

Abstract
Activity-based protein profiling (ABPP) has revolutionized the discovery and optimization of active-site ligands across distinct enzyme families, providing a robust platform for in-class selectivity profiling. Nonetheless, this approach is less straightforward for profiling reversible inhibitors and does not access proteins outside the ABPP probe’s target profile. While the active-site competitive acyl protein thioesterase 2 inhibitor ML349 (Ki = 120 nM) is highly selective within the serine hydrolase enzyme family, it could still interact with other cellular targets. Here we present a chemoproteomic workflow to enrich and profile candidate ML349-binding proteins. In human cell lysates, biotinylated-ML349 enriches a recurring set of proteins, including metabolite kinases and flavin-dependent oxidoreductases that are potentially enhanced by avidity-driven multimeric interactions. Confirmatory assays by native mass spectrometry and fluorescence polarization quickly rank-ordered these weak off-targets, providing justification to explore ligand interactions and stoichiometry beyond ABPP.
Keywords: Activity-based protein profiling, chemical proteomics, inhibitor selectivity, native mass spectrometry
Medicinal chemistry efforts primarily focus on achieving the highest potency ligands, which in the best-case scenario obviates any off-target engagement. In practice, many lead inhibitors have suboptimal potency, leaving open the possibility of off-target binding to other enzymes. In order to validate selectivity, medicinal chemistry campaigns typically assess inhibition against related enzymes that share the same catalytic mechanism. Particularly for kinases, hydrolases, and proteases, this workflow can include active-site competitive assays with reporter-linked covalent inhibitors.1 For example, fluorophosphonate probe labeling is occluded when an inhibitor covalently modifies or otherwise occupies an active site. Such competitive activity-based protein profiling (ABPP) methods enable evaluation of both potency and in-class selectivity either by in-gel fluorescence or affinity purification for mass spectrometry profiling.
Across the serine hydrolase enzyme family, ABPP methods have supported the development of several mechanism-based covalent scaffolds, including N-heterocyclic ureas,2 β-lactones,3 and activated carbamates.4−6 Since these scaffolds covalently modify their target enzymes, the competitive fluorophosphonate assay requires only enough probe and time to achieve complete labeling. Profiling reversible inhibitors is more complicated since each enzyme has a different rate of inactivation (Kinact), as well as different rates of inhibitor displacement (koff).7 Under such conditions, selectivity profiling is most representative of inhibition for enzymes with relatively slower Kinact constants, providing a kinetic window to profile target engagement. For example, the fluorophosphonate-TAMRA reacts extremely rapidly with acyl protein thioesterase 2 (APT2), which overwhelms much of the observable competition by the reversible active site inhibitor ML349.7 This can be addressed by tailoring the reactivity of the ABPP probe, such as using a less reactive N-heterocyclic urea. In this example, intraperitoneal injection of ML349 was chased with an alkynyl N-heterocyclic urea probe in living mice, allowing assessment of in vivo target engagement and partial in-class selectivity from isolated tissue homogenates.7
Due to these limitations, in-class selectivity profiling can also be carried out using reversible ligands linked to an affinity resin. Such affinity purification approaches have been used for decades for target enrichment and identification, particularly when coupled with quantitative shotgun proteomics methods. When adapted for on-bead competitive displacement assays, inhibitor selectivity can be quantitatively profiled for any enriched proteins. For example, a resin-linked kinase inhibitor cocktail (or kinome beads) allows multiplexed dose–response analysis of ligand selectivity across more than 200 enriched kinases.8 In addition, such native enrichment approaches also capture accessory proteins, or in some cases totally orthogonal enzyme classes. Using this approach, imatinib was found to bind the oxidoreductase NQO2 6-fold stronger than BCR-ABL, establishing unanticipated off-targets outside the targeted kinase family.8,9 More recently, cellular thermal shift assays (CETSA),10 drug affinity responsive target stability (DARTS),11 or CRISPR genome-wide approaches12 provide orthogonal routes to target discovery independent of affinity enrichment.
