

Abstract
IHVR-19029 (6) is a lead endoplasmic reticulum α-glucosidases I and II inhibitor, which efficiently protected mice from lethal Ebola and Marburg virus infections via injection route, but suffered from low bioavailability and off-target interactions with gut glucosidases when administered orally. In an effort to improve efficacious exposure levels and avoid side effects, we designed and synthesized ester prodrugs. Not only were the prodrugs stable in simulated gastric and intestinal fluids and were inactive against glucosidases but they also exhibited antiviral activities against dengue virus infection in a cell based assay. Further in vitro evaluation showed that the bioconversion of the prodrugs is species dependent: in mice, the prodrugs were converted to 6 in the plasma and liver; while in human, the conversion occurred mainly in liver. An in vivo pharmacokinetic study in mice demonstrated that the tetrabutyrate prodrug 8 achieved the most improved overall exposure of 6 upon both oral and intravenous administration.
Keywords: Ester prodrug, ER α-glucosidases I and II, antiviral, N-alkyldeoxynojirimycin
Iminosugars, such as deoxynojirimycin (1, DNJ) and its derivatives, act as broad spectrum antiviral agents due to their mimicry of d-glucose and inhibition of endoplasmic reticular (ER) glucosidases (Figure 1).1−3 The potency of DNJ could be improved by incorporating a group on the nitrogen atom of DNJ through a linker (2–6).4−6 We have discovered several classes of N-alkyl-deoxynojirimycins that inhibit the host ER α-glucosidases I and II and disrupt viral glycoprotein folding and virion assembly of multiple enveloped viruses in vitro and in vivo.7−12 A representative lead compound, IHVR-19029 (6), has been demonstrated to significantly protect mice from lethal infections of the Marburg and Ebola viruses when administrated via intraperitoneal injections. However, 6 has low oral bioavailability, partly due to poor absorption, as shown by a high efflux ratio in Caco-2 permeability experiment.9 In addition, like the other imino sugar analogues, 6 is a substrate mimetic that inhibits not only ER α-glucosidases but also the carbohydrate-metabolizing glucosidases in the gastrointestinal (GI) lumen. This leads to dose limiting osmotic diarrhea, especially following oral administration.
Figure 1.

Structure and activity of representative NADNJ glucosidase inhibitors against dengue virus infection on HEK293 cells.
To overcome these problems, we explored a prodrug approach. This strategy has been widely effective in the antiviral area with improved potency, selectivity, bioavailability, and solubility. This is exemplified by sofosbuvir for the hepatitis C virus,13 oseltamivir for influenza,14 and fosamprenavir15 and Glycovir (iminosugar NBDNJ)16 for the human immunodeficiency virus. We aimed to protect the hydroxy groups in the DNJ moiety because they are important for both on- and off-target effects against glucosidases. In addition, the modifications may also have the potential to overcome the efflux issue due to the changes in the polarities and sizes of the resulting DNJ prodrugs. Because the target glucosidases are bound to the ER membrane, it will be ideal if the prodrugs were metabolized into active competitive inhibitors in this region. Among the five families of carboxylesterases (CEs), two families, CES1 and CES2, are important for hydrolyzing esters into acid and alcohols. Both esterases can bind to the ER protein retention receptor and hydrolyze the ester prodrugs into their active forms there.17,18 Thus, we designed ester prodrugs of 6 with the expectation that these prodrugs would be stable against GI α-glucosidases and could be converted to the parent 6 after arrival at the intracellular ER membrane. Herein, we describe the synthesis, enzymatic and cell-based activities, stability, and pharmacokinetics (PK) of a family of ester prodrugs 7–9.
IHVR-19029 has four free hydroxy groups on the DNJ moiety that can be used for making esters. Three forms of tetra-esters, including acetate (7), butyrate (8), and isobutyrate (9), were synthesized through reactions with the corresponding acid anhydrides in pyridine with good to moderate yields (Scheme 1). Due to their different steric hindrance to the enzymatic cleavage, these three ester promoieties are expected to have different hydrolysis rates.
Scheme 1. Synthesis of Ester Prodrugs of IHVR-19029.

