

Abstract
When peripheral axons are damaged, neuronal injury signaling pathways induce transcriptional changes that support axon regeneration and consequent functional recovery. The recent development of bioinformatics techniques has allowed for the identification of many of the regeneration-associated genes that are regulated by neural injury, yet it remains unclear how global changes in transcriptome are coordinated. In this article, we review recent studies on the epigenetic mechanisms orchestrating changes in gene expression in response to nerve injury. We highlight the importance of epigenetic mechanisms in discriminating efficient axon regeneration in the peripheral nervous system and very limited axon regrowth in the central nervous system and discuss the therapeutic potential of targeting epigenetic regulators to improve neural recovery.
Keywords: axon regeneration, epigenetic regulation, histone modification, nerve injury
INTRODUCTION
Injury to the nervous system can cause the permanent loss of a number of neural functions, leading to cognitive defects, memory loss, paraplegia, and peripheral paralysis. The main factor affecting the degree of clinical severity is the distinct morphology of a neuron. A neuron develops a single axon that transmits electrical signals from the cell body to the target tissue over long distances. A human axon can be up to a meter in length in the case of peripheral axons in the legs. Traumatic damage to axon fibers can disrupt the neuronal wire and prevent the transmission of signals from the corresponding cell body (Stoll et al., 2002). Likewise, toxin exposure, genetic mutation, and metabolic impairment can also be detrimental to axonal connectivity by hindering cargo transport along axons or dismantling cytoskeletal structures, finally leading to axon loss (Coleman, 2005; De Vos et al., 2008; Wang et al., 2002).
The recovery of damaged axonal connections can be achieved through the regrowth of transected axons back to the original target cells, a process known as axon regeneration. However, functional recovery following axon regeneration only occurs in limited situations (Abe and Cavalli, 2008; Chen et al., 2007; Yiu and He, 2006). While developmental axon outgrowth is able to reach remote target tissue, the growth capacity of axons reduces as an organism ages and its synapses mature (Cai et al., 2001; Geoffroy et al., 2016). Therefore, tissue (e.g., muscle and skin) often degenerate before peripheral axons regenerate and rebuild synapses to the target (Gordon et al., 2011; Kobayashi et al., 1997). More importantly, axons in the central nervous system (CNS) rarely regenerate beyond the site of the lesion after exposure to injury. Damage to the CNS, such as spinal cord injuries, traumatic brain injuries, stroke, and various neurological disorders, typically results in regeneration failure, meaning that the prognosis is often poor for these patients (Coleman and Freeman, 2010; Yiu and He, 2006). In this article, we review recent efforts to understand the mechanisms that affect the regeneration efficiency of neurons, with a special focus on the epigenetic regulation of axon regeneration.
EXTRINSIC AND INTRINSIC FACTORS COORDINATING AXONAL REGENERATION CAPACITY
The low regeneration capacity of CNS axons is highlighted by the example of dorsal root ganglion (DRG) neurons (Fig. 1). The pseudo-unipolar DRG neuron develops a peripheral branch axon to receive sensory information and a central axon to deliver the signal to the CNS, and these two axon branches exhibit contrasting degrees of regeneration after transection. Following sciatic nerve axotomy (SNA) to damage the peripheral branches in mice, regenerated axons can synapse onto muscles in the lower hind limbs within two weeks (Magill et al., 2007; Shin et al., 2012). In contrast, central branches fail to regrow in the spinal cord after dorsal column axotomy (DCA; Fig. 1). Because the central and peripheral axons arise from a single DRG neuron, the difference in their respective regeneration ability was initially attributed to differences in the substrate composition of the CNS and PNS. Indeed, inhibitory molecules such as Nogo-A, oligodendrocyte-myelin glycoprotein (OMgp), and myelin-associated glycoprotein (MAG) are expressed by myelin in the extra-axonal environment of the injured CNS tissue (Chen et al., 2000; Mukhopadhyay et al., 1994; Wang, 2002). In addition, glial cells reactivated by a lesion form glial scars, where chondroitin-sulfate proteoglycans (CSPGs) act as a major barrier to the growing axons (McKeon et al., 1991). In these inhibitory surroundings, axon tips fail to build dynamic growth cones and dysmorphic axon terminals are formed instead, with the axons subsequently retracting (Bradke et al., 2012; Ertürk et al., 2007).