Here we evaluated the selectivity of the APT2 inhibitor ML349, comparing reported ABPP selectivity to traditional affinity-based target identification methods. APT2 is a widely expressed serine hydrolase with several reported substrates, including lysophospholipids,13 prostaglandin esters,14 and S-palmitoylated proteins.3 The active site competitive APT2 inhibitor ML349 was first identified from a high-throughput competitive activity-based fluorophosphonate assay in collaboration with the NIH Molecular Libraries Production Centers Network.16 Out of the 3 × 105 compounds tested, ML349 was identified as a promising competitive inhibitor with high selectivity across the serine hydrolase enzyme family. Even without further optimization, ML349 achieves target engagement and hydrolase selectivity in living mice.7,17
ML349 was later validated to modulate protein S-palmitoylation in cells. The S-palmitoylated tumor suppressor Scribble (Scrib) localizes to the plasma membrane in polarized epithelial cells to attenuate growth signals and promote contact inhibition.18 When polarized epithelial cells are induced to undergo epithelial-to-mesenchymal transition, Scrib relocates to the cytosol and is less S-palmitoylated.19 Treatment with submicromolar concentrations of ML349 increase the S-palmitoylation and membrane localization of Scrib, restoring plasma membrane localization to attenuate growth signals.19 As reported, ML349 only suppresses cell proliferation at significantly higher concentrations (100 × Ki), suggesting engagement of other potential off-target proteins.19 In the crystal structure of APT2 bound to ML349 (1.6 Å), ML349 binds to a hydrophobic channel extending across the enzyme active site, leaving only a solvent-exposed p-methoxy substituent to tether a reporter group.20 Since cell proliferation is only inhibited at concentrations well beyond concentrations required for APT2 inhibition, we set out to functionalize ML349 at the p-methoxy position for affinity-guided off-target identification.
Using an affinity purification strategy, we annotated a reproducible series of enriched targets by mass spectrometry. Some of these target proteins were individually profiled by native mass spectrometry, providing a direct assessment of ligand stoichiometry and affinity,21 particularly since several targets exist as homodimers. Confirmatory polarization assays with ML349-fluorescein ruled out other enriched targets, suggesting that resin avidity might enhance enrichment of otherwise low affinity targets. Overall, we report the target landscape of ML349, which includes APT2 and other weak cellular targets. Accordingly, while ABPP methods are streamlined for in-class selectivity of covalent inhibitors, classic ligand affinity purification methods access diverse cellular targets and circumvent potential false negatives missed in time-dependent competitive ABPP assays.
Previous structure–activity analysis demonstrated that interchanging the ML349 methoxy group from the para to ortho position abolished APT2 inhibition.7 Accordingly, we synthesized both p- and o-propargyl-ML349 derivatives and conjugated each to biotin-azide by copper catalyzed azide–alkyne cycloaddition (CuAAC) (Figure 1a). In a biochemical fluorogenic esterase assay, the p-substituted ML349-PEG3-biotin probe (ML349-biotin) lost approximately 9-fold affinity (Ki = 1100 nM), while the o-substituted probe (o-ML349-biotin) or the azide-PEG3-biotin linker alone were completely inactive (Figure 1b). Despite this reduction in affinity, we continued to explore if ML349-biotin could enrich APT2 or APT2-interacting proteins or reveal other protein targets. ML349-biotin was preassociated with streptavidin beads to decrease the sample processing time and limit dissociation of weak interacting proteins. The ML349-biotin/streptavidin resin was then incubated with HEK-293T whole cell lysate for 60 minutes and quickly transferred to spin columns for rapid washes. Since ML349 is a competitive inhibitor, incubation with fluorophosphonate-TAMRA displaces any ML349-bound serine hydrolases from the resin (Figure 2). By in-gel fluorescence, ML349-biotin exclusively enriches a ∼25 kDa hydrolase, corresponding to the molecular weight of APT2. Both o-ML349-biotin and unconjugated azide-PEG3-biotin did not enrich any hydrolases, providing two distinct controls to quantify ML349-binding proteins in cell lysates.
Figure 1.
