‐1 emerged (Figure 9C). The TUJ‐1+ cells increased with time and grew extensive neuronal processes (Figure 9C). Importantly, these cells were TUNEL negative when examined after 10 days in culture (Figure 9D), supporting the idea that viable neural progenitor cells and neurons existed in the ischemic core tissue.
To detect newly generated neuroblasts, cell cultures from Nestin‐GFP transgenic mice were similarly tested after ischemic stroke. In day‐10 cultures of the ischemic core tissue from this mouse, there were Tuj‐1+/Nestin‐GFP+ cells, and some of these cells also showed BrdU immunoreactivity (Figure 9E). These observations illustrates that a significant regenerative niche exists in the post‐stroke environment of the ischemic core region. Under favorable conditions, regenerative activities including cell proliferation and neural differentiation can take place in the core region.
The possibility of protecting neuronal and vasculature cells in the ischemic core
To delineate whether it was possible to promote cell survival in the ischemic core after stroke, we tested pharmacologically induced hypothermia (PIH) as a brain protective therapy 14, 30, 40, 41. The hypothermic compound neurotensin receptor 1 (NTR1) agonist HPI‐201 (2 mg/kg, i.p.) was injected 60 minutes after the onset of ischemia and the animal's body temperature was reduced from the normal 37°C to around 32°C in 30–60 minutes. The hypothermia was maintained for 6 h before it gradually returned to normal temperature (Figure 10A). Three days after stroke, the hypothermic treatment showed a significant effect of reducing the infarction volume measured in TTC staining assay (Figure 10B). Seven days after stroke, NeuN, Glut1 and TUNEL staining in brain sections showed that the hypothermia treatment markedly enhanced the survival of neuronal cells and endothelial cells in the ischemic core (Figure 10C,D).
Figure 10.
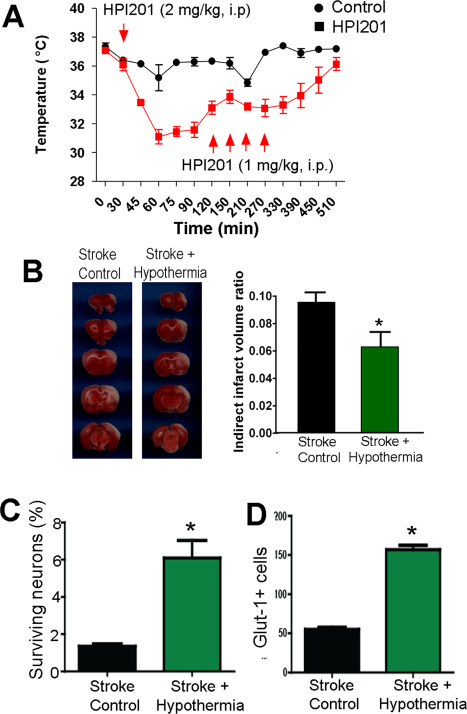
Protection of the neuronal and vascular cells in the ischemic core. A. The neurotensin receptor 1 agonist HPI‐201 was injected 60 minutes after the onset of ischemia. HPI‐201 injections effectively reduced the body temperature of the stroke animals from 37°C to 32°C for 6 h. B. TTC staining of the ischemic brain sections 3 days after focal ischemia. HPI‐201‐treated animals showed significantly reduced infarct volume compared to stroke/saline controls. N = 5 per group, *. P < 0.05 vs. stroke control. C and D. HPI‐201 treatment significantly increased surviving NeuN+ neurons (C) and endothelial cells (D) in the core 7 days after stroke. N = 7, *. P < 0.05 vs. stroke controls.