Permeability and stability in the gastrointestinal tract are important considerations for the efficient and safe delivery of iminosugar prodrugs via the oral route. Therefore, compounds 7–9 were evaluated using a stability assay in physiologically relevant pHs and an in vitro Caco-2 permeability assay. When these three compounds were tested in both simulated gastric fluid (SGF) and simulated intestinal fluid (SIF), good and moderate stabilities were observed with more than 89% and 72% of the prodrugs remaining, respectively (Table 1).19
Table 1. Stability of Prodrugs 7–9 and Conversion to 6 in Simulated Gastric Fluid (SGF) and Simulated Intestinal Fluid (SIF).
| stability in SGF (4 h, %) |
stability in
SIF (4 h, %) |
|||
|---|---|---|---|---|
| compd | remaining | converted to 6 | remaining | converted to 6 |
| 6 | 109 | 114 | ||
| 7 | 94 | 0 | 91 | 0 |
| 8 | 89 | 0 | 72 | 0 |
| 9 | 108 | 0 | 90 | 0 |
In the Caco-2 permeability test, although the acetate prodrug 7 demonstrated significantly improved permeability and efflux ratio compared to 6, low recoveries of all prodrugs from both A-B and B-A (<60%; 7 for example, 9% and 42%, respectively, Supporting Information) brought into question the reliability of these permeability values and suggested it was not a simple bidirectional passive diffusion for these prodrugs. Because esterases exist in Caco-2 cells, they can hydrolyze the ester prodrugs, raising a concern that these prodrugs were hydrolyzed while going across the intestine. Thus, we measured the percentages of prodrugs that remained and were converted to 6 after incubation with intestinal S9 for an hour. Although 6 was stable (76–95% remaining) in this assay, most prodrugs had less than half remaining in the order of 7 < 8 < 9 (Supporting Information) in accordance with the steric hindrance of the acyl groups. With only small percentages of the 6 present, most of the material was likely in a partially hydrolyzed state. As a result, it is a challenge to assess permeability and predict absorption of these prodrugs by Caco-2 measurements with confidence. However, it is evident that all prodrugs are more lipophilic than 6, and thus, it is reasonable to speculate that the prodrugs exhibit higher intrinsic permeability across cell membrane via passive diffusion.
To determine that the prodrugs can avoid interacting with α-glucosidases, we set up enzymatic assays using ER α-glucosidases I and II purified from pig liver and 4-methylumbelliferyl-α-d-glucoside (4-MU-α-d-glucoside) as a substrate.20,21 As expected, while 6 inhibited ER α-glucosidase I with submicromolar activity, all the prodrugs lost the ability to inhibit ER α-glucosidase I in enzymatic assays at concentrations up to 80 μM. Similar results were obtained in an ER α-glucosidase II assay (Figure 2A,B). Because iminosugar DNJ is a d-glucose substrate analogue, DNJ derivative 6 could be an inhibitor not only of the desired ER α-glucosidases but also of other α-glucosidases.22,23 The result shown in Figure 2 thus implies that these ester prodrugs would also have reduced activity against GI α-glucosidases and potentially overcome the off-target effects of the parent compound if they are stable in the GI.
Figure 2.

Characterization of iminosugar prodrugs with enzymatic and cell-based assays. (A,B) Evaluation of dose-dependent response of parent 6 and its prodrugs 7–9 in α-glucosidase I and II enzymatic assays (n = 3). (C) MDBK cells were infected with BVDV followed by treatment with concentrations of iminosugar 6 or its prodrugs 7–9. BVDV envelope glycoprotein E2 (BVDV E2) was examined in an immunoblot assay using β-actin as internal control. (D) Huh7.5 cells were infected with dengue virus (DENV) and treated with concentrations of iminosugar compound 6 and its prodrugs (n = 4). DENV RNAs were determined by qRT-PCR. EC50 and IC50 were calculated using Prism 5 (GraphPad Software, Inc.).