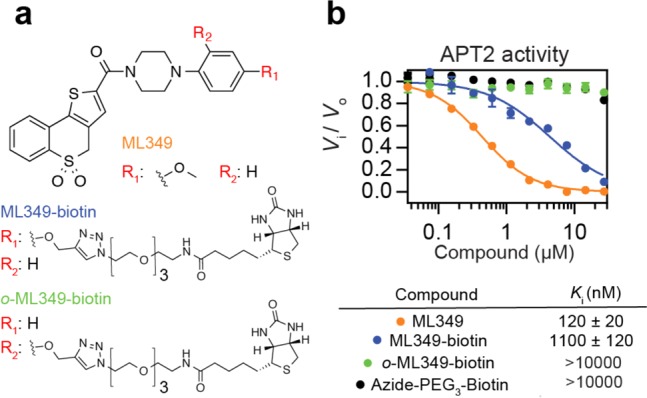
ML349 affinity probes inhibit APT2. (a) Chemical structures of ML349 and derivatives. (b) APT2 inhibition by ML349 and functionalized derivatives measured by resorufin acetate hydrolysis, reporting Ki values.
Figure 2.
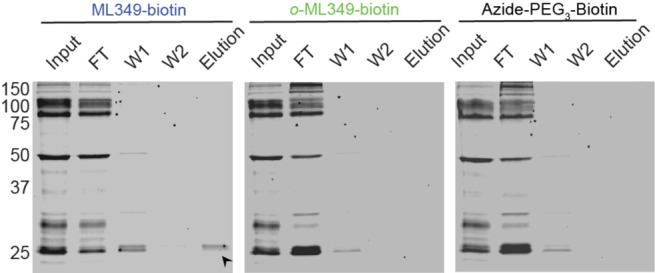
In-class selective APT2 enrichment by ML349-biotin. Elution of ML349-biotin enriched serine hydrolases from HEK-293T lysate and detection by FP-TAMRA in-gel fluorescence. A labeled band corresponding to the molecular weight of APT2 is highlighted with a black arrow. FT = flow through, W = wash.
Next, proteome-wide ML349-interacting proteins were eluted with excess ML349 and digested with trypsin for label-free mass spectrometry analysis. In our approach, we employed a data-independent acquisition workflow to maximize reproducibility, extracting precursor (MS1) peak area across replicates at high resolution (± 10 ppm), aligned retention times, and matched ion mobility drift times.22 In this method, once a peak is annotated in one data set, it can be cross-extracted to populate data across replicates. Each biological replicate was analyzed with three technical replicates for each probe, providing a dense data set for precise statistical analysis. In pairwise comparisons, ML349-biotin significantly enriched about 10 proteins (>5-fold) compared to o-ML349-biotin or azide-PEG3-biotin (Figure 3a,b and Tables S1 and S2). This list includes APT2 (LYPA2), as well as the metabolite kinases ADK, DCK, and PDXK, the oxidoreductase NQO2, and the flavin adenine dinucleotide synthase FAD1.
Figure 3.
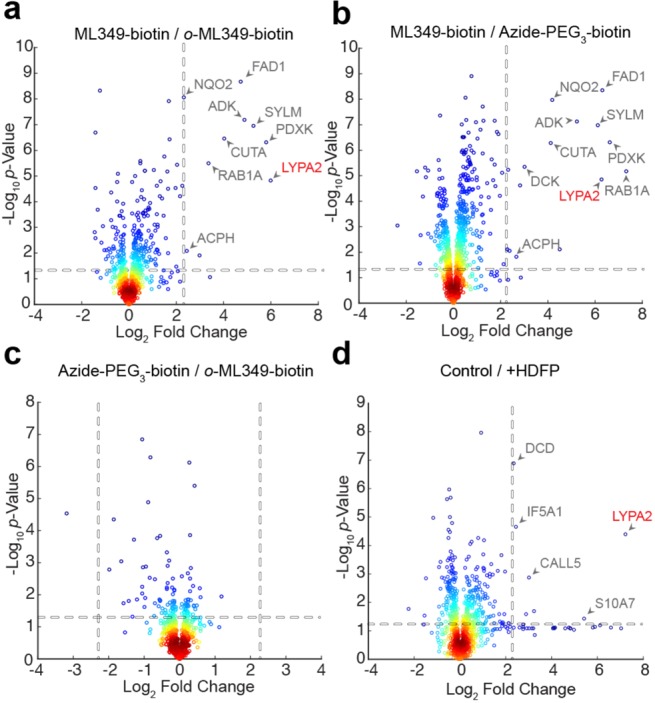
Label-free profiling of ML349-biotin enriched proteins. (a) ML349-biotin enrichment relative to o-ML349-biotin. (b) ML349-biotin enrichment relative to azide-PEG3-biotin. (c) o-ML349-biotin enrichment relative to azide-PEG3-biotin reports few o-ML349-biotin enriched proteins. (d) ML349-biotin enrichment with or without (control) APT2 covalent inhibition by HDFP. Protein identifiers (Uniprot accession code) are shown for significantly enriched proteins quantified by at least three peptides. Dotted lines indicate a minimum 5-fold change and p-value less than 0.05. Enriched binding proteins from HEK-293T cell lysates are shown.