Discussion
Different than the common focus on the peri‐infarct region, the present investigation focused on cell death, survival and regeneration in the ischemic core acutely and chronically after ischemic strokes. We show that a small percentage of neurons can survive in this region for at least 7 days after stroke. Moreover, many vascular endothelial cells and neurovascular structures in the core survive from the ischemic insult, and this may occur not only in relativity moderate ischemia but also after more severe stroke. Regenerative paracrine factors BDNF and VEGF remain at normal or even higher levels, regenerative activities such as proliferation of endothelial cells and formation/invasion of neural progenitor cells takes place in the core many days after stroke. As a result, extensive neurovascular networks were established in the ischemic core 14 days after stroke. Although the surviving cells in the core after stroke are few and the neurovascular structures may be imperfect or immature, they could provide a minimum but vital infrastructure for possible regeneration from endogenous mechanisms. It remains to be determined whether the surviving and regenerating neurons and vascular cells are functional cells and able to contribute to tissue repair. The LCBF measurement at 14 days after stroke, nevertheless, indicates that the vasculature existed in the core can carry blood flow into the region. For regenerative therapies using exogenous stem cells and neural progenitors, our data suggest that the microenvironment of ischemic core several days and weeks after stroke provides certain cellular support, blood flow supply and strong trophic factor support for cells to survive. Thus, it is possible that the remaining and regenerating neurovascular infrastructure could be utilized as therapeutic targets for repairing damaged neurovascular units and neuronal networks.
A short duration of LCBF decrease that reaches 30–70% of normal flow usually does not lead to neuronal cell death and infarct formation 2, 33. Infarct develops when LCBF decreases by more than 80–90% for a certain time 2, 33. Typically inside an ischemic core, LCBF is reduced to 15% or less of the control flow 33. In our focal ischemia model, the LCBF was reduced to around 11% of basal level during MCA/CCA occlusion, leading to lethal damage to the territory of the MCA branches. The autoradiography data illustrated a uniform reduction of the LCBF in the core region. The infarct is formed 1–3 days after stroke as in other stroke models 10. We thus conclude that the cerebral ischemia in our models was homogenous and severe enough to form infarction in the targeted brain tissue.
In the focal cortical ischemia model tested, the MCA was permanently occluded and paired with 10 minutes CCA ligation. The duration of the lethal ischemia induced by MCA/CCA occlusion seems to be relatively short compared to that for rat stroke models. Longer MCA/CCA occlusion was tested but resulted in excessive mortality, which was likely because mice is more sensitive than rats to cerebral ischemia 10. It is widely known that anesthesia has a great impact on the outcomes of ischemic stroke models. In our preliminary experiments, the MCA/CCA occlusion was able to be extended up to 20 minutes when 95% O2 gas was used instead of room air with isoflurane anesthesia. The high oxygen condition, however, resulted in large variations of the infarction volume in C57/BL mice and we decided to stay with regular air in the inhalant anesthesia. The long‐term cell survival in the core was also observed in a severe stroke model of a permanent MCA occlusion, suggesting the cell survival features may not be limited to one type of stroke. The pathological feature of surviving cells in the core of different strokes needs to be further compared and characterized. For example, it is possible that in the embolic stroke model, spontaneous thrombolysis might occur, leading to a transient and less severe ischemia. Although the large infarction formation does not support this assumption in the model tested here, a future investigation on permanent ischemia of filament insertion model may be examined to decisively eliminate this possibility.
It is important to point out that partial reperfusion due to incomplete (spontaneous and postthrombolytic) recanalization after an ischemic attack occurs in 30% and up to 70% of clinical cases at different times after the onset of ischemia 3, 31, 35, 59. For example, an MRI study on 82 stroke patients with MCA occlusion revealed that incomplete reconalization occurred in 39 patients, complete reconalization in 10, and persistent occlusion in 33 patients 59. In another CT/SPECT scan study on 354 stroke patients, the incidence of spontaneous reperfusion was 77% in patients with cortical infarcts 35. In this regard, the release of CCAs ligation while MCA was permanently occluded in our cortical stroke model resemble to a great extent a partial reperfusion condition seen in many clinical stroke cases.