Fig. 1. Different responses of the DRG neurons to dorsal column axotomy (DCA) and sciatic nerve axotomy (SNA).
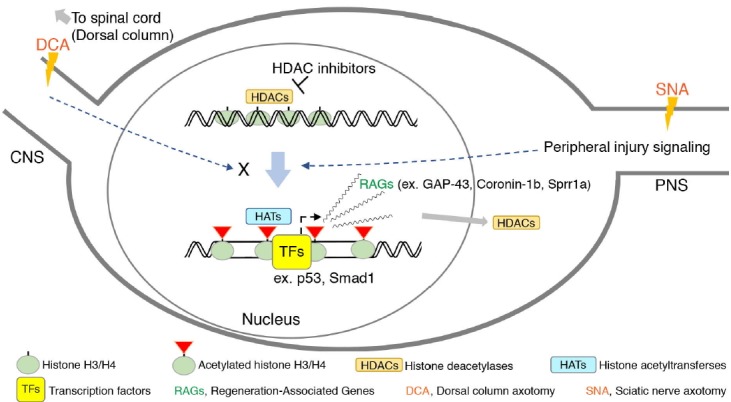
The axonal injury to the sciatic nerve induces specific signaling that activates the transcription of regeneration associated genes (RAGs) in the DRG neurons by harmonizing the function or activity of epigenetic regulators such as histone deacetylases (HDACs) and histone acetyltransferases (HATs), leading to a “regeneration program” being switched on. Transcription factors such as p53 and Smad1 can initiate gene transcription to promote regeneration after epigenetic changes take place. Most of the regulatory processes involved in epigenetic modulation fail to occur in the DRG neurons after DCA in the central nervous system. Due to this failure, an axonal injury in the central branch tends to be characterized by weak activation of the gene transcription required for a robust regeneration program, leading to regeneration failure in the central nervous system. The use of HDAC inhibitors in pharmacological applications to activate the epigenetic regulation of DRG neurons by enhancing the levels of histone acetylation has been reported. Green ellipse: histone complex. Green ellipse with a red triangle: post-translationally modified histone complex. HDACs: histone deacetylases. HATs: histone acetyltransferases. TFs: transcription factors. DCA: dorsal column axotomy. SNA: sciatic nerve axotomy. RAGs: regeneration-associated genes.
While these aforementioned extrinsic factors may influence regeneration ability, another important regulation mechanism involves neuron-intrinsic pathways (Abe and Cavalli, 2008; Liu et al., 2011). Interestingly, injured central axons in DRG neurons can regenerate in a hostile environment if the peripheral axons of the same neurons are also injured prior to the central lesion (Neumann and Woolf, 1999). This study supported the pioneering idea that damage to peripheral axons causes neurons to enter a pro-regenerative state, thereby improving axonal regrowth. Thus, a previous peripheral nerve lesion (known as a conditioning injury) can also improve regeneration ability in the PNS, as proven by the rapid regrowth of sciatic nerve fibers after two consecutive crush lesions (in vivo) and the enhanced neurite outgrowth in DRG neurons cultured from mice pre-injured by SNA (in vitro; Neumann et al., 2002; Pan et al., 2003; Shin et al., 2012). This conditioning injury effect indicates that there are neuron-intrinsic pathways that are induced by peripheral nerve lesions and that activate axon regeneration.
To date, many signaling pathways that convey axon injury signals have been identified, including the cAMP/PKA pathway, the PTEN/mTOR pathway, the gp130/Jak pathway, and the DLK/JNK pathway (Abe et al., 2010; Kenney and Kocsis, 1998; Neumann et al., 2002; Park et al., 2008; Qiu et al., 2002; Shin et al., 2012; Smith et al., 2009). Each of these pathways target a downstream transcription/translation regulator: CREB, ribosomal protein s6, STAT3, and c-Jun, respectively. Interrupting these transcriptional/translational pathways via genetic deletion or pharmacological intervention leads to the inhibition of peripheral axon regeneration and/or the conditioning injury effect (Bareyre et al., 2011; Gao et al., 2004; Raivich et al., 2004). These studies are consistent with an earlier report by Smith and Skene (1997), which showed that the application of a general transcription inhibitor, 5,6-dichlorobenzimidazole riboside (DRB), hinders the transition of neurite outgrowth from the slow “arborizing” stage to the fast “elongating” stage in adult DRG cultures from rats (Smith and Skene, 1997). Hence, the activation of axonal outgrowth is transcription-dependent.