Interestingly, NQO2 and ADK were both identified as binding proteins for active S-crizotinib stereoisomer,23 although the antiproliferative phenotype comes solely from inhibiting the oxidized nucleotide phosphatase MTH1. NQO2 is also inhibited by the BCR-ABL inhibitors imatinib (IC50 = 82 nM) and nilotinib (IC50 = 381 nM) in steady-state substrate assays.9 Additionally, the clinical PARP inhibitor niraparib inhibits DCK, preventing phosphorylation the chemotherapeutic nucleotide analogue cytarabine.24 Based on these reported examples, several ML349-binding proteins may be general targets of many drug-like molecules. Other enriched proteins include the small GTPase Rab1A, the secreted acetylcholinesterase binding protein CutA, and the mitochondrial tRNA ligase SYLM. Importantly, o-ML349-biotin showed only marginal ∼3-fold enrichment of NQO2 and FAD1 compared to azide-PEG3-biotin alone, confirming its use as an orthogonal control probe (Figure 3c and Table S3). Similar experiments were performed in MDCK cells lysates, where ML349 modulates Scrib S-palmitoylation.19 This analysis returned a similar profile of binding partners, recapitulating ML349-biotin enrichment of APT2, as well as multimeric nucleotide kinases and flavin-dependent oxidoreductases (Tables S5 and S6). Pretreatment with the generic lipase inhibitor hexadecylfluorophosphonate (HDFP)25 occludes the APT2 active site and blocks enrichment of APT2 (Figure 3d and Table S4). Since HDFP had no effect on ML349-binding protein enrichment, other enriched proteins are likely direct targets of ML349-biotin or ML349, and not APT2-interacting proteins.
In contrast to ABPP, affinity enrichment only returns proteins with some affinity for the ligand and does not report the fraction of enriched enzyme. Therefore, many of the enriched proteins could have marginal affinity for ML349-biotin, possibly influenced by the resin-enrichment strategy. On closer inspection, several of the enriched proteins from HEK-293T cells form oligomers, including NQO2 and PDXK. Based on this partial preference for oligomeric proteins, we wondered if avidity interactions contribute to the enrichment of weak ML349-interacting proteins.
Accordingly, we set out to validate and quantify ML349 binding to the recurring set of proteins. Recombinant APT2, NQO2, PDXK, and Rab1A were incubated with a fluorescein-conjugated ML349 to assay binding by fluorescence polarization.20 ML349-fluorescein (ML349-FL) (Figure S1a) interacted similarly with APT2 (Kd = 900 nM) and PDXK (Kd = 1.4 μM), but failed to interact with NQO2 or Rab1A in solution (Figure S1b). Either these interactions are disrupted by the conjugated fluorescein, or they derive some additional affinity from the PEG-biotin reporter group. In the competition experiments, ML349-FL was readily competed with ML349 for APT2 (IC50 = 1.1 μM) but showed marginal competition with PDXK (IC50 > 10 μM) (Figure S1c). Furthermore, ML349-biotin only weakly competed with ML349-FL for both APT2 and PDXK (IC50 > 10 μM). Therefore, unlike APT2, PDXK does not discriminate between ML349 and ML349-biotin.