Previous and current investigations have focused on the peri‐infarct or penumbra region of the ischemic brain. It is believed that only this region is savable due to moderate ischemia, less severe damage and the slow process of programmed cell death. This approach makes sense considering the majority of cells in this region survive the initial ischemic attack, providing a reasonable time window to rescue cells in following hours and days 1, 21, 28, 32, 38. Conversely, the ischemic core has been widely regarded as a pannecrotic tissue deteriorating soon after stroke 15, 58. Because of this historical focus on the peri‐infarct region, the temporal and spatial patterns of cell fating process in the ischemic core have been poorly defined and ignored in recent years. Our data indicate that up to 7 days after ischemic stroke, significant numbers of neurons and vascular cells are still surviving in the ischemic core. Considering that maximal infarction normally forms 3 days after permanent MCA occlusion 27, the survival of these cells persisted long after the maximal infarct formation.
In a few previous investigations, neurons have been reported to survive in ischemic tissue longer than 3–7 days 29. In an ischemic stroke model of rats, about 80% neurons became necrotic in the core region 7 days after stroke 26. Newly formed vessels were seen in the ischemic core after ischemic stroke and Nestin expression was detected with the vasculature‐associated cells 14 to 28 days after ischemia 17, 53. In a more recent investigation, McMarthy et al showed that about 5% NeuN+ cells could still be counted in the ischemic core 3 days after transient ischemic stroke in rats 52. Moreover, a recent investigation on biochemical parameters in the ischemic core and penumbra showed that protein synthesis was only partly inhibited by 36% at 3 days after transient ischemic stroke 8, suggesting that the cellular/molecular machinery of protein synthesis was largely preserved at this time. Unfortunately, these few reports did not give systematic evaluation of cell fate and related mechanisms in the core. It appears to us that a re‐visit to ischemic core is necessary and clinically important.
Different from the popular belief that neurons in the ischemic core die from necrosis, several early investigations on the cell death in the ischemic region indicated that many cells die with a shrunken cell body 22, which is strikingly different from the swollen cell body seen in glutamate/NMDA‐induced excitotoicity in vitro 12, 29. Garcia et al reported that neurons in the ischemic tissue showed early shrinkage and scalloping followed by appearance of Ghost neuron morphology observed up to 7 days after stroke 27. “Ghost cells” have a shadowy appearance in hematoxylin‐eosin (H&E) stained sections. Later, the Ghost cell was linked to apoptosis in sympathetic neurons 69. During the early hours after an ischemic insult morphological changes in the core include acute shrinkage, angularity, and homogeneous eosinophilia of the cytoplasm. The nucleus becomes shriveled, pyknotic and hyperchromatic 45, 46, 68. Many of these morphological features are inconsistent with necrosis but rather suggestive of apoptosis. There have been a few reports that observed apoptotic cell death in the core 49, 57.
The present investigation provides new information on the mechanisms of cell death inside the core. It is evident that ischemia induced cell death in the brain does not occur by typical necrosis or apoptosis as observed in vitro. In fact, cell death after cerebral ischemia in vivo is more likely a result from combinations of multiple injury mechanisms. Thus, mixed or hybrid features of both necrosis and apoptosis can be seen in a single cell 47, 72, 73, 76. For example, a dying neuron often has a swollen cytoplasm, deteriorated cellular membrane/organelles while its nucleus is highly condensed with involvement of caspase activation 72, 73, 76. These mixed cellular, molecular and ultrastructural features observed in the same cells have been termed “hybrid cell death” by us and has adopted a few different names with similar or various definitions by others 6, 11, 18, 47, 62, 72, 73, 76. Autophagy may contribute to ischemia‐induced neuronal cell death 18, 43, 62, 74. Whether this may happen in the ischemic core has not been shown before. Our report now provides the first evidence that neurons in the ischemic core bear the morphological and molecular features of necrosis, apoptosis as well as autophagy, leading to hybrid cell death.
GFAP‐positive astrocytes accumulated in the peri‐infarct area but do not move into the ischemic core. These cells formed the glial scar around 7 days after stroke and isolated the ischemic core from the surrounding area. Starting from hours after cerebral ischemia, activated microglial/microphages emerged and increased in the ischemic core 15, 42, 78. These cells are likely invaded from the circulation and proliferate in the core 19, 25, 50, 55. We noticed that many NeuN+ puncta inside the Iba‐1+ cells. These are likely the debris of neurons subjected to phagocytosis by microglia and microphages.
