

Abstract
Malignant gliomas are devastating tumours that frequently kill patients within 1 year of diagnosis. The major obstacle to a cure is diffuse invasion, which enables tumours to escape complete surgical resection and chemo- and radiation therapy. Gliomas use the same tortuous extracellular routes of migration that are travelled by immature neurons and stem cells, frequently using blood vessels as guides. They repurpose ion channels to dynamically adjust their cell volume to accommodate to narrow spaces and breach the blood-brain barrier through disruption of astrocytic endfeet, which envelop blood vessels. The unique biology of glioma invasion provides hitherto unexplored brain-specific therapeutic targets for this devastating disease.
Every year, more than 22,000 Americans are diagnosed with a malignant glioma. Current therapy for these primary brain cancers is inadequate, and approximately 95% of patients succumb to the disease within 5 years of diagnosis1. Treatment involves a three-pronged approach, which consists of maximal tolerable surgical resection followed by radiation and chemotherapy. Together, these typically add only months of additional survival. Major reasons for treatment failures include the challenge of delivering sufficient dosages of chemotherapeutics across the blood–brain barrier (BBB) and the diffuse invasion of tumour cells into the surrounding brain, which shelters them from surgery and radiation. Early radical surgical interventions attempted to remove the entire affected brain hemisphere only to witness recurrence from cells that had crossed into the other hemisphere2. Even now, in the era of modern microsurgical techniques, tumour recurrence is the norm, typically occurring within 1–2 cm of the original tumour border3. In light of the overall poor outcome from current therapies, a better understanding of glioma invasion is crucial for the future development of more effective interventions to contain this rapidly progressing disease. In this article, we review recent research, which offers hope that new strategies are emerging.
The past decade has witnessed important progress in our understanding of the genetic changes that characterize gliomas. Research from a consortium of scientists, The Cancer Genome Atlas Research Network, suggests that malignant gliomas comprise a genetically heterogeneous disease4, with core defects primarily in three signalling axes: the tyrosine kinase receptor pathway, the anti-apoptotic retinoblastoma pathway and the cell cycle regulatory (p53) axes. Not surprisingly, recent attention has focused on harnessing these molecular insights to tailor treatment to individual patients (for reviews, see REFS 5,6).
Another major advance has come from studies on the cells-of-origin from which gliomas derive. Gliomas may arise from adult neural stem cells or multipotent neural progenitor cells that persist in proliferative niches in the human CNS, namely the subventricular zone (SVZ) and the subgranular zone7 (for excellent recent reviews on this topic, see REFS 8,9). However, recent evidence indicates that the SVZ may be non-contributory in adults10. Gliomas may also arise from more differentiated lineages within the brain, including NG2 (neuron-glial antigen 2; also known as chondroitin sulphate proteoglycan 4 (CSPG4))-positive oligodendrocyte precursor cells11,12, astrocytes and even mature neurons13.
Despite their genetic differences, and possibly divergent cells-of-origin, all malignant gliomas share one conserved feature: aggressive invasiveness. Strikingly, unlike other high-grade solid cancers, malignant gliomas do not rely on intravascular or lymphatic metastasis to spread; instead, glioma cells actively migrate through the tortuous extracellular spaces of the brain, which leads to the formation of distant satellite tumours. Hence, they behave much more like non-malignant brain cells during embryonic development, or adult stem cells in the mature brain, which similarly migrate along extracellular routes, often exploiting the brain vasculature or, after injury, nerve bundles as guides (BOX 1). Other shared features — such as their responsiveness to neurotransmitters and neuropeptides, and their interactions with the extracellular matrix (ECM) and neighbouring cells — suggest that gliomas retain much of their neurobiological root. We review these brain-specific biological traits in greater detail here.
Box 1. Shared migratory traits between neural progenitor and glioma cells.
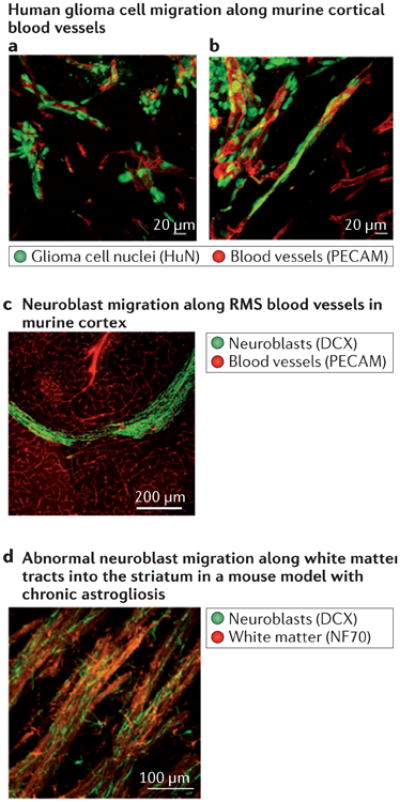
Migration along blood vessels and white matter tracts
Glioma cells actively seek out blood vessels41 and migrate along them82 (panel a of the figure). Human glioma cells (HuN) implanted intracranially into mice are shown invading along blood vessels (panel b of the figure). Indeed, the perivascular space is an important invasion pathway and is recognized as one of Scherer's secondary structures98 (BOX 2). Like glioma cells, neural progenitor cells or neuroblasts generated in the subependymal zone, one of the neurogenic zones still active in the adult brain of rodents, migrate over long distances. In rodents, progenitor cells migrate in tangentially orientated chains along the rostral migratory stream (RMS) into the olfactory bulb. The RMS contains a high density of parallel blood vessels that serve as a scaffold for neuroblast migration99100 (panel c of the figure). After injury, the brain seems to regain more plasticity, therefore subependymal and subgranular zone neurogenesis is increased and several cell types putatively migrate towards a lesion site in an attempt to restrict or repair tissue damage. Whereas astrocytes are usually stationary even after injury101, neuron-glial antigen 2 (NG2) cells — glial progenitor cells destined to form oligodendrocytes — readily migrate into lesion sites102,103. In addition, neuroblasts are recruited from the subependymal zone, and in contrast to NG2 cells, migrate over long distances to injury sites in the cortical grey or white matter using blood vessels or white matter tracts (panel d of the figure) as a substrate104.
Glutamate-mediated Ca2+ changes promotes motility
Gliomas assiduously release glutamate, which acts as a multifunctional growth factor that supports growth and invasion86. Glutamate acts as an autocrine and paracrine ligand to promote invasion by inducing oscillatory intracellular Ca2+ changes88 through activation of Ca2+-permeable AMPA receptors89. This mechanism is remarkably similar to that used by migratory neurons during the development of the cerebellum105, during which Ca2+ oscillations mediated by NMDA receptors guide granule cell migration.
Ca2+-activated K+ channels are essential for migration
One of the targets of these Ca2+ oscillations in migrating cells is the Ca2+-activated K+ channel KCa3.1. This channel is required for the migration of neuroblasts along the RMS106 but is equally utilized by invading gliomas50.
The similar biological traits of gliomas and neural stem cells or neural progenitor cells may provide further support for these being a likely cell-of-origin for gliomas. DCX, doublecortin; NF70, neuronal filament 70; PECAM, platelet endothelial cell adhesion molecule.
Gliomas rarely metastasize outside the brain
Malignant cancers spread in two phases. The first requires metastasis to another organ and typically occurs through haematogenous and lymphatic routes. The second phase involves local intra-organ invasion whereby cells infiltrate an organ to form a new tumour. Gliomas are exceedingly adept at infiltrating organs, but only 0.4–2% metastasize outside the brain14–16. This is in stark contrast to other solid cancers, including small-cell lung carcinoma, mammary ductal carcinoma, prostate cancer and colorectal cancers, which characteristically metastasize beyond the original organ. Several hypotheses have sought to explain the dearth of extracranial metastasis. First, although glioma cells associate with blood vessels, they may be unable to breach the basement membrane and enter the vasculature17. Second, the extraneural tissues may not contain the right milieu of growth factors that are capable of supporting glioma growth. Third, individuals with gliomas may not survive long enough for extracranial metastasis to become apparent. In support of this hypothesis, average postoperative survival time in patients with extracranial metastasis is 16–24 months18, which is longer than average. Although any or all of these explanations may be valid, gliomas are nevertheless exceedingly adept at intra-organ invasion, as are the neural precursor cells from which they are derived.