Regeneration-associated genes (RAG) are those genes whose expression are affected by injury signaling. Traditionally, RAGs were identified using in situ hybridization or RT-PCR experiments on a specific gene of interest in a nerve injury model. For example, GAP-43 was found to be upregulated following the appearance of a peripheral lesion (Tetzlaff et al., 1991) and transgenic co-overexpression of GAP-43 together with CAP-23, another RAG, resulted in the improved regeneration of spinal cord axons in peripheral nerve grafts (Bomze et al., 2001). With the recent development of various genetic techniques including viral vectors and CRISPR/Cas9, the definition of RAGs has narrowed to include genes that are not only affected by injury but also functionally required in the regeneration process (Ma and Willis, 2015).
One of the biggest obstacles when studying RAGs in the hope that therapeutics targeting functionally important RAGs will improve recovery after injury is that a large number of genes are affected by a nerve injury. Genome-wide studies using microarrays and deep-sequencing analyses have identified numerous genes that are upregulated or downregulated following a neural injury, with many of them overlapping between independent datasets (Ben-Tov Perry et al., 2012; Gong et al., 2016; Li et al., 2015; Michaelevski et al., 2010). The simultaneous manipulation of various important RAGs could be achieved by targeting common upstream regulatory factors instead of individual genes. To this end, rapidly increasing the understanding of the epigenetic mechanisms regulating regeneration will provide a novel platform for enhancing functional outcomes.
HISTONE ACETYLTRANSFERASES IN AXON REGENERATION
Epigenetic mechanisms regulating axon regeneration involve the acetylation and methylation of histone proteins and the methylation of DNA and microRNAs that affect the expression of RAGs. Epigenetic studies on axon regeneration only became common over the past decade following the discovery that acetyltransferases are involved in the process. Di Giovanni et al. (2006) found that the acetylation of the tumor suppressor protein p53 plays an important role in the promotion of neurite outgrowth in cultured rat cortical neurons. p53 regulates the expression of the previously identified RAGs Coronin-1b and Rab1, and this mechanism is dependent on p53 acetylation at the K320 position, which is known to be mediated by the histone acetyltransferase (HAT) PCAF (Bode and Dong, 2004). These findings lead to the testable hypothesis that HATs play a central role in switching on global axonal regeneration by catalyzing the acetylation of histones and p53.
It turns out that the acetyltransferases p300/CBP and PCAF are involved in neurite outgrowth in both a p53-dependent and p53-independent manner (Gaub et al., 2010, 2011; Tedeschi et al., 2009). p300/CBP and PCAF alter the acetylation state of p53 at positions K373 and K320, respectively. More importantly, these acetyltransferases induce the modification of histone in chromatin structures following neural injury, illustrating the involvement of epigenetic factors in axon regeneration (Lee and Workman, 2007). In adult retinal ganglial neurons in the CNS, the levels of p300 and acetylated histone H3 at K18 are downregulated compared to those on postnatal day 7, suggesting that these changes may be a factor in reduced axon outgrowth in adults (Gaub et al., 2011). As the HAT responsible for H3-K18, an euchromatin marker, overexpressing the p300 enzyme using an adenoviral vector increases the levels of acetyl H3-K18 in the retinal ganglial cells and improves the regeneration of damaged axons in the optic nerve. p300 has a dual function during axonal outgrowth, directly binding to RAG promoters including GAP-43, Coronin-1b, and Sprr1a as well as recruiting acetyl H3-K18 to those promoters. In addition, PCAF mediates the acetylation of H3 at K9, which leads to its binding to the promoters of important RAGs including BDNF, Galanin, and GAP-43 (Puttagunta et al., 2014). Along the same lines, a loss-of-function mutation of PCAF in mice inhibits the effect of a conditioning injury (i.e., SNA) in promoting regeneration following DCA in the spinal cord. At the same time, overexpressing PCAF in spinal cord axons improves central axon regeneration, highlighting the functional importance of HAT in the regulation of axonal regrowth. Overall, open chromatin structures resulting from histone acetylation are associated with increased RAG expression and the promotion of axon regeneration.