A recent study showed that the expression of VEGF receptor 3 (VEGFR‐3) could be detected in perivascular cells in the ischemic core 3–7 days after stroke 67. The existence of neurovascular structures and the remaining levels of neurotrohpic factors in the core chronically after stroke should play significant roles in regenerative stroke therapies. Nestin expression within the ischemic tissue increased as early as 6 h and peaking at 7 days. This expression persisted for at least 4 weeks after transient ischemic stroke 44, 66. The Nestin expression was observed in neuronal and non‐neuronal cells. In a microarray analysis of gene expression after permanent MCA occlusion in rats, there were 2882 genes upregulated and 2835 genes downregulated in the ischemic core at 3 days after stroke 63. The affected genes were involved with the inflammation, cell death/growth/proliferation/migration, DNA replication/recombination/repair, cellular assembly/organization, cell, signaling and neurovascular unit development. The Nestin expression and the broad spectrum gene regulation in the core imply very active regenerative/repair processes occurring inside the core even after the severe permanent ischemia.
Using pharmacological hypothermia treatment that provides a global brain protection, we demonstrate that cell death in the core region can be significantly ameliorated by a therapeutic intervention. Pharmacological hypothermia was maintained for 6 h in these experiments. The optimal hypothermia duration and whether the protection on core neurons is a long lasting effect remain to be determined, especially in aged animals 16, 64. Cell death in the core is often not examined in previous and current investigations. We recommend that protective effects consisting of reducing cell death in neuronal and endothelial cell populations in the ischemic core should be specifically evaluated in experimental stroke treatments. Promoting the endogenous regenerative mechanisms inside the core should be included as a therapeutic target in future investigations.
Conflict of Interests: All authors claim no conflict of interest related to this investigation.
Acknowledgment
This work was supported by NIH grants NS075338 (LW/SPY), NS062097 (LW), NS091585 (LW), NS073378 (SPY) and VA National Merit grant RX000666 (SPY). We acknowledge the Emory Electron Microscope Core supported by NIH S10 grant (S10 RR025679 01). This study was also supported by the O. Wayne Rollins Endowment to SPY.
References
- 1. Aronowski J, Cho KH, Strong R, Grotta JC (1999) Neurofilament proteolysis after focal ischemia; when do cells die after experimental stroke? J Cereb Blood Flow Metab 19:652–660. [DOI] [PubMed] [Google Scholar]
- 2. Back T (1998) Pathophysiology of the ischemic penumbra–revision of a concept. Cell Mol Neurobiol 18:621–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barber PA, Davis SM, Infeld B, Baird AE, Donnan GA, Jolley D, Lichtenstein M (1998) Spontaneous reperfusion after ischemic stroke is associated with improved outcome. Stroke 29:2522–2528. [DOI] [PubMed] [Google Scholar]
- 4. Baron JC, Yamauchi H, Fujioka M, Endres M (2014) Selective neuronal loss in ischemic stroke and cerebrovascular disease. J Cereb Blood Flow Metab 34:2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barone FC (2009) Ischemic stroke intervention requires mixed cellular protection of the penumbra. Curr Opin Investig Drugs 10:220–223. [PubMed] [Google Scholar]
- 6. Benchoua A, Guegan C, Couriaud C, Hosseini H, Sampaio N, Morin D, Onteniente B (2001) Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci 21:7127–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bereczki D, Wei L, Otsuka T, Acuff V, Pettigrew K, Patlak C, Fenstermacher J (1993) Hypoxia increases velocity of blood flow through parenchymal microvascular systems in rat brain. J Cereb Blood Flow Metab 13:475–486. [DOI] [PubMed] [Google Scholar]