Where do glioma cells migrate?
The pathways through which glioma cells migrate can be roughly divided into two compartments: the perivascular space and the brain parenchyma. These spaces differ with regard to inherent mechanical and physical constraints. The perivascular space is fluid-filled, continuous with the subarachnoid space and surrounds all blood vessels, including penetrating arteries, arterioles and veins. The parenchyma contains neuronal and glial cell bodies and their processes, so extracellular spaces in the parenchyma are narrow and tortuous, and provide considerably greater physical resistance than the perivascular space. These compartments provide constitutive trails for glioma cell migration19.
Extracellular space and the importance of the ECM
Both the perivascular space and the interstitial spaces of the parenchyma contain a combination of ECM molecules, some of which constitute basement membranes, which separate functional domains. On the whole, the brain lacks the stiff fibrillar collagen matrix that is typical of other tissues, explaining its gelatinous consistency19. The interstitial spaces in the parenchyma are filled with a matrix mainly composed of proteoglycans (lectican family; also known as the hyalectan family) and their binding partners, hyaluronan and tenascins20. These water-binding ECM molecules are primarily produced by astrocytes and oligodendrocytes and form a gellike filling throughout the extracellular brain spaces. Many of these molecules — for example, neurocan and brevican (lectins), and phosphocan (a CSPG) — are brain-specific and particularly suited to support cell migration21. Tenascins form a family of CSPGs that bind to other ECM components and to cell surface receptors, most notably integrins. Interestingly, gliomas lay down their own pro-migratory ECM proteins and secrete brevican and tenascins to increase their invasiveness21–23. Tenascin C, which plays an important part in embryonic cell migration, is also produced by invading gliomas24. In addition, both tenascin C23 and tenascin W localize to blood vessels that are occupied by gliomas and are believed to stimulate vessel sprouting or angiogenesis25,26.
Basement membranes are specialized and highly organized self-aggregating sheets composed of ECM molecules that are 50–100 nm thick and surround blood vessels in all tissues. In the brain, parts of the vascular tree, including the meninges, large vessels and postcapillary venules, are encircled by two basement membranes, whereas only one composite basement membrane surrounds capillaries27,28. Basement membranes are rich in fibronectin and vitronectin, molecules that increase glioma cell motility29–31. The basement membrane functions as a physical and biochemical barrier that separates mesoderm-derived epithelial cells from ectoderm-derived neurons and glia. It is also an important component of the BBB, serving as a second physical barrier, and in addition signals from the basement membrane induce tight junction formation, which is necessary to maintain the BBB. Disruption of the basement membrane in pathological states has been linked to dysfunction of the BBB32.
Invading glioma cells interact with the ECM
Cell movement is an orchestrated biological process that requires a coordinated sequence of adhesion of the leading edge of a migrating cell, anchoring to the ECM, and last, detachment of the trailing end33. Actin–myosin molecular motors provide the main contractile force, and myosin II is specifically important in glioma invasion through narrow spaces34. Cell attachment is mediated by cell–cell and cell–matrix receptors, such as integrins, cadherins and neural cell adhesion molecules, whereas detachment requires the activity of proteases that also degrade ECM components, such as matrix metalloproteinases (MMPs)35–37.
Integrins are transmembrane receptors involved in cell–cell and cell–matrix interactions and enable cells to sense their environment and adjust their behaviour to environmental cues35. Integrins are heterodimers composed of one of 18 α- and one of 8 β-subunits, and the combination determines substrate specificity and signalling modality. Upregulation of the β1 subunit is associated with increased glioma invasion37,38, whereas αvβ3 and αvβ5 support tumour-induced angiogenesis37,39. The integrin αvβ5 can be selectively blocked with a synthetic peptide, cilengitide, which demonstrated modest anti-tumour activity in a Phase II trial40 but failed to increase overall survival in a large randomized Phase III trial.
Although the ECM molecules of the basement membrane are important for attachment of cell processes, the dense matrix that fills the extracellular space can be an obstacle for migrating glioma cells. To overcome this, glioma cells express a large number of secreted proteases, including the MMPs membrane type MMP1 (also known as MMP14), MMP2 and MMP9, the serine protease uPA and cell surface proteases, including ADAMs (disintegrin and metalloproteinases; also known as adamalysins). In addition, they recruit microglia, astrocytes and endothelial cells to also secrete proteases. The combined activity of these proteases remodels the ECM to favour tumour invasion while also regulating the activity of growth factors and chemokines that increase glioma proliferation and migration21,37. The use of MMP inhibitors to halt glioma invasion seems to be a promising therapeutic strategy, but clinical trials carried out thus far have not achieved clinical success.
Glioma cells migrate along the vasculature
As already described by Scherer (BOX 2), gliomas frequently populate and migrate along existing brain structures, including nerve tracts, blood vessels and the meninges. Whether they accidentally reach these structures or purposefully seek these out is important to understand. Our knowledge regarding this question is limited, in part, because it is difficult to dynamically assess cell migration within these structures. However, several biochemical and histological studies that use a combination of in vivo and ex vivo preparations are beginning to shed light on this question.
Box 2. A historical perspective: Scherer's structures.
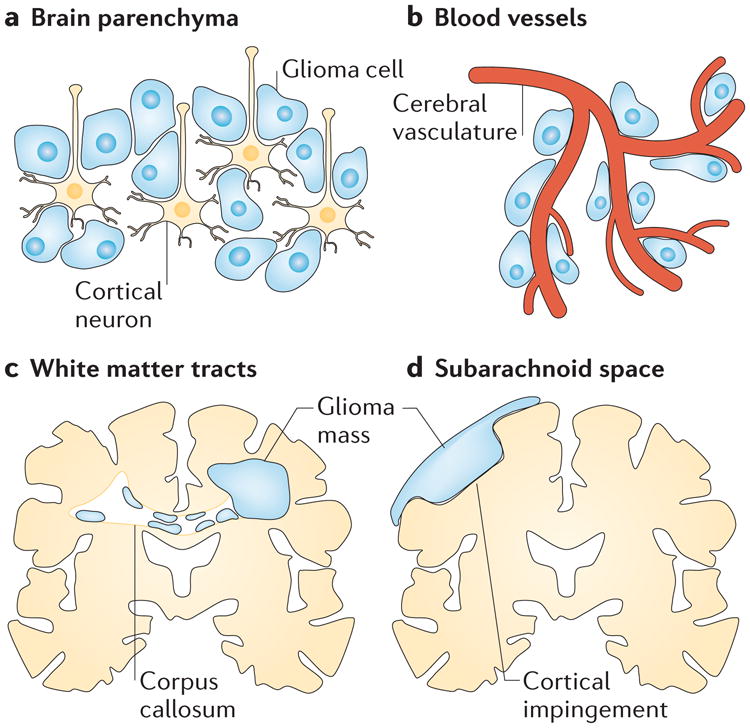
The extensive invasion of gliomas within the brain has fascinated pathologists and neurosurgeons for the past century. In 1938, Hans Joachim Scherer, a German neuropathologist, published a widely referenced manuscript in which he serially sectioned the brains of 100 patients with glioma and carefully examined the tumour and the surrounding brain98. He concluded that gliomas migrate along existing brain structures that he called ‘secondary structures’, which are formed by the interaction of glioma cells with the neural microenvironment. In appreciation of his pioneering work, these are now frequently called ‘Scherer's structures’ and include the following pathways of glioma cell invasion: the brain parenchyma (part a of the figure), pre-existing blood vessels (part b of the figure), white matter tracts (either perifascicularly intrafascicularly or interfibrillary) (part c of the figure) and the subarachnoid space below the meningeal covering of the brain (part d of the figure).
Importantly, Scherer suggested that invading glioma cells assume the physical shape of the structure they occupy rather than each cell having a characteristic form107. Hence, cells from the same tumour do not form a single type of Scherer's structure; instead, cells can appear very different in shape, depending on whether they migrate along blood vessels, nerve tracts or the subarachnoid space. This is an important observation that is of relevance to the recently discovered hydrodynamic mechanism of cell movement discussed in this Review.