HISTONE DEACETYLASES IN AXON REGENERATION
The level of histone acetylation is primarily regulated by the balance between HATs and histone deacetylases (HDACs). HDACs thus represent another important component of the epigenetic pathway that regulates regeneration by counteracting HAT activity (Cho et al., 2013; Gaub et al., 2010; Rivieccio et al., 2009). They can be categorized into several structural classes; HDACs from these classes (I, IIa and IIb, III and IV) are involved in the regenerative transcriptional response, suggesting that there might be independent mechanisms regulating HDAC activity following neural injury (Cho and Cavalli, 2014). Both inhibiting the expression of HDAC6 and pharmacologically blocking HDAC6 activity using selective inhibitors can improve neurite regrowth in rat cortical neurons on MAG-coated dishes or with the application of CSPG (Rivieccio et al., 2009). Notably, the positive effects of HDAC6 inhibition are limited to improvement in axon growth suppressed by the inhibitory circumstances; inhibiting HDAC6 in permissive conditions cannot enhance growth, which supports the belief that HDAC6 represses regenerative growth rather than governing general axon extension in vitro. HDAC6 is also upregulated under the oxidative stress generated by homocysteic acid (HCA), and inhibiting HDAC6 reduces cell death via injury. Therefore, HDAC6 seems to respond to broader forms of neural injury beyond axon transection.
HDACs generally work by maintaining a closed chromatin structure. Hence, the modulation of HDAC function by injury signaling is thought to inhibit HDAC activity, resulting in an increase in acetylated histone levels. Trichostatin A (TSA), a general class I/II HDAC inhibitor, and MS-275, a selective inhibitor for HDAC1 (a class I HDAC), similarly induce the binding of acetyl H4 to RAG promoter regions (Finelli et al., 2013). This binding rate increases following injury to the sciatic nerve (a 40% concordance between SNA and MS-275 treatment), suggesting that a peripheral lesion might elevate gene expression via inhibition of HDAC1. These inhibitors consequently improve neurite outgrowth in cultured murine DRG neurons. SNA also promotes the export of HDAC5, a class IIa HDAC, together with HDAC3, leading to the upregulation of acetyl histone H3 in the nuclei of injured DRG neurons (Cho et al., 2013). HDAC5 can efficiently transport in and out of the nucleus while its enzymatic activity is relatively low compared to other HDAC classes (Cho and Cavalli, 2014). It is thought that the nuclear export of HDAC5 induced by SNA drives the concomitant export of HDAC3, a class I HDAC which is responsible for the deacetylation of histones in the basal state. HDAC5 nuclear export is thus required for the regeneration of transected axons in the sciatic nerve (Cho et al., 2013). Cho et al.’s (2013) study also identifies genes whose expression are both injury- and HDAC5-dependent. Using cultured DRG neurons overexpressing a HDAC5 mutant that is forced to remain in the nucleus, microarray data demonstrate that transcriptome-level changes are indeed regulated via HDAC function following injury.
How then does HAT activity coordinate with HDAC localization/function following injury? A study on HDAC1 provides evidence that the presence of HDAC in RAG promoter regions is negatively correlated with the enrichment of p300 (Finelli et al., 2013). Gaub et al. (2011) also demonstrated that the pharmacological inhibition of class I/II HDACs by TSA increases the CBP- and PCAF-dependent acetylation of H3-K9-14 and the binding of acetylated H3 to the promoters of CBP/p300 and PCAF themselves, leading to the up-regulation of HAT expression levels (Gaub et al., 2010). As a result, the repression of HDAC activity by injury signaling appears not only to recruit the HAT complex to the upstream regulatory region of the RAGs (e. g. GAP-43 and Coronin-1b) but also to enlarge the pool of available HAT complexes, equipping the injured neuron with a readily accessible chromatin state.