- 8. Bonova P, Burda J, Danielisova V, Nemethova M, Gottlieb M (2013) Development of a pattern in biochemical parameters in the core and penumbra during infarct evolution after transient MCAO in rats. Neurochem Int 62:8–14. [DOI] [PubMed] [Google Scholar]
- 9. Broughton BR, Reutens DC, Sobey CG (2009) Apoptotic mechanisms after cerebral ischemia. Stroke 40:e331–e339. [DOI] [PubMed] [Google Scholar]
- 10. Carmichael ST (2005) Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx 2:396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cho YS (2014) Perspectives on the therapeutic modulation of an alternative cell death, programmed necrosis (review). Int J Mol Med 33:1401–1406. [DOI] [PubMed] [Google Scholar]
- 12. Choi DW (1987) Ionic dependence of glutamate neurotoxicity. J Neurosci 7:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi DW (1992) Excitotoxic cell death. J Neurobiol 23:1261–1276. [DOI] [PubMed] [Google Scholar]
- 14. Choi KE, Hall CL, Sun JM, Wei L, Mohamad O, Dix TA, Yu SP (2012) A novel stroke therapy of pharmacologically induced hypothermia after focal cerebral ischemia in mice. FASEB J 26:2799–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Claus HL, Walberer M, Simard ML, Emig B, Muesken SM, Rueger MA et al (2013) NG2 and NG2‐positive cells delineate focal cerebral infarct demarcation in rats. Neuropathology 33:30–38. [DOI] [PubMed] [Google Scholar]
- 16. Corbett D, Thornhill J (2000) Temperature modulation (hypothermic and hyperthermic conditions) and its influence on histological and behavioral outcomes following cerebral ischemia. Brain Pathol 10:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DeGirolami U, Crowell RM, Marcoux FW (1984) Selective necrosis and total necrosis in focal cerebral ischemia. Neuropathologic observations on experimental middle cerebral artery occlusion in the macaque monkey. J Neuropathol Exp Neurol 43:57–71. [DOI] [PubMed] [Google Scholar]
- 18. Fayaz SM, Suvanish Kumar VS, Rajanikant GK (2014) Necroptosis: who knew there were so many interesting ways to die? CNS Neurol Disord Drug Targets 13:42–51. [DOI] [PubMed] [Google Scholar]
- 19. Felger JC, Abe T, Kaunzner UW, Gottfried‐Blackmore A, Gal‐Toth J, McEwen BS et al (2010) Brain dendritic cells in ischemic stroke: time course, activation state, and origin. Brain Behav Immun 24:724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferrer I (2006) Apoptosis: future targets for neuroprotective strategies. Cerebrovasc Dis 21 Suppl 2:9–20. [DOI] [PubMed] [Google Scholar]
- 21. Ferrer I, Planas AM (2003) Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol 62:329–339. [DOI] [PubMed] [Google Scholar]
- 22. Garcia JH, Kamijyo Y (1974) Cerebral infarction. Evolution of histopathological changes after occlusion of a middle cerebral artery in primates. J Neuropathol Exp Neurol 33:408–421. [DOI] [PubMed] [Google Scholar]
- 23. Garcia JH, Lassen NA, Weiller C, Sperling B, Nakagawara J (1996) Ischemic stroke and incomplete infarction. Stroke 27:761–765. [DOI] [PubMed] [Google Scholar]
- 24. Garcia JH, Liu KF, Ho KL (1995) Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke 26:636–642; discussion 643. [DOI] [PubMed] [Google Scholar]
- 25. Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ (1994) Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat). Am J Pathol 144:188–199. [PMC free article] [PubMed] [Google Scholar]
- 26. Garcia JH, Wagner S, Liu KF, Hu XJ (1995) Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 26:627–634; discussion 35. [DOI] [PubMed] [Google Scholar]