These early observations were derived from static images, which do not permit examination of the dynamic changes that occur during the natural history of disease. As such, they leave several important questions regarding mechanisms of glioma cell migration unanswered. For example, is this migration a stochastic process, with cells randomly reaching these ‘secondary structures’, or do these structures actively attract gliomas and provide a distinct advantage for their invasion? Is it possible that these structures simply provide the path of least resistance? As discussed in this Review, glioma cells respond to a range of chemical cues, including growth factors, peptides, chemokines and extracellular matrix components, making it likely that they actively seek out a preferential microenvironment for invasion.
Blood vessels in particular are a critical substratum for glioma cell migration. In situ studies of human glioma cells demonstrate that when injected into the brain, the vast majority (>85%) of glioma cells move into contact with a blood vessel41 (FIG. 1). The recruitment of glioma cells to blood vessels occurs through bradykinin, which acts as a chemotactic signalling peptide41. Bradykinin is produced in vascular endothelial cells by enzymatic cleavage of high-molecular-weight kininogen by kallikrein. Binding of bradykinin to bradykinin 2 receptors (B2Rs) activates these G protein-coupled receptors (GPCRs) and causes inositol-1,4,5-trisphosphate receptor 3 (IP3R3)-dependent increases in intracellular Ca2+ concentration ([Ca2+]i) in glioma cells42. The resulting cyclic changes in [Ca2+]i activate downstream ion channels that support cell shape and volume changes that are necessary for cell invasion. Pharmacological inhibition or genetic elimination of B2Rs on gliomas impairs their homing onto blood vessels41. As a result, preclinical studies are evaluating the use of the bradykinin receptor inhibitor icatibant (Firazyr; Shire) as a treatment option. In the United States, icatibant was approved by the US Food and Drug Administration (FDA) in 2011 for the treatment of acute attacks of hereditary angioedema, an autosomal dominant disease that is characterized by unrestrained bradykinin activity. In rodent models of glioma, icatibant significantly reduced the percentage of glioma cells associated with blood vessels from 77% to 19%, which resulted in blunted glioma cell migration through the cerebral parenchyma and smaller tumour volumes41. Although bradykinin is probably not the only chemoattractant that guides glioma cells to blood vessels, it does seem to be the predominant molecule (responsible for approximately 75% of glioma cells on blood vessels). These data suggest that preventing the association of glioma cells with the vasculature could be an important therapeutic strategy to curtail glioma dispersion. Further clinical evaluation of this FDA-approved drug for the treatment of gliomas is warranted.
Figure 1. Glioma cells associate with blood vessels in the brain.
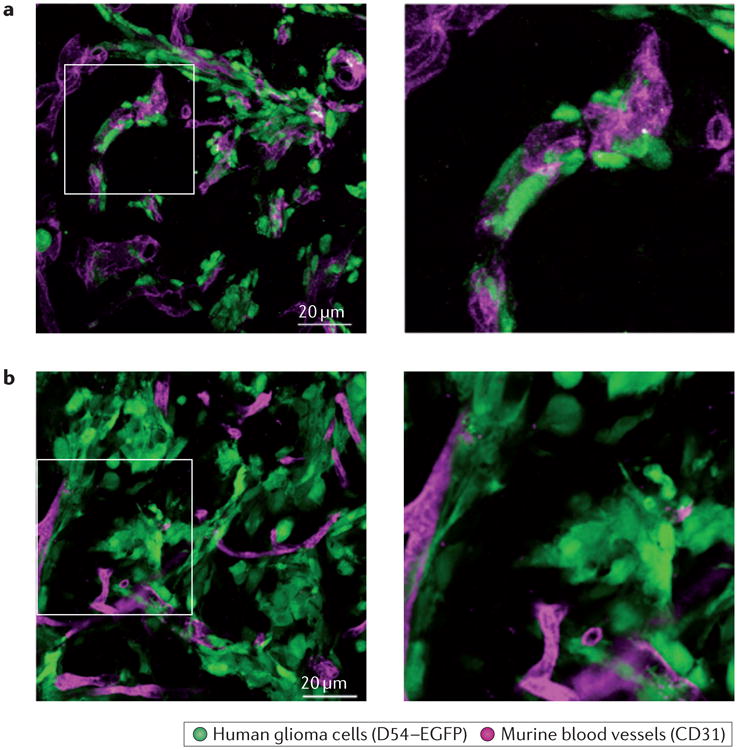
Most glioma cells that have migrated away from the main tumour mass into the brain parenchyma can be found on blood vessels. Examples of the patient-derived glioma line GBM22 (part a) and of the human glioma cell line D54 (part b) are shown, with high-magnification zooms of the boxed areas on the right. In both examples, glioma cells closely associate with blood vessels in the brain. Human glioma cells are labelled with enhanced green fluorescent protein (EGFP), and murine vessels are labelled with a CD31-specific antibody.
Located along larger brain vessels, the Virchow–Robin space is continuous with the subarachnoid space and is separated from the parenchyma by the basement membrane created by astrocytes along their endfeet. The cerebrospinal fluid (CSF) contained in this compartment comprises the glymphatic system43 in which the CSF is emptied into collecting veins along the meninges, leading to the clearance of parenchymal deposits such as amyloid. Hence, it stands to reason that for cells entering this space, convection towards the brain surface and into the subarachnoid space could influence cellular movement. The perivascular space provides little physical resistance to glioma migration. It thus serves as an accessible route for glioma invasion. Whether cells take advantage of the convective forces of the CSF or instead move through guided migration along the basement membrane remains to be seen.
How do glioma cells migrate?
Whether glioma cells migrate along the basement membrane of blood vessels, intraparenchymally or follow white matter tracts, they are challenged by the brain's limited extracellular space. Measurements in vivo in the murine neocortex suggest that the width of the undisturbed extracellular space in the brain is 38–64 nm44, which is vastly smaller than the size of migrating glioma cells. In light of these spatial constraints, it is remarkable that cells can move at all.
Hydrodynamic mode of cell invasion
Time-lapse imaging studies conducted in vivo and ex vivo show that invading glioma cells undergo remarkable shape and volume changes34,45,46. Using quantitative, three-dimensional imaging in vivo, these studies demonstrate that enhanced green fluorescent protein-expressing human gliomas implanted into mice show periodic changes in cellular volume (as much as ∼33%) as cells cycle between extension and retraction of processes46. Interestingly, volume changes of similar magnitude occurred as cells migrated across Transwell barriers over a range of barrier sizes (3–8 μm), suggesting that once a cell encounters a barrier it reduces its volume by the maximal extent possible. In glioma cells, this equates to about a 33% volume decrease and required the invading cells to shed essentially all ‘free cytoplasmic water’ that was not bound to proteins, nucleic acids and macromolecules46. Given that at least 50% of a mammalian cell is occupied by organelles, this decrease in volume by 33% is close to the theoretical limit.
Cytoplasmic water fluxes across the cell membrane through aquaporin 1 (AQP1) or AQP4 (REFS 47–49) and follows the osmotic force provided by ions that flux through channels. The emerging hydrodynamic model then suggests that gliomas repurpose Cl− and K+ channels (which are normally used to regulate the membrane potential) to regulate their shape and cell volume (FIG. 2). The underlying channels have been molecularly identified and belong to the ClC family of voltage-gated Cl− channels and the KCa family of Ca2+-activated K+ channels50.
Figure 2. A hydrodynamic model of glioma cell migration.

a | Glioma cells encounter spatial barriers (for example, other cells) as they migrate along blood vessels through the brain. Gliomas express various channels, including Cl− channel protein 3 (ClC3; a voltage-gated Cl− channel), Ca2+-activated K+ channel KCa3.1 and aquaporins. b | Upon stimulation of G protein-coupled receptors (GPCRs), there is a rise in intracellular Ca2+ concentration ([Ca2+]i) in glioma cells, leading to Ca2+-dependent activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII)-dependent ClC3 and KCa3.1 channel opening. This leads to the efflux of Cl− and K+, obligating water to flow down its osmotic gradient and leave the cell. This volume decrease enables glioma cells to decrease cytoplasmic volume and squeeze into small spaces. c | After passing a spatial barrier, glioma cells can regain volume through ion influx through the Na+–K+–Cl− cotransporter 1 (NKCC1) and acid-sensing ion channel 1 (ASIC1). BK, bradykinin.