SIGNALING MECHANISMS REGULATING HISTONE ACETYLATION IN INJURED NEURONS
The signaling pathways modulating HATs and HDACs are largely unknown for nerve injury. HATs and HDACs are mainly localized in the nucleus while axonal injury typically occurs far from the cell body (Rishal and Fainzilber, 2014). Therefore, how an axon lesion transmits stress signals to the molecules in the nucleus is puzzling. Following SNA, both the upregulation of PCAF and its binding to RAG promoters require ERK MAPK activity. Phosphorylated ERK levels are also elevated by SNA in DRG cell bodies, leading to the conclusion that injury-induced ERK signaling regulates PCAF activity (Puttagunta et al., 2014). Meanwhile, cytoplasmic relocalization of HDAC5 following SNA utilizes the rapid back-propagation of the calcium ion influx from the site of the injury to the cell body, a process which is dependent on the voltage-gated calcium channels and internal calcium stores (Cho et al., 2013). The calcium signaling-dependent phosphorylation of PKCμ is sufficient for its entry into the nucleus, and the subsequent phosphorylation of nuclear HDAC5 by active PKCμ leads to the translocation of phosphorylated HDAC5 to the cytoplasmic region. Because PKCμ is known to target other class IIa HDACs, such as HDAC4/7/9 (Wang, 2006), it is possible that the calcium wave generated after a peripheral injury activates the expression of pro-regenerative genes via the export of these additional class IIa HDACs. Further studies delineating the pathways that regulate histone acetylation-dependent responses in the regenerative transcriptome would improve the likelihood of developing therapeutics that could enhance the recovery process after a neural injury.
METHYLATION OF DNA AND HISTONES
The methylation of histone proteins or DNA itself is an important and frequently occurring component of epigenetic regulation in many biological processes (Jones, 2012). However, little is known about its contribution to regenerative gene expression after axonal injury. The relationship between chromatin methylation and axon regeneration as it is currently understood is rather descriptive and correlative. The genes upregulated after SNA (e.g., GAP-43, BDNF and Galanin) show a lower affinity for H3-K9me2 in their promoter sequences. Thus, H3-K9me2 and H3-K27me3 are classified as histone modifiers that repress gene expression after injury (Puttagunta et al., 2014). Nonetheless, the correlation between gene expression level and histone methylation level is weak because many of the genes examined in the study exhibit little concordance between methylation levels and changes in expression after injury to either PNS or CNS axons.
DNA methylation following neuronal injury has been analyzed in a bioinformatics study that allows for the further analysis of their dataset in a user-based platform (Lindner et al., 2014). This study presents a microarray dataset for gene promoters and CpG islands that were immunoprecipitated by 5-methylcytosine antibodies. However, a comparative analysis of differentially methylated genes in SNA versus DCA injury models has found only a slight correlation between gene expression and methylation. Therefore, the functional involvement of DNA methylation in regeneration is still an open question that needs further investigation.
EPIGENETIC FACTORS DIFFERENTIATING CNS AND PNS RESPONSES TO AXON INJURY
At the transcription level, the failure to switch on an effective regeneration program after CNS axon injury is indicated by the lack of change in the expression of RAGs. For example, Sprr1a, GAP-43 and Galanin are strongly upregulated by SNA but not by DCA (Puttagunta et al., 2014). Chandran et al. (2016) performed an extensive bioinformatics analysis on previously published microarray data to establish RAG co-expression modules following PNS injury, which was validated using eight independent PNS datasets. The co-expression modules exhibited weaker correlations in CNS datasets, demonstrating that the PNS expression network is not observed in the CNS transcriptome. Therefore, the difference in the regulation of transcription between PNS and CNS is not limited to a subset of RAGs. Instead, there are substantial PNS-to-CNS differences in injury-induced changes to gene expression.
Epigenetic responses to axon injury differ between PNS and CNS, suggesting that they may work as a major factor affecting axonal regeneration in these two systems. A peripheral lesion on the facial motor nerve induces CBP and acetylated p53 at K373, whereas these factors are inhibited by an optic nerve crush injury in the CNS (Gaub et al., 2011). Similarly, the phosphorylation of PKCμ and acetylated H3, which are the kinase and the downstream target of HDAC5, respectively, are induced in injured neurons after SNA but not by the crushing of the optic nerve (Cho et al., 2013). The DRG injury paradigm allows more direct comparisons between the effects of SNA and DCA, both of which damage the same set of neurons. Indeed, the upregulation of PCAF, acetyl H4 and acetyl H3-K9 are induced in DRG neurons by SNA but not by DCA (Fig. 1). Their affinity for the RAG promoter regions is also specifically elevated by SNA (Finelli et al., 2013; Puttagunta et al., 2014). Likewise, the activation of some transcription factors also display a SNA-specific pattern; the nuclear levels of phospho-Smad1 and Hif1α increase only following SNA but not a central lesion (Cho et al., 2015; Finelli et al., 2013). This growing body of evidence supports the hypothesis that the regulatory mechanisms activating epigenetic regenerative responses are not functional in the CNS, thus preventing the gene expression changes essential for robust axonal regrowth. The differences in the activation of the regulatory signals in the CNS and PNS has yet to be determined for each of the pathways involved (e.g., calcium and MAPK signaling pathways).