- 27. Garcia JH, Yoshida Y, Chen H, Li Y, Zhang ZG, Lian J et al (1993) Progression from ischemic injury to infarct following middle cerebral artery occlusion in the rat. Am J Pathol 142:623–635. [PMC free article] [PubMed] [Google Scholar]
- 28. Ghobrial GM, Chalouhi N, Zohra M, Dalyai RT, Ghobrial ML, Rincon F et al (2014) Saving the ischemic penumbra: endovascular thrombolysis versus medical treatment. J Clin Neurosci 21:2092–2095. [DOI] [PubMed] [Google Scholar]
- 29. Goldberg MP, Choi DW (1993) Combined oxygen and glucose deprivation in cortical cell culture: calcium‐dependent and calcium‐independent mechanisms of neuronal injury. J Neurosci 13:3510–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gu X, Wei ZZ, Espinera A, Lee JH, Ji X, Wei L et al (2015) Pharmacologically induced hypothermia attenuates traumatic brain injury in neonatal rats. Exp Neurol 267:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hakim AM, Pokrupa RP, Villanueva J, Diksic M, Evans AC, Thompson CJ et al (1987) The effect of spontaneous reperfusion on metabolic function in early human cerebral infarcts. Ann Neurol 21:279–289. [DOI] [PubMed] [Google Scholar]
- 32. Heiss WD (2011) The ischemic penumbra: correlates in imaging and implications for treatment of ischemic stroke. The Johann Jacob Wepfer award 2011. Cerebrovasc Dis 32:307–320. [DOI] [PubMed] [Google Scholar]
- 33. Hossmann KA (2006) Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol 26:1057–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jay TM, Lucignani G, Crane AM, Jehle J, Sokoloff L (1988) Measurement of local cerebral blood flow with [14C]iodoantipyrine in the mouse. J Cereb Blood Flow Metab 8:121–129. [DOI] [PubMed] [Google Scholar]
- 35. Jorgensen HS, Sperling B, Nakayama H, Raaschou HO, Olsen TS (1994) Spontaneous reperfusion of cerebral infarcts in patients with acute stroke. Incidence, time course, and clinical outcome in the Copenhagen Stroke Study. Arch Neurol 51:865–873. [DOI] [PubMed] [Google Scholar]
- 36. Jovin TG, Yonas H, Gebel JM, Kanal E, Chang YF, Grahovac SZ et al (2003) The cortical ischemic core and not the consistently present penumbra is a determinant of clinical outcome in acute middle cerebral artery occlusion. Stroke 34:2426–2433. [DOI] [PubMed] [Google Scholar]
- 37. Kato H, Kogure K (1999) Biochemical and molecular characteristics of the brain with developing cerebral infarction. Cell Mol Neurobiol 19:93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kidwell CS, Alger JR, Saver JL (2003) Beyond mismatch: evolving paradigms in imaging the ischemic penumbra with multimodal magnetic resonance imaging. Stroke 34:2729–2735. [DOI] [PubMed] [Google Scholar]
- 39. Koennecke HC (2003) Editorial comment–challenging the concept of a dynamic penumbra in acute ischemic stroke. Stroke 34:2434–2435. [DOI] [PubMed] [Google Scholar]
- 40. Lee JH, Wei L, Gu X, Wei Z, Dix TA, Yu SP (2014) Therapeutic effects of pharmacologically induced hypothermia against traumatic brain injury in mice. J Neurotrauma 31:1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee JH, Wei L, Gu X, Won S, Wei ZZ, Dix TA, Yu SP (2016) Improved therapeutic benefits by combining physical cooling with pharmacological hypothermia after severe stroke in rats. Stroke 47:1907–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H, Zhang N, Lin HY, Yu Y, Cai QY, Ma L, Ding S (2014) Histological, cellular and behavioral assessments of stroke outcomes after photothrombosis‐induced ischemia in adult mice. BMC Neurosci 15:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li WL, Yu SP, Chen D, Yu SS, Jiang YJ, Genetta T, Wei L (2013) The regulatory role of NF‐kappaB in autophagy‐like cell death after focal cerebral ischemia in mice. Neuroscience 244:16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Y, Chopp M (1999) Temporal profile of nestin expression after focal cerebral ischemia in adult rat. Brain Res 838:1–10. [DOI] [PubMed] [Google Scholar]