The most interesting aspect of this model is that the energetic driving force for the movement of the cytosol comes from an unusual outwardly directed gradient for Cl−. In mature neurons, [Cl−]i is around 10 mM and is at electrochemical equilibrium; hence, Cl− will not move upon opening of ion channels. Glioma cells, by contrast, accumulate Cl−, which increases [Cl−]i to ∼100 mM, well above the equilibrium predicted by the resting potential51.
This is achieved by the sustained activity of the Na+–K+–Cl− cotransporter 1 (NKCC1; also known as SLC12A2), which imports Cl− against its concentration gradient by harnessing the transmembrane Na+ gradient. As a result of the high [Cl−]i, glioma cells are ‘primed’ to shrink their volume upon the opening of any Cl− channel in the membrane, and the insertion of a recombinant ligand-gated Cl− channel proved that Cl− efflux is indeed sufficient to achieve the volume decrease observed in in vivo experiments51. NKCC1 is constitutively expressed in gliomas, and this expression positively correlates with invasiveness52. Importantly, NKCC1 activity is regulated through phosphorylation by the protein kinase WNK3 (REF. 53), which is in turn regulated by the epidermal growth factor (EGF) through AKT (also known as protein kinase B)52. In addition to accumulating Cl−, NKCC1 can regulate migration speed through interaction with the actin cytoskeleton, thereby enhancing contractility as glioma cells migrate over a substratum52. These findings suggest that NKCC1 is a multimodal, anti-invasive target. Indeed, bumetanide (Bumex; Hoffmann-La Roche), an FDA-approved diuretic that inhibits NKCC1, significantly reduced glioma invasion in preclinical studies52,54.
ClC3 is in turn responsible for the efflux of Cl−, which is followed by the efflux of water; this enables glioma cells to adjust their shape as they move. Although voltage-gated, increases in [Ca2+]i are sufficient to activate ClC3 after Ca2+/calmodulin-dependent protein kinase II-dependent phosphorylation55. If these Cl− channels are inhibited by chlorotoxin (also known as TM-601), a 36-amino-acid peptide produced in the venom of the Israeli dessert scorpion (Leiurus quinquestriatus), glioma cell migration is reduced56,57. Importantly, not only does chlorotoxin block glioma cell migration through inhibition of ClC3 but it also specifically binds to human glioma cells and not to surrounding neural tissue56,58. Evaluation of intracavitary administration of chlorotoxin in a Phase I/II clinical trial (NCT00040573) in 18 adult patients demonstrated that the drug was well tolerated, with no significant toxicities, and bound specifically to the glioma for the entire 5-day monitoring period59. Because of the specific binding, chlorotoxin is also being used in several studies to delineate tumour tissue from surrounding normal brain tissue, both in whole-brain imaging to measure tumour extent and intraoperatively to facilitate resection of malignant tissue60,61. Development of similar inhibitors of other ion channels that are expressed by gliomas may be a worthy therapeutic strategy to restrict the spread of glioma cells.
To maintain electroneutrality, the ClC3-mediated Cl− flux must be coupled with a cation conductance. Glioma cells express multiple members of the KCa channel family, which are also activated upon increases in [Ca2+]i. In glioma cells, only KCa1.1 and KCa3.1 (also known as SK4 and the Gardos channel) form functional channels50,62–64. Both KCa1.1 and KCa3.1 have an important role in maintaining a cation conductance and in balancing Cl− efflux, and are crucially important for glioma cell migration, which can be induced by chemokines. Inhibition of either channel reduces glioma invasion in vitro and in vivo50,64–66. For KCa3.1, the specific inhibitor senicapoc has been evaluated for treatment of sickle cell disease and could be explored in future glioma trials.
Ligands increase migration by increasing Ca2+-dependent activation of ion channels
Both Cl− and K+ channels implicated in glioma invasion are sensitive to [Ca2+]i. Therefore, any [Ca2+]i increase results in a simultaneous increase in the conduction of Cl− and K+, which in turn causes the hypothesized volume changes.
Interestingly, many ligands that increase glioma cell migration also increase [Ca2+]i. For example, bradykinin, which is responsible for chemotactic attraction of gliomas to blood vessels (see above), raises [Ca2+]i in glioma cells through the GPCR B2R41,42. Cl− and K+ channels expressed by glioma cells are activated by bradykinin, and inhibition of these channels is sufficient to block bradykinin-induced migration and tumour expansion50,66. Specifically, gliomas express a splice isoform of the channel KCa1.1, which has increased sensitivity to Ca2+ (REFS 67,68). Given that KCa1.1 channels localize to the same lipid-raft microdomains as IP3R, the interaction between these proteins may play an important part in ligand-activated migration62. Aside from bradykinin, several other ligands, including lysophosphatidic acid (LPA), thrombin and sphingosine 1-phosphate, also bind to GPCRs, leading to IP3R-dependent [Ca2+]i increases, thereby converging on a pathway that potentiates glioma cell migration through ion channel activation42,69.
One of the most common groups of mutations in gliomas affects the gene encoding the EGF receptor (EGFR). In primary glioblastomas, ∼40% have EGFR amplifications, ∼60% have EGFR overexpression and ∼30% have EGFR mutations that result in a gain of function70. EGFR is a receptor tyrosine kinase that is activated by EGF and, in a similar manner to GPCRs, its activation leads to an IP3R-dependent increase in [Ca2+]i in human glioma cells42. EGF also leads to downstream activation of transient receptor potential canonical channel 1 (TRPC1), a non-selective cation channel that is permeable to Ca2+ (REF. 71). Ca2+ influx through TRPC1 activates glioma Cl− channels, which match the electrophysiological characteristics of ClC3, and inhibition of either significantly hampers migration71,72. The ability of glioma cells to respond to EGF is similar to that of other migratory cells in the adult brain73,74. These studies demonstrate that an important mechanism by which chemotactic ligands increase migration is by promoting Ca2+-dependent activation of ion channels.
Thus, a coordinated cascade that begins with ligand-induced activation of ion channels, and which leads to the movement of Cl− and K+ ions along with water, enables glioma cells to adjust their cell shape and volume to navigate the narrow extracellular spaces of the brain (FIG. 2).
Invading glioma cells disrupt brain function
In the healthy brain, astrocyte endfeet cover ∼99% of the vascular surface75,76, and the interaction between astrocytes and the vasculature is of functional importance for brain homeostasis, the coupling of blood flow and neuronal activity, and the BBB. The astrocyte–vascular interface is composed of endothelial cells forming the vascular walls, one or two basement membranes anchoring endothelial cells and astrocyte endfeet, which are also attached to the basement membrane. In addition, contractile cells are embedded between endothelial cells and astrocytes, either in direct contact with the vessel (pericytes at capillaries) or located between basement membranes (vascular smooth muscle cells at arterioles and arteries)77.
Many astrocytic functions depend on the polarized membrane domain that is typical of astrocyte endfeet78. This polarity is characterized by the enriched localization of several proteins in the endfoot. For example, the water channel AQP4 builds a complex with the inward rectifier K+ channel Kir4.1. Both are anchored into the membrane by the dystrophin-associated glycoprotein complex78,79 and are involved in K+ and water homeostasis80. Glucose transporters localize to endfeet, where they take up glucose, which serves as an energy substrate for astrocytes and nearby neurons after conversion to lactate81.
Disruption of astroglial function by abluminal glioma cell migration
When human glioma cells are implanted into the rodent brain, a large majority of glioma cells associate with blood vessels and move along their surface to invade the unaffected brain45,82,83. In this process, glioma cells surround blood vessels and seem to ‘lift up’ astrocytic processes as they degrade the perivascular basement membrane45,82,83. In response to glioma-induced displacement, astrocytes shift into a reactive phenotype and withdraw processes from the vasculature82,84. This perturbation of the physical interaction between vascular endothelial cells and astrocytes has two important consequences. First, it leads to a breakdown of the BBB and, second, it disrupts the neurovascular unit (FIGS 3,4). Although a leaky BBB is commonly observed in tumour masses in which newly generated vessels lack tight junction proteins, the focal breach of the BBB owing to local invasion is surprising and unexpected. Whether the displacement of astrocytic endfeet is responsible for a downregulation of tight junctional proteins such as claudin 5 and zonula occludens 1 (REF. 82) or whether it is the presence of the glioma cell per se remains to be shown. Also, whether this opening is transient or permanent is unknown. The consequence, however, is both troubling and potentially exciting. This focal breach enables motogens (thrombin, LPA and bradykinin, as discussed above), potentially toxic molecules and immune cells in the bloodstream to enter the brain, but it also may permit the targeted delivery of anti-invasive chemotherapeutic drugs to invading cells, which hitherto had been thought to be protected from chemotherapeutics by the BBB.