Furthermore, to confirm that these prodrugs can be converted into active inhibitors of glucosidases in cells, we performed a cell-based surrogate assay in which an immunoblot assay approach was used to analyze the Bovine Viral Diarrhea virus (BVDV) glycosylated envelop protein (BVDV E2 protein) in Madin–Darby Bovine Kidney Epithelial Cells (MDBK) infected with BVDV. As others and we have previously shown, inhibition of ER glucosidases causes unprocessed N-glycans to occur on N-glycoproteins, such as the BVDV E2 protein. BVDV E2 containing an unprocessed N-glycan migrates more slowly than BVDV E2 with a complex (mature) N-glycan does and is more rapidly degraded. This change can be detected with SDS PAGE of infected cell lysates and has been used as a surrogate marker of intracellular activity of ER glucosidase inhibition.7−12 The slower mobility rate and reduced intensity were clearly observed when MDBK cells were infected with BVDV, followed by treatment with various concentrations of either 6 or its prodrugs, compared to the untreated cells (Figure 2C).
Dengue virus is also sensitive to ER glucosidase inhibition, in vitro and in vivo, and has served as a convenient bioreporter for the efficacy and activity of ER glucosidase inhibitors.7−12 We therefore determined the ability of the prodrugs to inhibit dengue virus growth in tissue. The prodrugs consistently demonstrated antiviral activities, though less potently than parent 6, in a cell based assay against dengue virus infection, indicating that at least part of the parent 6 molecules were released inside the cells (Figure 2D).
We further investigated the stability and conversion of the prodrugs in pooled liver microsomes and plasma (Tables 2 and 3). When the three prodrugs were incubated in liver microsomes (LM), more than 88% of the prodrugs degraded within 60 min regardless of the presence or absence of cofactor (Table 2). Higher conversion (>52%) into 6 was observed when there was no cofactor in both human and mouse microsomes, indicating that the prodrugs are more easily converted into 6 by esterases or under nonoxidative metabolism. In the human liver microsomes, the order of conversion to 6 in the absence of cofactor was 8 ≫ 7 ≫ 9; while in the mouse liver microsomes, it was 9 > 8 > 7, albeit at similar rates. Among the three prodrugs, the tetrabutyrate 8 can be efficiently hydrolyzed into 6 (>95%) in both human and mouse microsomes by esterases.
Table 2. Prodrug Stability and Conversion to 6 in Pooled Human (H) and Mouse (M) Liver Microsomes (LM).
| |
remaining in LM (%, 60 min) |
converted to 6 in LM (%, 60 min) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
T1/2 (min) |
H |
M |
H |
M |
||||||
| compd | H | M | +cofactor | –cofactor | +cofactor | –cofactor | +cofactor | –cofactor | +cofactor | –cofactor |
| 6 | 104 | 94 | 68 | 99 | 63 | 96 | ||||
| 7 | 3 | 2 | 0 | 0 | 0 | 0 | 22 | 74 | 25 | 93 |
| 8 | 14 | 20 | 4 | 4 | 10 | 12 | 20 | 99 | 30 | 96 |
| 9 | 10 | 14 | 2 | 2 | 5 | 7 | 13 | 53 | 23 | 101 |
Table 3. Stability in Human and Mouse Plasmas.
| remaining in
plasma (%,
2 h) |
conversion
to 6 in plasma (%, 2 h) |
|||
|---|---|---|---|---|
| compd | H | M | H | M |
| 6 | 109 | 93 | ||
| 7 | 84 | 0 | 0 | 55 |
| 8 | 106 | 0 | 0 | 85 |
| 9 | 100 | 0 | 0 | 66 |
In mouse plasma, 85% of 8 was converted into 6, which was the highest rate among the three prodrugs, while 9 and 7 had 66% and 55% conversions, respectively, after 120 min (Table 3). In contrast, in human plasma and even in whole blood (data not shown), less than 2% of conversion into 6 was observed, and the result is consistent with the fact that human esterases are mainly distributed in various tissues and are less prevalent in blood. These results suggested that the ester prodrugs could be cleaved into parent 6 in both mice (plasma and liver) and humans (mainly in liver and other tissues), supporting the translatability of in vivo efficacy studies in mice to humans.
Interestingly, we noticed that under several conditions where the prodrugs were not efficiently converted to the parent 6, there were very low amounts of the original prodrugs remaining, suggesting that partially hydrolyzed intermediates are accumulating in those conditions. Indeed, further analysis of potential intermediates of prodrug 8 at different time points after incubation with human liver microsomes by LC–MS/MS showed that several products consistent with being mono- and diester protection on the DNJ head were generated transiently, and they were gradually converted to 6 (Figure 3). This observation has led to our interest in the prodrugs with partial protections, which are currently under investigation.