- 45. Li Y, Chopp M, Powers C (1997) Granule cell apoptosis and protein expression in hippocampal dentate gyrus after forebrain ischemia in the rat. J Neurol Sci 150:93–102. [DOI] [PubMed] [Google Scholar]
- 46. Little JR, Sundt TM Jr, Kerr FW (1974) Neuronal alterations in developing cortical infarction. An experimental study in monkeys. J Neurosurg 40:186–198. [DOI] [PubMed] [Google Scholar]
- 47. Liu CL, Siesjo BK, Hu BR (2004) Pathogenesis of hippocampal neuronal death after hypoxia‐ischemia changes during brain development. Neuroscience 127:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Majid A, He YY, Gidday JM, Kaplan SS, Gonzales ER, Park TS et al (2000) Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke 31:2707–2714. [DOI] [PubMed] [Google Scholar]
- 49. Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J et al (2003) Reperfusion differentially induces caspase‐3 activation in ischemic core and penumbra after stroke in immature brain. Stroke 34:207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, Chuai M et al (2008) Accumulation of macrophage‐like cells expressing NG2 proteoglycan and Iba1 in ischemic core of rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab 28:149–163. [DOI] [PubMed] [Google Scholar]
- 51. Mattson MP (2003) Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med 3:65–94. [DOI] [PubMed] [Google Scholar]
- 52. McCarthy CA, Vinh A, Broughton BR, Sobey CG, Callaway JK, Widdop RE (2012) Angiotensin II type 2 receptor stimulation initiated after stroke causes neuroprotection in conscious rats. Hypertension 60:1531–1537. [DOI] [PubMed] [Google Scholar]
- 53. Mennel HD, El‐Abhar H, Schilling M, Bausch J, Krieglstein J (2000) Morphology of tissue damage caused by permanent occlusion of middle cerebral artery in mice. Exp Toxicol Pathol 52:395–404. [DOI] [PubMed] [Google Scholar]
- 54. Mergenthaler P, Dirnagl U, Meisel A (2004) Pathophysiology of stroke: lessons from animal models. Metab Brain Dis 19:151–167. [DOI] [PubMed] [Google Scholar]
- 55. Moxon‐Emre I, Schlichter LC (2010) Evolution of inflammation and white matter injury in a model of transient focal ischemia. J Neuropathol Exp Neurol 69:1–15. [DOI] [PubMed] [Google Scholar]
- 56. Nagahiro S, Uno M, Sato K, Goto S, Morioka M, Ushio Y (1998) Pathophysiology and treatment of cerebral ischemia. J Med Invest 45:57–70. [PubMed] [Google Scholar]
- 57. Nakashima K, Yamashita K, Uesugi S, Ito H (1999) Temporal and spatial profile of apoptotic cell death in transient intracerebral mass lesion of the rat. J Neurotrauma 16:143–151. [DOI] [PubMed] [Google Scholar]
- 58. Nedergaard M (1988) Mechanisms of brain damage in focal cerebral ischemia. Acta Neurol Scand 77:81–101. [DOI] [PubMed] [Google Scholar]
- 59. Neumann‐Haefelin T, du Mesnil de Rochemont R, Fiebach JB, Gass A, Nolte C, Kucinski T et al (2004) Effect of incomplete (spontaneous and postthrombolytic) recanalization after middle cerebral artery occlusion: a magnetic resonance imaging study. Stroke 35:109–114. [DOI] [PubMed] [Google Scholar]
- 60. Niizuma K, Endo H, Chan PH (2009) Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem 109 Suppl 1:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ohab JJ, Fleming S, Blesch A, Carmichael ST (2006) A neurovascular niche for neurogenesis after stroke. J Neurosci 26:13007–13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Puyal J, Ginet V, Clarke PG (2013) Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol 105:24–48. [DOI] [PubMed] [Google Scholar]