Figure 3. Glioma cells break down the blood–brain barrior.

a | In the healthy brain, astrocytic endfeet surround blood vessels. Vascular endothelial cells form the blood–brain barrier (BBB) though tight junctions, trapping cells and serum components in the vascular lumen. b | Glioma cells migrate along blood vessels, leading to displacement of astrocytic endfeet, degradation of tight junctions on endothelial cells and breakdown of the basement membrane surrounding blood vessels. In turn, serum components leak into the neural parenchyma.
Figure 4. Glioma cells displace astrocytic endfeet from blood vessels.
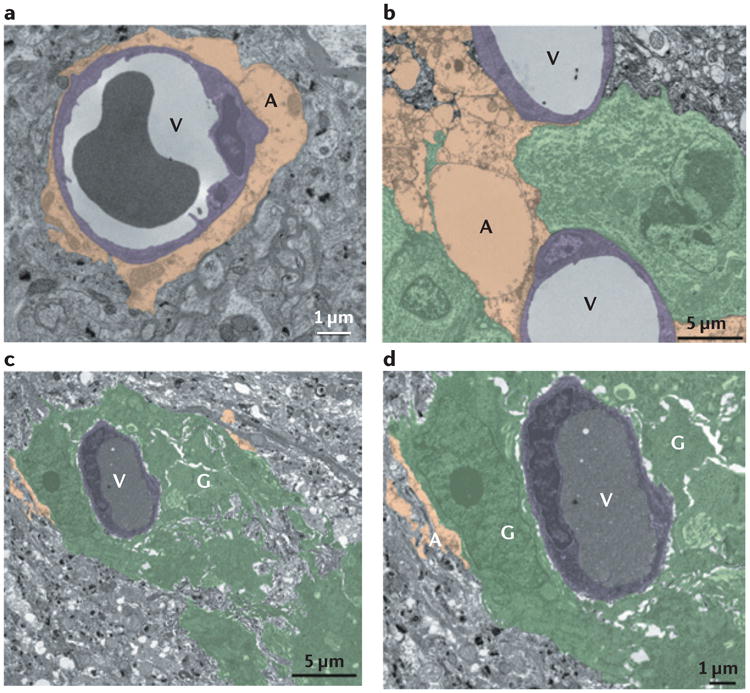
Electron micrographs of blood vessels (V) that are normally covered by astrocyte endfeet (A) (part a) can be partially (part b) or completely (parts c and d) ensheathed by patient-derived glioma cell lines GBM14 (part b) and GBM22 (parts c and d) glioma cells (G).
In addition, the displacement of astrocytic endfeet from the vessel wall abolished the functional regulation of vessel diameter by astrocytes. Astrocytes are responsible for a local activity-dependent adjustment of blood flow, whereby the astrocytes translate neuronal activity into vasodilation or vasoconstriction85. However, vessels encased by glioma cells were rendered unresponsive to astrocyte-derived vasoactive substances that regulate blood flow82. Interestingly, no harm seems to be done to the underlying smooth muscle, as vessels remain fully responsive to direct smooth muscle stimulation. Nonetheless, the absence of astrocyte-derived signals to the smooth muscle on tumour-ensheathed vessels may compromise adequate delivery of oxygen and glucose to neurons.
The biological advantage gained by glioma cells migrating along vessels and displacing astrocytic endfeet is probably multifactorial. We propose that glioma cells at the tumour border seek this interaction to facilitate proliferation and invasion while simultaneously disrupting astrocyte physiological functions that are crucial for brain homeostasis. Glioma cells probably seek the perivascular space to better position themselves to extract important nutrients from the bloodstream. While inhabiting this space, glioma cell-driven displacement of astrocytic endfeet can interrupt several astrocyte–vasculature processes. These include the astrocyte–neuron lactate shuttle, arteriolar cerebral blood flow regulation and BBB maintenance.
Glutamate in glioma biology
Glutamate has been shown to have many important roles in glioma biology: for example, acting as a multifaceted growth factor86, a space-vacating excitotoxin87 and a motogenic stimulant88,89. In the vicinity of a tumour, extracellular glutamate can reach neurotoxic concentrations90. Glutamate is produced by the tumour from glutamine and released through the system xc− cysteine–glutamate antiporter, which is highly expressed by human gliomas90–92. The sustained toxic increases in extracellular glutamate concentration lead to neuronal hyperexcitability and eventual death, and recent studies suggest that increased extracellular glutamate concentration is responsible for the tumour-associated seizures93,94 that occur in up to 80% of patients with primary brain tumours. With regards to glioma invasion, the extracellular release of glutamate through system xc− also has an autocrine effect, which promotes invasiveness. Specifically, glioma cells lack the GluR2 subunit of the AMPA receptor, which renders the channels permeable to Ca2+ (REFS 88,89). Thus, glutamate released from glioma cells activates these Ca2+-permeable AMPA channels, leading to oscillations in [Ca2+]i (REF. 88), which drive cell motility. When either system xc−-mediated glutamate release or Ca2+-permeable AMPA channels were inhibited, both Ca2+ oscillations and migration through spatial barriers were inhibited88. In addition, when AMPA channels were made impermeable to Ca2+, glioma cells invaded the brain less robustly, leading to smaller tumour volume89,95. Although it is clear that glutamate-dependent Ca2+ signalling is critical for the preservation of a pro-migratory phenotype, it is still unknown whether this Ca2+ signalling is necessary for system xc− -mediated glutamate release. Taken together, these data suggest that glutamate released by glioma cells not only has extrinsic effects on neuronal excitability but also intrinsically promotes glioma invasiveness in a Ca2+-dependent manner.
Glutamate release can be inhibited using sulfasalazine, an FDA-approved drug for the treatment of inflammatory bowel disease, through inhibition of system xc− (REFS 88,96). In rodent models of gliomas, sulfasalazine reduced the frequency of electrographic epileptic activity through inhibition of glutamate extrusion93,94. Sulfasalazine also decreased the autocrine pro-migratory effects of glutamate on glioma cells by interfering with Ca2+-permeable AMPA receptor-dependent cell invasion and restrained in vivo tumour spreading by 50% (REF. 88). Thus, sulfasalazine should be considered as a therapeutic agent for gliomas, both to reduce seizure activity and to control invasive growth in patients. However, a preliminary study of ten patients with recurrent gliomas suffering from advanced disease found that sulfasalazine administration lacked clinical efficacy97. Regrettably, this study was under-powered and performed in patients who were already in very poor health. Future studies should evaluate sulfasalazine as an adjuvant therapy to prevent tumour-associated seizures in patients who have not already progressed to end-stage disease, and one such study is underway (NCT01577966).
Conclusions and future challenges
Recent genomic analysis from The Cancer Genome Atlas Research Network and others illustrates a much broader molecular heterogeneity of malignant gliomas than previously appreciated4. This should have probably been expected in light of the multiple lineages from which gliomas can arise in the adult brain. However, the propensity of glioma cells to move and invade the brain via the same extracellular routes used by neural cells along existing structures such as blood vessels seems to be shared among all of the malignant gliomas. Despite being cancerous, numerous biological traits inherited from their neural ancestors remain active and important in the disease process. These biological traits and the interactions with neurons, other glia and endothelial cells have thus far not been exploited therapeutically, but these uniquely neural biological traits of gliomas are ripe for further exploration, and several of the aforementioned targets along with available drugs are summarized in TABLE 1. Gliomas notoriously develop drug resistance and/or upregulate compensatory pathways in response to monotherapies; hence, use of these agents as adjuvants or as a part of a drug cocktail may meet greater clinical success. Targeting ion channels and transporters that mediate hydrodynamic shape and volume changes, which enable cells to navigate the extracellular space of the brain efficiently, is proof of concept that anti-invasive strategies are possible. The finding that invading glioma cells focally breach the BBB suggests a role for early administration of specific anti-invasive drugs. Similarly, chemotaxis towards bradykinin could be exploited by blocking B2Rs using specific inhibitors, such as icatibant.