Figure 3.

Analysis of the intermediates in the conversion of 8 to 6 in human liver microsomes.
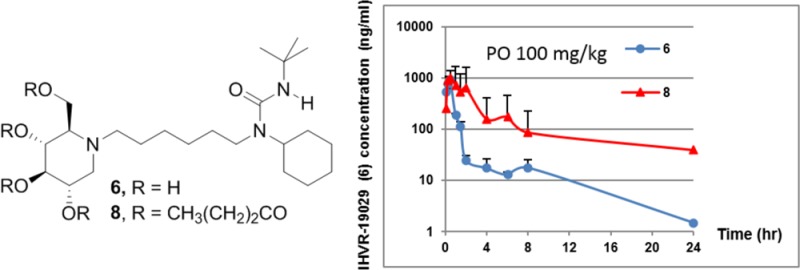
We validated the benefit of this prodrug approach with an in vivo experimentation. The pharmacokinetics of these prodrugs were investigated in mice, and, as shown in Table 4, oral (PO) bioavailability of 6 is low, consistent with what was reported previously.9 All the prodrugs quickly disappeared in all the study groups at the first sampling time point (5 min post dosing) except for 9, which remained detectable for up to 4 h postdosing (data not shown). Plasma exposure of 6 was determined in male BALB/c mice dosed with the prodrugs. The Tmax of prodrugs, measured by the formation of compound 6, is about 15–30 min, suggesting that the prodrugs are rapidly absorbed and converted after the oral administration. The relative exposure after intravenous injection (IV) was 8 > 9 > 7, while the order became 8 > 7 > 9 when administered orally. As expected, improved oral exposure of 6 was observed when dosing the prodrugs 7 and 8. Especially when dosing at 138.1 μmol/kg (100 mg/kg) orally, about a 2-fold and 4-fold increase in exposure (AUCinf) was observed, respectively, compared to dosing the same milligram amount of the parent 6. Moreover, at 138.1 μmol/kg (100 mg/kg) oral dosing, prodrug 8 provided elevated levels (>1 μM) of the parent 6 for more than 3 h, which is comparable to the serum level achieved by an antiviral efficacious dose of 169.3 μmol/kg (75 mg/kg) via intraperitoneal injection.9 As shown in Table 4, the improved exposure likely resulted from more sustained drug levels in the plasma. One can assume that the parent drug 6 was converted from the prodrugs in plasma and liver in a controlled fashion, leading to slow metabolism of the parent by liver enzymes. Even with the IV dosing at 6.9 μmol/kg (5 mg/kg), it is encouraging to observe that prodrug 8 provided about a 2-fold increase of exposure AUC. The overall superior PK profile of 8 is likely attributed to its higher conversion percentage in plasma, liver, and other tissues. It is also interesting to note that both 7 and 8 showed higher than proportional increases in PO exposure (AUCinf) from low dose (40.9 and 34.5 μmol/kg) to high dose (163.4 and 138.1 μmol/kg, Table 4). This may reflect different kinetics of redistribution from tissue, as evidenced by the multiple intermittent increases in exposure. Alternatively, this may be caused by the saturation of drug metabolizing enzymes or Pgp transporters in the gut. Although the exact mechanism remains to be determined, the improvement of systemic exposure demonstrates the potential of this ester prodrug approach in an in vivo setting.