Since the ML349-FL polarization experiments had limited target scope, we set out to evaluate ligand binding and stoichiometry by native mass spectrometry. Putative ML349-binding proteins were mixed in an equal ratio with ML349 and directly infused for electrospray ionization native mass spectrometry. Using nanoelectrospray ionization, proteins are transferred from native buffer into the gas-phase in a gentle fashion to maintain native-like conformations and noncovalent interactions with small molecules. The signals generated using this method have been shown to be reflective of solution-phase equilibria, providing a direct assay to profile protein–protein and protein–ligand interactions.26 Since native MS analysis reports on electrosprayed protein–ligand complexes, we predict some deviation from solution-phase affinity measurements due to high local concentration of ligands within electrospray droplets. Nonetheless, this assay provides a direct measure of relative binding stoichiometry. As a proof of principle, ML349 and ML349-biotin did not form significant amounts of complex with the homologous hydrolase APT1, but formed a stoichiometric complex with APT2 with no observable dimer formation (Figure 4a,b). In agreement with 9-fold increase in Ki, ML349-biotin displayed substoichiometric binding to APT2 (Figures 4a and S2). This approach provides a rapid assessment of the relation between homodimerization and the stoichiometry of ML349 or ML349-biotin association (Figures 4a and S2). As expected, Rab1A was observed solely as a monomer, yet did not associate with ML349. PDXK and NQO2 were detected as homodimers, yet with sub-stoichiometrtic association with ML349 and ML349-biotin. NQO2 can associate with two ML349 ligands per homodimer, yet ML349-biotin preferentially binds only one site per homodimer. Conversely, PDXK binds only one ML349 or ML349-biotin per dimer. Therefore, in contrast to NQO2, PDXK enrichment is unlikely biased by avidity effects. Nonetheless, on-resin interactions are likely quite different than those in solution or in the gas-phase. Overall, ML349 demonstrates significantly weaker affinity for Rab1A, NQO2, and PDXK, suggesting low-micromolar concentrations of ML349 achieve selective APT2 engagement in cells.
Figure 4.
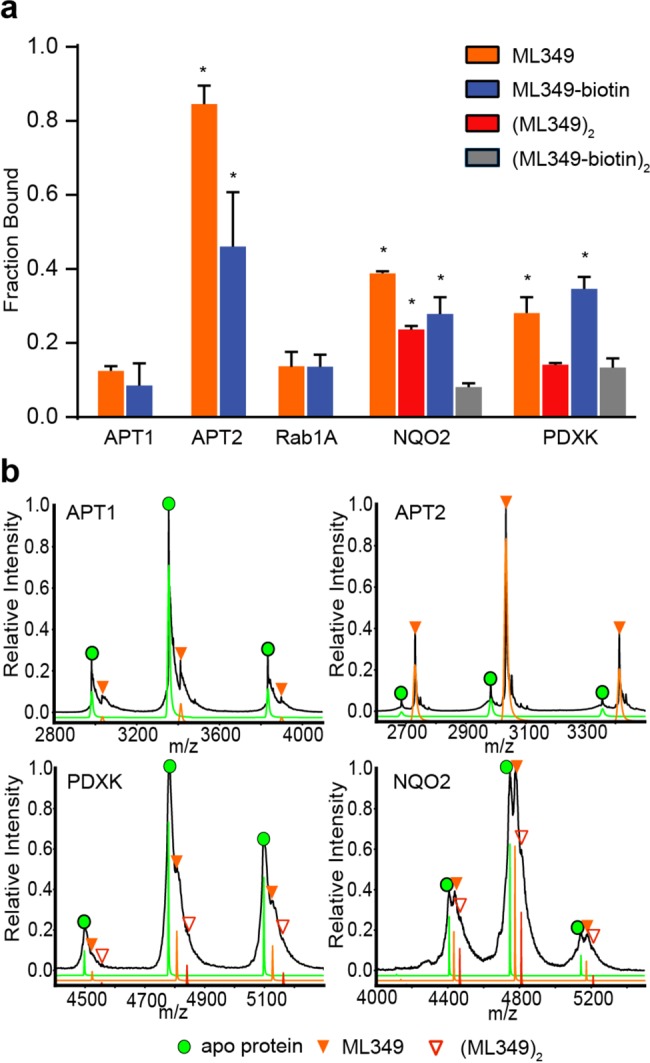
Native mass spectrometry profiling of inhibitor engagement and stoichiometry. (a) Graphical distribution of ligand–protein complexes derived from native mass spectrometry analysis. NQO2 and PDXK formed observable dimers, forming two potential ligand binding sites per complex. Red bars represent occupancy at both binding sites, representing two ML349 molecules per dimer. Gray bars represent complete occupancy of ML349-biotin in NQO2 and PDXK. Asterisks (*) indicate a p-value less than 0.05, as compared to the nonspecific binding to APT1 (n = 3, standard deviation). (b) Representative mass/charge spectra with three charge states used for quantitation of proteins binding to ML349 before (black trace) and after deconvolution (green = apo protein, orange = ML349 bound protein, red = 2xML349 bound protein) are shown for each protein.