Table 1. Possible neural targets for clinical management of malignant gliomas.
| Protein | Normal function in nervous system |
Presumed mechanism in glioma biology |
Drug | Trial |
|---|---|---|---|---|
| MMP2, MMP3, MMP9 | Synaptic plasticity108; neuroblast migration along RMS109 | Digestion of extracellular matrix |
|
|
| Integrin | Neuroblast, oligodendrocyte, migroglial and astrocytic migration (for a review, see REF. 110); cell signalling | Cell–cell and cell–matrix contact | Cilengitide | Phase III |
| AQP1–AQP4 | Water and ion homeostasis80; production of CSF (for a review, see REF. 111) | Cell volume regulation | ||
| ClC3 | Regulation of LTP112 and GABAergic quantal size113 | Cell volume regulation | Chlorotoxin | Phase I/II |
| KCa1.1 | Repolarization after action potential114,115 | Volume change, pro-invasive | ||
| KCa3.1 | Neuroblast migration along RMS106 | Volume change, pro-invasive | Senicapoc | Phase II (sickle cell) |
| NKCC1 | Neuronal [Cl−]i regulation116; glial volume regulation117 | Cl− accumulation serves as energy for volume changes for invasion and proliferation | Bumetanide | FDA-approved diuretic |
| Bradykinin receptor 2 | Vasodilation | Blood vessel association, chemotaxis | Icatibant | Orphan drug status, Phase I (angioedema) |
| System xc− | Glial oxidative homeostasis (for a review, see REF. 118) | Autocrine glutamate signalling | Sulfasalazine | Phase 0 |
AQP, aquaporin; [Cl−]i, intracellular Cl− concentration; ClC3, Cl− channel protein 3; CSF, cerebrospinal fluid; FDA, US Food and Drug Administration; KCa, Ca2+-activated K+ channel; LTP, long-term potentiation; MMP, matrix metalloproteinase; NKCC1, Na+–K+–Cl− cotransporter 1; RMS, rostral migratory stream.
The vast majority of current clinical trials approach gliomas from a traditional oncological perspective, focusing on the tumour per se. Future therapies should be more neurocentric and consider the brain-specific interaction of gliomas with their host organ. Maintenance of the BBB could reduce leakage of serum components into parenchyma, thereby controlling oedema. Also, prevention of glutamate release through system xc− may decrease excitotoxic neuronal death, leading to less space for tumour expansion. For a sea-change in our treatment of gliomas to occur, novel aspects of glioma biology must be targeted.
Acknowledgments
This work was supported by US National Institutes of Health (NIH) research grants 2RO1-NS036692, 5RO1NS031234, 1RO1-NS082851 and 5RO1-NS052634 to H.S., V.A.C. (F31NS073181) and S.W. (F31NS074597) were supported by Ruth L. Kirschstein National Research Service Awards. S.R. received funding from the German Research Foundation (DFG), the Epilepsy Foundation and the American Brain Tumor Association (ABTA).
Footnotes
Competing interests statement: The authors declare no competing interests.
References
- 1.Central Brain Tumor Registry of the United States. CBTRUS Statistical report: primary brain and central nervous system tumors diagnosed in the United States2004–2006. CBTRUS. 2010 [online], http://www.cbtrus.org/2010-NPCR-SEER/CBTRUS-WEBREPORT-Final-3-2-10.pdf.
- 2.Dandy WE. Removal of right cerebral hemisphere for certain tumors with hemiplegia: preliminary report. JAMA. 1928;90:823–825. [Google Scholar]
- 3.Hou LC, Veeravagu A, Hsu AR, Tse VC. Recurrent glioblastoma multiforme: a review of natural history and management options. Neurosurg Focus. 2006;20:E5. doi: 10.3171/foc.2006.20.4.2. [DOI] [PubMed] [Google Scholar]
- 4.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. This paper shows that gliomas can be molecularly classified into four subtypes (pro-neural, neural, classical and mesenchymal) that respond differently to therapeutic intervention. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature Rev Cancer. 2010;10:319–331. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. 2012;149:36–47. doi: 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- 8.Stiles CD, Rowitch DH. Glioma stem cells: a midterm exam. Neuron. 2008;58:832–846. doi: 10.1016/j.neuron.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 9.Zong H, Verhaak RG, Canoll P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev Mol Diagn. 2012;12:383–394. doi: 10.1586/erm.12.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanai N, et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature. 2011;478:382–386. doi: 10.1038/nature10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugiarto S, et al. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20:328–340. doi: 10.1016/j.ccr.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu C, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedmann-Morvinski D, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338:1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beauchesne P. Extra-neural metastases of malignant gliomas: myth or reality? Cancers. 2011;3:461–477. doi: 10.3390/cancers3010461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamilton JD, et al. Glioblastoma multiforme metastasis outside the CNS: three case reports and possible mechanisms of escape. J Clin Oncol. 2014 doi: 10.1200/JCO.2013.48.7546. http://dx.doi.org/10.1200/JCO.2013.48.7546. [DOI] [PubMed]
- 16.Lun M, Lok E, Gautam S, Wu E, Wong ET. The natural history of extracranial metastasis from glioblastoma multiforme. J Neurooncol. 2011;105:261–273. doi: 10.1007/s11060-011-0575-8. [DOI] [PubMed] [Google Scholar]
- 17.Bernstein JJ, Woodard CA. Glioblastoma cells do not intravasate into blood vessels. Neurosurgery. 1995;36:124–132. doi: 10.1227/00006123-199501000-00016. [DOI] [PubMed] [Google Scholar]
- 18.Slowik F, Balogh I. Extracranial spreading of glioblastoma multiforme. Zentralbl Neurochir. 1980;41:57–68. [PubMed] [Google Scholar]
- 19.Gritsenko PG, Ilina O, Friedl P. Interstitial guidance of cancer invasion. J Pathol. 2012;226:185–199. doi: 10.1002/path.3031. [DOI] [PubMed] [Google Scholar]
- 20.Zimmermann DR, Dours-Zimmermann MT. Extracellular matrix of the central nervous system: from neglect to challenge. Histochem Cell Biol. 2008;130:635–653. doi: 10.1007/s00418-008-0485-9. [DOI] [PubMed] [Google Scholar]
- 21.Mentlein R, Hattermann K, Held-Feindt J. Lost in disruption: role of proteases in glioma invasion and progression. Biochim Biophys Acta. 2012;1825:178–185. doi: 10.1016/j.bbcan.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Kelly G, Zerillo C, Jaworski DM, Hockfield S. Expression of a cleaved brain-specific extracellular matrix protein mediates glioma cell invasion in vivo. J Neurosci. 1998;18:2370–2376. doi: 10.1523/JNEUROSCI.18-07-02370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brosicke N, van Landeghem FK, Scheffler B, Faissner A. Tenascin-C is expressed by human glioma in vivo and shows a strong association with tumor blood vessels. Cell Tissue Res. 2013;354:409–430. doi: 10.1007/s00441-013-1704-9. [DOI] [PubMed] [Google Scholar]
- 24.Joester A, Faissner A. The structure and function of tenascins in the nervous system. Matrix Biol. 2001;20:13–22. doi: 10.1016/s0945-053x(00)00136-0. [DOI] [PubMed] [Google Scholar]
- 25.Martina E, et al. Tenascin-W is a specific marker of glioma-associated blood vessels and stimulates angiogenesis in vitro. FASEB J. 2010;24:778–787. doi: 10.1096/fj.09-140491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alves TR, et al. Tenascin-C in the extracellular matrix promotes the selection of highly proliferative and tubulogenesis-defective endothelial cells. Exp Cell Res. 2011;317:2073–2085. doi: 10.1016/j.yexcr.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Sixt M, et al. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. 2001;153:933–946. doi: 10.1083/jcb.153.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207–217. doi: 10.1083/jcb.34.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nature Rev Cancer. 2003;3:422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 30.Fukushima Y, Tamura M, Nakagawa H, Itoh K. Induction of glioma cell migration by vitronectin in human serum and cerebrospinal fluid. J Neurosurg. 2007;107:578–585. doi: 10.3171/JNS-07/09/0578. [DOI] [PubMed] [Google Scholar]
- 31.Ohnishi T, et al. Fibronectin-mediated cell migration promotes glioma cell invasion through chemokinetic activity. Clin Exp Metastasis. 1997;15:538–546. doi: 10.1023/a:1018422926361. [DOI] [PubMed] [Google Scholar]