The next question is whether the activation of epigenetic regulation is sufficient for the successful regeneration of injured CNS axons. In rodents, the overexpression of p300 and the pharmacological inhibition of different HDACs can enhance peripheral regeneration after SNA and improve central regeneration after DCA (Cho et al., 2013; Finelli et al., 2013; Gaub et al., 2011). As a result, identifying selective pharmacological interventions that manipulate epigenetic modifiers in the axon regeneration pathway may accelerate the development of effective and safe therapeutics for neural recovery after injury.
CONCLUSION
Injury to axons can cause devastating neural impairment which is often permanent when the CNS is damaged. Currently, no therapy for the recovery of damaged neuronal connections exists because effective targets that can modulate or work in parallel with the neuron-intrinsic regeneration program remain to be identified. Epigenetic regulators integrate injury signals into global gene expression patterning, thereby providing a group of potential drug candidates that can interact with transcriptome to promote efficient axon regeneration.
ACKNOWLEDGMENTS
This research was supported by the Korea Research Fellowship to J.E.S. (2015H1D3A1066313) and NRF-2015R1A5A1009024 and NRF-2016R1C1B2006675 to Y.C. funded by the Ministry of Science, ICT and Future Planning through the National Research Foundation of Korea.
REFERENCES
- Abe N., Cavalli V. Nerve injury signaling. Curr Opin Neurobiol. 2008;18:276–283. doi: 10.1016/j.conb.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe N., Borson S.H., Gambello M.J., Wang F., Cavalli V. Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J Biol Chem. 2010;285:28034–28043. doi: 10.1074/jbc.M110.125336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareyre F.M., Garzorz N., Lang C., Misgeld T., Büning H., Kerschensteiner M. In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc Natl Acad Sci USA. 2011;108:6282–6287. doi: 10.1073/pnas.1015239108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Tov Perry R., Doron-Mandel E., Iavnilovitch E., Rishal I., Dagan S.Y., Tsoory M., Coppola G., McDonald M.K., Gomes C., Geschwind D.H., et al. Subcellular knockout of importin β1 perturbs axonal retrograde signaling. Neuron. 2012;75:294–305. doi: 10.1016/j.neuron.2012.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode A.G., Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- Bomze H.M., Bulsara K.R., Iskandar B.J., Caroni P., Skene J.H. Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat Neurosci. 2001;4:38–43. doi: 10.1038/82881. [DOI] [PubMed] [Google Scholar]
- Bradke F., Fawcett J.W., Spira M.E. Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat Rev Neurosci. 2012;13:183–193. doi: 10.1038/nrn3176. [DOI] [PubMed] [Google Scholar]
- Cai D., Qiu J., Cao Z., McAtee M., Bregman B.S., Filbin M.T. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21:4731–4739. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V., Coppola G., Nawabi H., Omura T., Versano R., Huebner Ea Zhang A., Costigan M., Yekkirala A., Barrett L., et al. A systems-level analysis of the peripheral nerve intrinsic axonal growth program. Neuron. 2016;89:956–970. doi: 10.1016/j.neuron.2016.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.S., Huber A.B., van der Haar M.E., Frank M., Schnell L., Spillmann A.A., Christ F., Schwab M.E. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434–439. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- Chen Z.-L., Yu W.-M., Strickland S. Peripheral regeneration. Annu Rev Neurosci. 2007;30:209–233. doi: 10.1146/annurev.neuro.30.051606.094337. [DOI] [PubMed] [Google Scholar]