As chemical proteomics methods become widely adopted, it is increasingly recognized that small molecule inhibitors often have multiple unanticipated binding partners in cells. In contrast to biochemical affinity-based methods, photoaffinity groups provide another sensitive approach to identify ligand targets in living cells.27,28 This approach retrieves a snapshot of live-cell engagement of putative interacting proteins, although quantifying the interaction affinity requires titrations, competitions, or additional biochemical validation. This becomes even more challenging for well-validated inhibitors like (+)-JQ1, where photoaffinity profiling identified an additional >100 additional cellular binding partners.29 Clearly ligand space for many inhibitors is broader than previously envisioned. Other emerging approaches that leverage thermal stabilization may also miss many relevant interactions when the target is not sufficiently stabilized. Overall, various target identification methods each have different caveats, and selectivity from one assay can present different targets from another assay. ML349 is highly selective by competitive fluorophosphonate ABPP, yet still weakly binds several hydrolases by ML349-biotin enrichment. Therefore, affinity-enrichment is likely more sensitive for initial target discovery, but does not provide information on the relative level of target occupancy. Importantly, many of the ML349-enriched targets are common to other chemoproteomic analyses, implying that many drug-like molecules share common promiscuous protein targets. Future efforts combining different target enrichment strategies will provide the most robust approach for understanding ligand selectivity across the proteome and provide new starting points for lead discovery across putative off-targets.
In summary, we present a detailed profile of the proteome-wide target landscape of the reversible APT2 inhibitor ML349. While fluorophosphonate ABPP was critical for the initial discovery and characterization of ML349, we now demonstrate that narrow in-class ABPP can overlook the extent of ligand targets outside of the tailored enzyme family. In this example, ML349-biotin/streptavidin resin enriches about a dozen targets outside of the serine hydrolase enzyme family, although confirmatory experiments suggest many of these may reflect multivalent interactions and are unlikely valid cellular targets at working inhibitor concentrations. Nonetheless, these efforts present a new affinity probe for profiling ligand engagement across this series of targets, providing a path to future competitive biochemical screens for proteins beyond the serine hydrolase enzyme family.
Acknowledgments
We would like to thank Dahvid Davda for helpful advice on interpretation of kinetic data.
Glossary
ABBREVIATIONS
- ABPP
activity-based protein profiling
- APT
acyl protein thioesterase
- FP
fluorophosphonate
- TAMRA
tetramethyl-6-carboxyrhodamine
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00441.
Author Contributions
S.J.W. and F.S.C. synthesized compounds. S.J.W. performed affinity purification proteomic experiments. S.J.W. and J.D.M. processed and analyzed LC–MS data. S.J.W. purified recombinant proteins. S.J.W. and S.Y.H. performed enzyme assays. J.D.E. and B.T.R. performed native mass spectrometry and interpreted the data. S.J.W. and B.R.M. designed experiments and wrote the paper.
Financial support for these studies was provided by the National Institutes of Health R00 CA151460, DP2 GM114848, the American Heart Association 14POST20420040 (J.D.M.), and the University of Michigan.
The authors declare no competing financial interest.