- 32.Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood–brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- 33.Ridley AJ, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 34.Beadle C, et al. The role of myosin II in glioma invasion of the brain. Mol Biol Cell. 2008;19:3357–3368. doi: 10.1091/mbc.E08-03-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolfenson H, Lavelin I, Geiger B. Dynamic regulation of the structure and functions of integrin adhesions. Dev Cell. 2013;24:447–458. doi: 10.1016/j.devcel.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70:217–228. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- 37.Kwiatkowska A, Symons M. Signaling determinants of glioma cell invasion. Adv Exp Med Biol. 2013;986:121–141. doi: 10.1007/978-94-007-4719-7_7. [DOI] [PubMed] [Google Scholar]
- 38.Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009;15:4589–4599. doi: 10.1158/1078-0432.CCR-09-0575. [DOI] [PubMed] [Google Scholar]
- 39.Scaringi C, Minniti G, Caporello P, Enrici RM. Integrin inhibitor cilengitide for the treatment of glioblastoma: a brief overview of current clinical results. Anticancer Res. 2012;32:4213–4223. [PubMed] [Google Scholar]
- 40.Reardon DA, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26:5610–5617. doi: 10.1200/JCO.2008.16.7510. [DOI] [PubMed] [Google Scholar]
- 41.Montana V, Sontheimer H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci. 2011;31:4858–4867. doi: 10.1523/JNEUROSCI.3825-10.2011. In this paper, bradykinin is shown to bind to the B2R on glioma cells, attracting the majority of glioma cells to blood vessels and increasing migration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang SS, et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010;70:1173–1183. doi: 10.1158/0008-5472.CAN-09-2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iliff JJ, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thorne RG, Nicholson C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci USA. 2006;103:5567–5572. doi: 10.1073/pnas.0509425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farin A, et al. Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia. 2006;53:799–808. doi: 10.1002/glia.20334. [DOI] [PubMed] [Google Scholar]
- 46.Watkins S, Sontheimer H. Hydrodynamic cellular volume changes enable glioma cell invasion. J Neurosci. 2011;31:17250–17259. doi: 10.1523/JNEUROSCI.3938-11.2011. This study shows that the cellular volume of human glioma cells migrating through cerebral parenchyma oscillates by 33%, enabling these cells to fit through narrow spaces in the brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy E, Sontheimer H. Expression and function of water channels (Aquaporins) in migrating malignant astrocytes. Glia. 2007;55:1034–1043. doi: 10.1002/glia.20524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding T, et al. Role of aquaporin-4 in the regulation of migration and invasion of human glioma cells. Int J Oncol. 2011;38:1521–1531. doi: 10.3892/ijo.2011.983. [DOI] [PubMed] [Google Scholar]
- 49.McCoy ES, Haas BR, Sontheimer H. Water permeability through aquaporin-4 is regulated by protein kinase C and becomes rate-limiting for glioma invasion. Neuroscience. 2009;168:971–981. doi: 10.1016/j.neuroscience.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cuddapah VA, Turner KL, Seifert S, Sontheimer H. Bradykinin-induced chemotaxis of human gliomas requires the activation of KCa3.1 and ClC-3. J Neurosci. 2013;33:1427–1440. doi: 10.1523/JNEUROSCI.3980-12.2013. A study showing that bradykinin promotes migration by activating ion channels that are involved in volume regulation in glioma cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Habela CW, Ernest NJ, Swindall AF, Sontheimer H. Chloride accumulation drives volume dynamics underlying cell proliferation and migration. J Neurophysiol. 2009;101:750–757. doi: 10.1152/jn.90840.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garzon-Muvdi T, et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 2012;10:e1001320. doi: 10.1371/journal.pbio.1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haas BR, et al. With-No-Lysine Kinase 3 (WNK3) stimulates glioma invasion by regulating cell volume. Am J Physiol Cell Physiol. 2011;301:C1150–C1160. doi: 10.1152/ajpcell.00203.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haas BR, Sontheimer H. Inhibition of the sodium-potassium-chloride cotransporter isoform-1 reduces glioma invasion. Cancer Res. 2010;70:5597–5606. doi: 10.1158/0008-5472.CAN-09-4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuddapah VA, Sontheimer H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J Biol Chem. 2010;285:11188–11196. doi: 10.1074/jbc.M109.097675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soroceanu L, Manning TJ, Jr, Sontheimer H. Modulation of glioma cell migration and invasion using Cl–and K+ ion channel blockers. J Neurosci. 1999;19:5942–5954. doi: 10.1523/JNEUROSCI.19-14-05942.1999. This study introduces a hydrodynamic model of cell invasion and shows that K+ and Cl− channels promote glioma cell invasion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lui VC, Lung SS, Pu JK, Hung KN, Leung GK. Invasion of human glioma cells is regulated by multiple chloride channels including ClC-3. Anticancer Res. 2010;30:4515–4524. [PubMed] [Google Scholar]
- 58.Soroceanu L, Gillespie Y, Khazaeli MB, Sontheimer H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998;58:4871–4879. [PubMed] [Google Scholar]
- 59.Mamelak AN, et al. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J Clin Oncol. 2006;24:3644–3650. doi: 10.1200/JCO.2005.05.4569. This Phase I trial is the first to demonstrate that a Cl− channel inhibitor (chlorotoxin) specifically binds to gliomas in patients. [DOI] [PubMed] [Google Scholar]
- 60.Hockaday DC, et al. Imaging glioma extent with 131I-TM-601. J Nucl Med. 2005;46:580–586. [PubMed] [Google Scholar]
- 61.Veiseh M, et al. Tumor paint: a chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007;67:6882–6888. doi: 10.1158/0008-5472.CAN-06-3948. [DOI] [PubMed] [Google Scholar]
- 62.Weaver AK, Bomben VC, Sontheimer H. Expression and function of calcium-activated potassium channels in human glioma cells. Glia. 2006;54:223–233. doi: 10.1002/glia.20364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McFerrin MB, Turner KL, Cuddapah VA, Sontheimer H. Differential role of IK and BK potassium channels as mediators of intrinsic and extrinsic apoptotic cell death. Am J Physiol Cell Physiol. 2012;303:C1070–C1078. doi: 10.1152/ajpcell.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.D'Alessandro G, et al. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis. 2013;4:e773. doi: 10.1038/cddis.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sciaccaluga M, et al. CXCL12-induced glioblastoma cell migration requires intermediate conductance Ca2+-activated K+ channel activity. Am J Physiol Cell Physiol. 2010;299:C175–C184. doi: 10.1152/ajpcell.00344.2009. [DOI] [PubMed] [Google Scholar]
- 66.Ransom CB, Sontheimer HBK. Channels in human glioma cells. J Neurophysiol. 2001;85:790–803. doi: 10.1152/jn.2001.85.2.790. [DOI] [PubMed] [Google Scholar]
- 67.Ransom CB, Liu X, Sontheimer H. BK channels in human glioma cells have enhanced calcium sensitivity. Glia. 2002;38:281–291. doi: 10.1002/glia.10064. [DOI] [PubMed] [Google Scholar]
- 68.Liu X, Chang Y, Reinhart PH, Sontheimer H, Chang Y. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J Neurosci. 2002;22:1840–1849. doi: 10.1523/JNEUROSCI.22-05-01840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manning TJ, Jr, Parker JC, Sontheimer H. Role of lysophosphatidic acid and Rho in glioma cell motility. Cell Motil Cytoskeleton. 2000;45:185–199. doi: 10.1002/(SICI)1097-0169(200003)45:3<185::AID-CM2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 70.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 71.Bomben VC, Turner KL, Barclay TT, Sontheimer H. Transient receptor potential canonical channels are essential for chemotactic migration of human malignant gliomas. J Cell Physiol. 2011;226:1879–1888. doi: 10.1002/jcp.22518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cuddapah VA, Turner KL, Sontheimer H. Calcium entry via TRPC1 channels activates chloride currents in human glioma cells. Cell Calcium. 2013;53:187–194. doi: 10.1016/j.ceca.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Caric D, et al. EGFRs mediate chemotactic migration in the developing telencephalon. Development. 2001;128:4203–4216. doi: 10.1242/dev.128.21.4203. [DOI] [PubMed] [Google Scholar]