- Cho Y., Cavalli V. HDAC signaling in neuronal development and axon regeneration. Curr Opin Neurobiol. 2014;27:118–126. doi: 10.1016/j.conb.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y., Sloutsky R., Naegle K.M., Cavalli V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell. 2013;155:894–908. doi: 10.1016/j.cell.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y., Shin J.E., Ewan E.E., Oh Y.M., Pita-Thomas W., Cavalli V. Activating injury-responsive genes with hypoxia enhances axon regeneration through neuronal HIF-1α. Neuron. 2015;88:720–734. doi: 10.1016/j.neuron.2015.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman M.P., Freeman M.R. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk A., Hellal F., Enes J., Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli M.J., Wong J.K., Zou H. Epigenetic regulation of sensory axon regeneration after spinal cord injury. J Neurosci. 2013;33:19664–19676. doi: 10.1523/JNEUROSCI.0589-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Deng K., Hou J., Bryson J.B., Barco A., Nikulina E., Spencer T., Mellado W., Kandel E.R., Filbin M.T. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron. 2004;44:609–621. doi: 10.1016/j.neuron.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Gaub P., Tedeschi a Puttagunta R., Nguyen T., Schmandke a., Di Giovanni S. HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 2010;17:1392–1408. doi: 10.1038/cdd.2009.216. [DOI] [PubMed] [Google Scholar]
- Gaub P., Joshi Y., Wuttke A., Naumann U., Schnichels S., Heiduschka P., Di Giovanni S. The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain. 2011;134:2134–2148. doi: 10.1093/brain/awr142. [DOI] [PubMed] [Google Scholar]
- Geoffroy C.G., Meves J.M., Zheng B. The age factor in axonal repair after spinal cord injury: A focus on neuron-intrinsic mechanisms. Neurosci Lett. 2016 doi: 10.1016/j.neulet.2016.11.003. doi.org/10.1016/j.neulet.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni S., Knights C.D., Rao M., Yakovlev A., Beers J., Catania J., Avantaggiati M.L., Faden A.I. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006;25:4084–4096. doi: 10.1038/sj.emboj.7601292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L., Wu J., Zhou S., Wang Y., Qin J., Yu B., Gu X., Yao C. Global analysis of transcriptome in dorsal root ganglia following peripheral nerve injury in rats. Biochem Biophys Res Commun. 2016;478:206–212. doi: 10.1016/j.bbrc.2016.07.067. [DOI] [PubMed] [Google Scholar]
- Gordon T., Tyreman N., Raji M.A. The basis for diminished functional recovery after delayed peripheral nerve repair. J Neurosci. 2011;31:5325–5334. doi: 10.1523/JNEUROSCI.6156-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Kenney a M., Kocsis J.D. Peripheral axotomy induces long-term c-Jun amino-terminal kinase-1 activation and activator protein-1 binding activity by c-Jun and junD in adult rat dorsal root ganglia In vivo. J Neurosci. 1998;18:1318–1328. doi: 10.1523/JNEUROSCI.18-04-01318.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi J., Mackinnon S.E., Watanabe O., Ball D.J., Gu X.M., Hunter D.A., Kuzon W.M., Jr The effect of duration of muscle denervation on functional recovery. Muscle Nerve. 1997;20:858–866. doi: 10.1002/(sici)1097-4598(199707)20:7<858::aid-mus10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Lee K.K., Workman J.L. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- Li S., Xue C., Yuan Y., Zhang R., Wang Y., Wang Y., Yu B., Liu J., Ding F., Yang Y., et al. The transcriptional landscape of dorsal root ganglia after sciatic nerve transection. Sci Rep. 2015;5:16888. doi: 10.1038/srep16888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner R., Puttagunta R., Nguyen T., Di Giovanni S. DNA methylation temporal profiling following peripheral versus central nervous system axotomy. Sci Data. 2014;1:140038. doi: 10.1038/sdata.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K., Tedeschi A., Park K.K., He Z. Neuronal intrinsic mechanisms of axon regeneration. Annu Rev Neurosci. 2011;34:131–152. doi: 10.1146/annurev-neuro-061010-113723. [DOI] [PubMed] [Google Scholar]
- Ma T.C., Willis D.E. What makes a RAG regeneration associated? Front Mol Neurosci. 2015;8:43. doi: 10.3389/fnmol.2015.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill C.K., Tong A., Kawamura D., Hayashi A., Hunter Da Parsadanian A., Mackinnon S.E., Myckatyn T.M. Reinnervation of the tibialis anterior following sciatic nerve crush injury: a confocal microscopic study in transgenic mice. Exp Neurol. 2007;207:64–74. doi: 10.1016/j.expneurol.2007.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon R.J., Schreiber R.C., Rudge J.S., Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelevski I., Segal-Ruder Y., Rozenbaum M., Medzihradszky K.F., Shalem O., Coppola G., Horn-Saban S., Ben-Yaakov K., Dagan S.Y., Rishal I., et al. Signaling to transcription networks in the neuronal retrograde injury response. Sci Signal. 