Supplementary Material
References
- Niphakis M. J.; Cravatt B. F. Enzyme Inhibitor Discovery by Activity-Based Protein Profiling. Annu. Rev. Biochem. 2014, 83, 341–377. 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- Adibekian A.; Martin B. R.; Wang C.; Hsu K. L.; Bachovchin D. A.; Niessen S.; Hoover H.; Cravatt B. F. Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat. Chem. Biol. 2011, 7, 469–478. 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker F. J.; Rocks O.; Vartak N.; Menninger S.; Hedberg C.; Balamurugan R.; Wetzel S.; Renner S.; Gerauer M.; Scholermann B.; Rusch M.; Kramer J. W.; Rauh D.; Coates G. W.; Brunsveld L.; Bastiaens P. I.; Waldmann H. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat. Chem. Biol. 2010, 6, 449–456. 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- Li W.; Blankman J. L.; Cravatt B. F. A Functional Proteomic Strategy to Discover Inhibitors for Uncharacterized Hydrolases. J. Am. Chem. Soc. 2007, 129, 9594–9595. 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- Cognetta A. B. 3rd; Niphakis M. J.; Lee H. C.; Martini M. L.; Hulce J. J.; Cravatt B. F. Selective N-Hydroxyhydantoin Carbamate Inhibitors of Mammalian Serine Hydrolases. Chem. Biol. 2015, 22, 928–937. 10.1016/j.chembiol.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. W.; Cognetta A. B.; Niphakis M. J.; Cravatt B. F. Proteome-Wide Reactivity Profiling Identifies Diverse Carbamate Chemotypes Tuned for Serine Hydrolase Inhibition. ACS Chem. Biol. 2013, 8, 1590–1599. 10.1021/cb400261h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adibekian A.; Martin B. R.; Chang J. W.; Hsu K. L.; Tsuboi K.; Bachovchin D. A.; Speers A. E.; Brown S. J.; Spicer T.; Fernandez-Vega V.; Ferguson J.; Hodder P. S.; Rosen H.; Cravatt B. F. Confirming target engagement for reversible inhibitors in vivo by kinetically tuned activity-based probes. J. Am. Chem. Soc. 2012, 134, 10345–10348. 10.1021/ja303400u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- Winger J. A.; Hantschel O.; Superti-Furga G.; Kuriyan J. The structure of the leukemia drug imatinib bound to human quinone reductase 2 (NQO2). BMC Struct. Biol. 2009, 9, 7. 10.1186/1472-6807-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina D. M.; Jafari R.; Ignatushchenko M.; Seki T.; Larsson E. A.; Dan C.; Sreekumar L.; Cao Y.; Nordlund P. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 2013, 341, 84–87. 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Lomenick B.; Hao R.; Jonai N.; Chin R. M.; Aghajan M.; Warburton S.; Wang J.; Wu R. P.; Gomez F.; Loo J. A.; Wohlschlegel J. A.; Vondriska T. M.; Pelletier J.; Herschman H. R.; Clardy J.; Clarke C. F.; Huang J. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 21984–21989. 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O.; Sanjana N. E.; Hartenian E.; Shi X.; Scott D. A.; Mikkelsen T. S.; Heckl D.; Ebert B. L.; Root D. E.; Doench J. G.; Zhang F. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda T.; Sugimoto H.; Yamashita S. Sequence, expression in Escherichia coli, and characterization of lysophospholipase II1. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1999, 1437, 182–193. 10.1016/S1388-1981(99)00007-4. [DOI] [PubMed] [Google Scholar]
- Manna J. D.; Wepy J. A.; Hsu K.-L.; Chang J. W.; Cravatt B. F.; Marnett L. J. Identification of the Major Prostaglandin Glycerol Ester Hydrolase in Human Cancer Cells. J. Biol. Chem. 2014, 289, 33741–33753. 10.1074/jbc.M114.582353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adibekian A.; Martin B. R.; Chang J. W.; Hsu K. L.; Tsuboi K.; Bachovchin D. A.; Speers A. E.; Brown S. J.; Spicer T.; Fernandez-Vega V.; Ferguson J.; Cravatt B. F.; Hodder P.; Rosen H.. Characterization of a Selective, Reversible Inhibitor of Lysophospholipase 2 (LYPLA2). In Probe Reports from the NIH Molecular Libraries Program; National Institutes of Health: Bethesda, MD, 2010. [PubMed]