- 74.Lindberg OR, Persson A, Brederlau A, Shabro A, Kuhn HG. EGF-induced expansion of migratory cells in the rostral migratory stream. PLoS ONE. 2012;7:e46380. doi: 10.1371/journal.pone.0046380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nature Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 76.Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. 2010;58:1094–1103. doi: 10.1002/glia.20990. [DOI] [PubMed] [Google Scholar]
- 77.Yousif LF, Di Russo J, Sorokin L. Laminin isoforms in endothelial and perivascular basement membranes. Cell Adh Migr. 2013;7:101–110. doi: 10.4161/cam.22680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335:75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- 79.Wolburg H, Noell S, Wolburg-Buchholz K, Mack A, Fallier-Becker P. Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood-brain barrier. Neuroscientist. 2009;15:180–193. doi: 10.1177/1073858408329509. [DOI] [PubMed] [Google Scholar]
- 80.Nagelhus EA, et al. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26:47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 81.Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. 1999;283:496–497. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- 82.Watkins S, et al. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nature Commun. doi: 10.1038/ncomms5196. (in the press). This recent study demonstrates that invading glioma cells disrupt physiological interactions between astrocytes and blood vessels, hijack control of vascular tone and break down the BBB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zagzag D, et al. Vascular apoptosis and involution in gliomas precede neovascularization: a novel concept for glioma growth and angiogenesis. Lab Invest. 2000;80:837–849. doi: 10.1038/labinvest.3780088. [DOI] [PubMed] [Google Scholar]
- 84.Nagano N, Sasaki H, Aoyagi M, Hirakawa K. Invasion of experimental rat brain tumor: early morphological changes following microinjection of C6 glioma cells. Acta Neuropathol. 1993;86:117–125. doi: 10.1007/BF00334878. [DOI] [PubMed] [Google Scholar]
- 85.Attwell D, et al. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.de Groot J, Sontheimer H. Glutamate and the biology of gliomas. Glia. 2010;59:1181–1189. doi: 10.1002/glia.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–4391. This study is the first to show that inhibition of glutamate release from glioma cells (and thus of the resulting glutamate excitotoxicity) decreases neurotoxicity. [PubMed] [Google Scholar]
- 88.Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67:9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ishiuchi S, et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nature Med. 2002;8:971–978. doi: 10.1038/nm746. This study shows that glutamate acts at Ca2+-permeable AMPA receptors in glioma cells to promote migration. [DOI] [PubMed] [Google Scholar]
- 90.Marcus HJ, Carpenter KL, Price SJ, Hutchinson PJ. In vivo assessment of high-grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J Neurooncol. 2010;97:11–23. doi: 10.1007/s11060-009-9990-5. [DOI] [PubMed] [Google Scholar]
- 91.Takeuchi S, et al. Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery. 2013;72:33–41. doi: 10.1227/NEU.0b013e318276b2de. [DOI] [PubMed] [Google Scholar]
- 92.Chung WJ, et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Buckingham SC, et al. Glutamate release by primary brain tumors induces epileptic activity. Nature Med. 2011;17:1269–1274. doi: 10.1038/nm.2453. A study showing that gliomas release glutamate through system xc−. This leads to epileptic activity, which can be inhibited by sulfasalazine, an FDA-approved drug. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Campbell SL, Buckingham SC, Sontheimer H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia. 2012;53:1360–1370. doi: 10.1111/j.1528-1167.2012.03557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ishiuchi S, et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci. 2007;27:7987–8001. doi: 10.1523/JNEUROSCI.2180-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chung WJ, Sontheimer H. Sulfasalazine inhibits the growth of primary brain tumors independent of nuclear factor-κB. J Neurochem. 2009;110:182–193. doi: 10.1111/j.1471-4159.2009.06129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Robe PA, et al. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer. 2009;9:372. doi: 10.1186/1471-2407-9-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scherer HJ. Structural development in gliomas. Am J Cancer. 1938;34:333–351. This visionary description by Scherer demonstrates the most common sites for glioma invasion, now known as Scherer's structures. [Google Scholar]
- 99.Bozoyan L, Khlghatyan J, Saghatelyan A. Astrocytes control the development of the migration-promoting vasculature scaffold in the postnatal brain via VEGF signaling. J Neurosci. 2012;32:1687–1704. doi: 10.1523/JNEUROSCI.5531-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Saghatelyan A. Role of blood vessels in the neuronal migration. Semin Cell Dev Biol. 2009;20:744–750. doi: 10.1016/j.semcdb.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 101.Bardehle S, et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nature Neurosci. 2013;16:580–586. doi: 10.1038/nn.3371. [DOI] [PubMed] [Google Scholar]
- 102.Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nature Neurosci. 2013;16:668–676. doi: 10.1038/nn.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jablonska B, et al. Chordin-induced lineage plasticity of adult SVZ neuroblasts after demyelination. Nature Neurosci. 2010;13:541–550. doi: 10.1038/nn.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cayre M, Canoll P, Goldman JE. Cell migration in the normal and pathological postnatal mammalian brain. Prog Neurobiol. 2009;88:41–63. doi: 10.1016/j.pneurobio.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Komuro H, Rakic P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron. 1996;17:275–285. doi: 10.1016/s0896-6273(00)80159-2. [DOI] [PubMed] [Google Scholar]
- 106.Turner KL, Sontheimer H. KCa3.1 modulates neuroblast migration along the rostral migratory stream (RMS) in vivo. Cereb Cortex. 2013 doi: 10.1093/cercor/bht090. http://dx.doi.org/10.1093/cercor/bht090. [DOI] [PMC free article] [PubMed]
- 107.Scherer HJ. A critical review: the pathology of cerebral gliomas. J Neurol Psychiatry. 1940;3:147–177. doi: 10.1136/jnnp.3.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meighan SE, et al. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J Neurochem. 2006;96:1227–1241. doi: 10.1111/j.1471-4159.2005.03565.x. [DOI] [PubMed] [Google Scholar]
- 109.Bovetti S, Bovolin P, Perroteau I, Puche AC. Subventricular zone-derived neuroblast migration to the olfactory bulb is modulated by matrix remodelling. Eur J Neurosci. 2007;25:2021–2033. doi: 10.1111/j.1460-9568.2007.05441.x. [DOI] [PubMed] [Google Scholar]
- 110.Clegg DO, Wingerd KL, Hikita ST, Tolhurst EC. Integrins in the development, function and dysfunction of the nervous system. Front Biosci. 2003;8:d723–d750. doi: 10.2741/1020. [DOI] [PubMed] [Google Scholar]
- 111.Venero JL, Vizuete ML, Machado A, Cano J. Aquaporins in the central nervous system. Prog Neurobiol. 2001;63:321–336. doi: 10.1016/s0301-0082(00)00035-6. [DOI] [PubMed] [Google Scholar]
- 112.Farmer LM, Le BN, Nelson DJ. CLC-3 chloride channels moderate long-term potentiation at Schaffer collateral-CA1 synapses. J Physiol. 2013;591:1001–1015. doi: 10.1113/jphysiol.2012.243485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Riazanski V, et al. Presynaptic CLC-3 determines quantal size of inhibitory transmission in the hippocampus. Nature Neurosci. 2011;14:487–494. doi: 10.1038/nn.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Du W, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nature Genet. 2005;37:733–738. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- 115.Brenner R, et al. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nature Neurosci. 2005;8:1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- 116.Yamada J, et al. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.MacVicar BA, Feighan D, Brown A, Ransom B. Intrinsic optical signals in the rat optic nerve: role for K+ uptake via NKCC1 and swelling of astrocytes. Glia. 2002;37:114–123. doi: 10.1002/glia.10023. [DOI] [PubMed] [Google Scholar]
- 118.McBean GJ. Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci. 2002;23:299–302. doi: 10.1016/s0165-6147(02)02060-6. [DOI] [PubMed] [Google Scholar]