Table 4. Pharmacokinetics Profile of Three Ester Prodrugs (Determine the Parent Compound 6 in Plasma; NC, not calculated).
| compd | route | dose μmol/kg (mg/kg) | compd measured | Tmax (h) | Cmax (ng/mL) | T1/2 (h) | Cl (mL/h/kg) | Vss (mL/kg) | AUCinf (h·ng·mL) |
|---|---|---|---|---|---|---|---|---|---|
| 6 | IV | 11 (5) | 6 | 2843 | 0.98 | 4197 | 2133 | 1191 | |
| PO | 56.4 (25) | 6 | 0.25 | 184 | 5.19 | NC | NC | 150 | |
| PO | 225.4 (100) | 6 | 0.5 | 915 | 5.46 | NC | NC | 996 | |
| 7 | IV | 8.2 (5) | 6 | 0.25 | 463 | 1.11 | NC | NC | 368 |
| PO | 40.9 (25) | 6 | 0.25 | 375 | 1.40 | NC | NC | 359 | |
| PO | 163.4 (100) | 6 | 0.25 | 1597 | 0.72 | NC | NC | 1912 | |
| 8 | IV | 6.9 (5) | 6 | 0.083 | 15733 | 0.84 | NC | NC | 2288 |
| PO | 34.5 (25) | 6 | 0.25 | 319 | 1.72 | NC | NC | 529 | |
| PO | 138.1 (100) | 6 | 0.5 | 998 | 2.29 | NC | NC | 3884 | |
| 9 | IV | 6.9 (5) | 6 | 0.083 | 1213 | 1.25 | NC | NC | 569 |
| PO | 34.5 (25) | 6 | 0.50 | 104 | 0.58 | NC | NC | 197 | |
| PO | 138.1 (100) | 6 | 0.5 | 195 | 1.18 | NC | NC | 362 |
In conclusion, to minimize the off-target interaction with α-glucosidases within the gastrointestinal tract and increase the plasma exposure of the IHVR-19029 (6) following oral administration, ester prodrugs 7–9 were prepared and evaluated in α-glucosidase assays and cell-based antiviral assays. These prodrugs are inactive against isolated enzymes but active when absorbed inside the cells, which suggests that the ester prodrugs are converted into parent 6 inside the cell. These prodrugs were stable in SGF and SIF, demonstrating good chemical stability at physiological pHs. Additional plasma stability tests and assessments in liver microsome assays indicated that the parent 6 could be released from the ester prodrugs in plasma and liver for mice but primarily in the liver for humans. Although this seems to be a disadvantage for rapid systemic circulation of the active compound in human, it should be beneficial for liver distribution of the active compound for liver tropical viral infections, including many types of hemorrhagic fever viruses, such as Ebola virus, which was discovered to replicate quickly in the spleens and livers of cynomolgus monkeys.24 Finally, prodrugs are readily converted to parent 6 in mice upon both IV and PO administration. Tetrabutyrate 8 displays significantly increased overall exposure than that of direct administration of the parent compound in both IV and PO routes. Prodrug 8 and its partially hydrolyzed mono- and dibutyrate analogues have been selected for further evaluation, for which data will be reported in due course.
Acknowledgments
The authors thank Dr. Terry Butters (Oxford University, UK) for his advice and discussion. We would also like to thank Dr. Bonnie L. Mai of SRI Biosciences Division and Dr. Chunyan Han of Pharmaron for the helpful discussions. We are very grateful to Professor Donna Huryn, the editor for this paper, for her thoughtful and critical comments and suggestions.
Glossary
ABBREVIATIONS
- GI
gastrointestinal
- DNJ
deoxynojirimycin
- ER
endoplasmic reticular
- BVDV
bovine viral diarrhea virus
- SGF
simulated gastric fluid
- SIF
simulated intestinal fluid
- LM
liver microsome
- IV
intravenous
- PO
per os
- MDBK
Madin–Darby Bovine Kidney Epithelial Cells
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00332.
Experimental details for the synthesis of prodrugs 7–9, enzymatic assay, in vitro evaluation, and in vivo PK protocols (PDF)
This project is supported by NIH grant (NIH AI104636). We acknowledge the support from NIAID nonclinical and preclinical service program to support the pharmacokinetic study in mice. This study was also partially supported by the Hepatitis B Foundation and the Commonwealth of Pennsylvania.
The authors declare no competing financial interest.