- Davda D.; Martin B. R. Acyl protein thioesterase inhibitors as probes of dynamic S-palmitoylation. MedChemComm 2014, 5, 268–276. 10.1039/C3MD00333G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Zheng B.; DeRan M.; Jarugumilli G. K.; Fu J.; Brooks Y. S.; Wu X. ZDHHC7-mediated S-palmitoylation of Scribble regulates cell polarity. Nat. Chem. Biol. 2016, 12, 686–693. 10.1038/nchembio.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J., Davda D.; Cheung S.; Kit M.; Majmudar J. D.; Won S. J.; Gang M.; Pasupuleti S.; Choi A.; Bartkowiak C.; Martin B. R.. APT2 inhibition restores Scribble localization and S-palmitoylation in Snail-transformed cells. Cell Chem. Biol. DOI: 10.1016/j.chembiol.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won S. J.; Davda D.; Labby K. J.; Hwang S. Y.; Pricer R. E.; Majmudar J. D.; Armacost K. A.; Rodriguez L. A.; Rodriguez C. L.; Chong F. S.; Torossian K. A.; Palakurthi J.; Hur E. S.; Meagher J. L.; Brooks C. L.; Stuckey J. A.; Martin B. R. Molecular mechanism for isoform-selective inhibition of acyl protein thioesterases 1 and 2 (APT1 and APT2). ACS Chem. Biol. 2016, 11 (12), 3374–3382. 10.1021/acschembio.6b00720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstadler S. A.; Sannes-Lowery K. A. Applications of ESI-MS in drug discovery: interrogation of noncovalent complexes. Nat. Rev. Drug Discovery 2006, 5, 585–595. 10.1038/nrd2083. [DOI] [PubMed] [Google Scholar]
- Distler U.; Kuharev J.; Navarro P.; Levin Y.; Schild H.; Tenzer S. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 2013, 11, 167–170. 10.1038/nmeth.2767. [DOI] [PubMed] [Google Scholar]
- Huber K. V. M; Salah E.; Radic B.; Gridling M.; Elkins J. M.; Stukalov A.; Jemth A.-S.; Gokturk C.; Sanjiv K.; Stromberg K.; Pham T.; Berglund U. W.; Colinge J.; Bennett K. L.; Loizou J. I.; Helleday T.; Knapp S.; Superti-Furga G. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. 10.1038/nature13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic C. E.; Wright G.; Remsing Rix L. L.; Kim W.; Kuenzi B. M.; Luo Y.; Watters J. M.; Koomen J. M.; Haura E. B.; Monteiro A.N.; Radu C.; Lawrence H. R.; Rix U. Proteome-wide Profiling of Clinical PARP Inhibitors Reveals Compound-Specific Secondary Targets. Cell Chem. Biol. 2016, 10.1016/j.chembiol.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B. R.; Wang C.; Adibekian A.; Tully S. E.; Cravatt B. F. Global profiling of dynamic protein palmitoylation. Nat. Methods 2012, 9, 84–89. 10.1038/nmeth.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jecklin M. C.; Touboul D.; Jain R.; Toole E. N.; Tallarico J.; Drueckes P.; Ramage P.; Zenobi R. Affinity Classification of Kinase Inhibitors by Mass Spectrometric Methods and Validation Using Standard IC50 Measurements. Anal. Chem. 2009, 81, 408–419. 10.1021/ac801782c. [DOI] [PubMed] [Google Scholar]
- Ranjitkar P.; Perera B. G. K.; Swaney D. L.; Hari S. B.; Larson E. T.; Krishnamurty R.; Merritt E. A.; Villén J.; Maly D. J. Affinity-Based Probes Based on Type II Kinase Inhibitors. J. Am. Chem. Soc. 2012, 134, 19017–19025. 10.1021/ja306035v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; Zhang C.-J.; Chen G. Y. J.; Yao S. Q. Cell-Based Proteome Profiling of Potential Dasatinib Targets by Use of Affinity-Based Probes. J. Am. Chem. Soc. 2012, 134, 3001–3014. 10.1021/ja208518u. [DOI] [PubMed] [Google Scholar]
- Li Z.; Wang D.; Li L.; Pan S.; Na Z.; Tan C. Y. J.; Yao S. Q. Minimalist” Cyclopropene-Containing Photo-Cross-Linkers Suitable for Live-Cell Imaging and Affinity-Based Protein Labeling. J. Am. Chem. Soc. 2014, 136, 9990–9998. 10.1021/ja502780z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.