- 63. Ramos‐Cejudo J, Gutierrez‐Fernandez M, Rodriguez‐Frutos B, Exposito Alcaide M, Sanchez‐Cabo F, Dopazo A, Diez‐Tejedor E (2012) Spatial and temporal gene expression differences in core and periinfarct areas in experimental stroke: a microarray analysis. PLoS One 7:e52121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sandu RE, Buga AM, Balseanu AT, Moldovan M, Popa‐Wagner A (2016) Twenty‐four hours hypothermia has temporary efficacy in reducing brain infarction and inflammation in aged rats. Neurobiol Aging 38:127–140. [DOI] [PubMed] [Google Scholar]
- 65. Sharp FR, Lu A, Tang Y, Millhorn DE (2000) Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab 20:1011–1032. [DOI] [PubMed] [Google Scholar]
- 66. Shin YJ, Kim HL, Park JM, Cho JM, Kim SY, Lee MY (2013) Characterization of nestin expression and vessel association in the ischemic core following focal cerebral ischemia in rats. Cell Tissue Res 351:383–395. [DOI] [PubMed] [Google Scholar]
- 67. Shin YJ, Park JM, Cho JM, Cha JH, Kim SY, Lee MY (2013) Induction of vascular endothelial growth factor receptor‐3 expression in perivascular cells of the ischemic core following focal cerebral ischemia in rats. Acta Histochem 115:170–177. [DOI] [PubMed] [Google Scholar]
- 68. Steinberg GK, Gelb AW, Lam AM, Manninen PH, Peerless SJ, Rassi Neto A, Floyd P (1986) Correlation between somatosensory evoked potentials and neuronal ischemic changes following middle cerebral artery occlusion. Stroke 17:1193–1197. [DOI] [PubMed] [Google Scholar]
- 69. Tomkins CE, Edwards SN, Tolkovsky AM (1994) Apoptosis is induced in post‐mitotic rat sympathetic neurons by arabinosides and topoisomerase II inhibitors in the presence of NGF. J Cell Sci 107:1499–1507. [DOI] [PubMed] [Google Scholar]
- 70. Wang LL, Chen D, Lee J, Gu X, Alaaeddine G, Li J et al (2014) Mobilization of endogenous bone marrow derived endothelial progenitor cells and therapeutic potential of parathyroid hormone after ischemic stroke in mice. PLoS One 9:e87284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wei L, Craven K, Erinjeri J, Liang GE, Bereczki D, Rovainen CM et al (1998) Local cerebral blood flow during the first hour following acute ligation of multiple arterioles in rat whisker barrel cortex. Neurobiol Dis 5:142–150. [DOI] [PubMed] [Google Scholar]
- 72. Wei L, Han BH, Li Y, Keogh CL, Holtzman DM, Yu SP (2006) Cell death mechanism and protective effect of erythropoietin after focal ischemia in the whisker‐barrel cortex of neonatal rats. J Pharmacol Exp Ther 317:109–116. [DOI] [PubMed] [Google Scholar]
- 73. Wei L, Ying DJ, Cui L, Langsdorf J, Yu SP (2004) Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res 1022:54–61. [DOI] [PubMed] [Google Scholar]
- 74. Wei N, Yu SP, Gu XH, Chen DD, Whalin MK, Xu GL et al (2013) The involvement of autophagy pathway in exaggerated ischemic brain damage in diabetic mice. CNS Neurosci Ther 19:753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Whitaker VR, Cui L, Miller S, Yu SP, Wei L (2007) Whisker stimulation enhances angiogenesis in the barrel cortex following focal ischemia in mice. J Cereb Blood Flow Metab 27:57–68. [DOI] [PubMed] [Google Scholar]
- 76. Xiao AY, Wei L, Xia S, Rothman S, Yu SP (2002) Ionic mechanism of ouabain‐induced concurrent apoptosis and necrosis in individual cultured cortical neurons. J Neurosci 22:1350–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zeng XJ, Yu SP, Zhang L, Wei L (2010) Neuroprotective effect of the endogenous neural peptide apelin in cultured mouse cortical neurons. Exp Cell Res 316:1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang Z, Chopp M, Powers C (1997) Temporal profile of microglial response following transient (2 h) middle cerebral artery occlusion. Brain Res 744:189–198. [DOI] [PubMed] [Google Scholar]