2010;3:ra53. doi: 10.1126/scisignal.2000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay G., Doherty P., Walsh F.S., Crocker P.R., Filbin M.T. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–767. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- Neumann S., Woolf C.J. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/s0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Neumann S., Bradke F., Tessier-Lavigne M., Basbaum A.I. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–893. doi: 10.1016/s0896-6273(02)00702-x. [DOI] [PubMed] [Google Scholar]
- Pan Y.A., Misgeld T., Lichtman J.W., Sanes J.R. Effects of neurotoxic and neuroprotective agents on peripheral nerve regeneration assayed by time-lapse imaging in vivo. J Neurosci. 2003;23:11479–11488. doi: 10.1523/JNEUROSCI.23-36-11479.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K.K., Liu K., Hu Y., Smith P.D., Wang C., Cai B., Xu B., Connolly L., Kramvis I., Sahin M., et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttagunta R., Tedeschi A., Sória M.G., Hervera A., Lindner R., Rathore K.I., Gaub P., Joshi Y., Nguyen T., Schmandke A., et al. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat Commun. 2014;5:3527. doi: 10.1038/ncomms4527. [DOI] [PubMed] [Google Scholar]
- Qiu J., Cai D., Dai H., Mcatee M., Hoffman P.N., Bregman B.S., Filbin M.T. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/s0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- Raivich G., Bohatschek M., Da Costa C., Iwata O., Galiano M., Hristova M., Nateri A.S., Makwana M., Riera-Sans L., Wolfer D.P., et al. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 2004;43:57–67. doi: 10.1016/j.neuron.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Rishal I., Fainzilber M. Axon-soma communication in neuronal injury. Nat Rev Neurosci. 2014;15:32–42. doi: 10.1038/nrn3609. [DOI] [PubMed] [Google Scholar]
- Rivieccio M.A., Brochier C., Willis D.E., Walker B.A., D’Annibale M.A., McLaughlin K., Siddiq A., Kozikowski A.P., Jaffrey S.R., Twiss J.L., et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci USA. 2009;106:19599–19604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.E., Cho Y., Beirowski B., Milbrandt J., Cavalli V., DiAntonio A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74:1015–1022. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.S., Skene J.H. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P.D., Sun F., Park K.K., Cai B., Wang C., Kuwako K., Martinez-Carrasco I., Connolly L., He Z. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009;64:617–623. doi: 10.1016/j.neuron.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G., Jander S., Myers R.R. Degeneration and regeneration of the peripheral nervous system: from Augustus Waller’s observations to neuroinflammation. J Peripher Nerv Syst JPNS. 2002;7:13–27. doi: 10.1046/j.1529-8027.2002.02002.x. [DOI] [PubMed] [Google Scholar]
- Tedeschi A., Nguyen T., Puttagunta R., Gaub P., Di Giovanni S. A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ. 2009;16:543–554. doi: 10.1038/cdd.2008.175. [DOI] [PubMed] [Google Scholar]
- Tetzlaff W., Alexander S.W., Miller F.D., Bisby M.A. Response of facial and rubrospinal neurons to axotomy: changes in mRNA expression for cytoskeletal proteins and GAP-43. J Neurosci. 1991;11:2528–2544. doi: 10.1523/JNEUROSCI.11-08-02528.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos K.J., Grierson A.J., Ackerley S., Miller C.C.J. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Wang K.C. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. 2002;417:941–944. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- Wang Q.J. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol Sci. 2006;27:317–323. doi: 10.1016/j.tips.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Wang M.S., Davis Aa Culver D.G., Glass J.D. WldS mice are resistant to paclitaxel (taxol) neuropathy. Ann Neurol. 2002;52:442–447. doi: 10.1002/ana.10300. [DOI] [PubMed] [Google Scholar]
- Yiu G., He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]