Supplementary Material
References
- Chang J.; Guo J.-T.; Du Y.; Block T. M. Imino sugar glucosidase inhibitors as broadly active anti-filovirus agents. Emerging Microbes Infect. 2013, 2, e77. 10.1038/emi.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.; Block T. M.; Guo J.-T. Antiviral therapies targeting host ER alpha-glucosidases: Current status and future directions. Antiviral Res. 2013, 99, 251–260. 10.1016/j.antiviral.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.; Block T. M.; Guo J.-T. Viral resistance of MOGS-CDG patients implies a broad-spectrum strategy against acute virus infections. Antiviral Ther. 2015, 20, 257–9. 10.3851/IMP2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A.; Ouzounov S.; Jordan R.; Simsek E.; Lu X.; Moriarty R. M.; Jacob G.; Dwek R. A.; Block T. M. Imino sugars that are less toxic but more potent as antivirals, in vitro, compared with N-n-nonyl DNJ. Antiviral Chem. Chemother. 2002, 13, 299–304. 10.1177/095632020201300505. [DOI] [PubMed] [Google Scholar]
- Gu B.; Mason P.; Wang L.; Norton P.; Bourne N.; Moriarty R.; Mehta A.; Despande M.; Shah R.; Block T. Antiviral profiles of novel iminocyclitol compounds against bovine viral diarrhea virus, West Nile virus, dengue virus and hepatitis B virus. Antiviral Chem. Chemother. 2007, 18, 49–59. 10.1177/095632020701800105. [DOI] [PubMed] [Google Scholar]
- Chang J.; Wang L.; Ma D.; Qu X.; Guo H.; Xu X.; Mason P. W.; Bourne N.; Moriarty R.; Gu B.; Guo J.-T.; Block T. M. Novel imino sugar derivatives demonstrate potent antiviral activity against flaviviruses. Antimicrob. Agents Chemother. 2009, 53, 1501–8. 10.1128/AAC.01457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y.; Ye H.; Guo F.; Wang L.; Gill T.; Khan S.; Cuconati A.; Guo J.-T.; Block T. M.; Chang J.; Xu X. Design and synthesis of N-alkyldeoxynojirimycin derivatives with improved metabolic stability as inhibitors of BVDV and Tarcaribe virus. Bioorg. Med. Chem. Lett. 2013, 23, 4258–4262. 10.1016/j.bmcl.2013.04.052. [DOI] [PubMed] [Google Scholar]
- Du Y.; Ye H.; Gill T.; Wang L.; Guo F.; Cuconati A.; Guo J.-T.; Block T. M.; Chang J.; Xu X. N-Alkyldeoxynojirimycin derivatives with novel terminal tertiary amide substitution for treatment of bovine viral diarrhea virus (BVDV), Dengue, and Tarcaribe virus infections. Bioorg. Med. Chem. Lett. 2013, 23, 2172–2176. 10.1016/j.bmcl.2013.01.108. [DOI] [PubMed] [Google Scholar]
- Chang J.; Warren T. K.; Zhao X.; Gill T.; Guo F.; Wang L.; Comunale M. A.; Du Y.; Alonzi D. S.; Yu W.; Ye H.; Liu F.; Guo J.-T.; Mehta A.; Cuconati A.; Butters T. D.; Bavari S.; Xu X.; Block T. M. Small molecule inhibitors of ER α-glucosidases are active against multiple hemorrhagic fever viruses. Antiviral Res. 2013, 98, 432–440. 10.1016/j.antiviral.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.; Gill T.; Wang L.; Du Y.; Ye H.; Qu X.; Guo J.; Cuconati A.; Zhao K.; Block T. M.; Xu X.; Chang J. Design, Synthesis, and Biological Evaluation of N-Alkylated Deoxynojirimycin (DNJ) Derivatives for the Treatment of Dengue Virus Infection. J. Med. Chem. 2012, 55, 6061–6075. 10.1021/jm300171v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.; Schul W.; Yip A.; Xu X.; Guo J.-T.; Block T. M. Competitive inhibitor of cellular α-glucosidases protects mice from lethal dengue virus infection. Antiviral Res. 2011, 92, 369–371. 10.1016/j.antiviral.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.; Schul W.; Butters T. D.; Yip A.; Liu B.; Goh A.; Lakshminarayana S. B.; Alonzi D.; Reinkensmeier G.; Pan X.; Qu X.; Weidner J. M.; Wang L.; Yu W.; Borune N.; Kinch M. A.; Rayahin J. E.; Moriarty R.; Xu X.; Shi P.-Y.; Guo J.-T.; Block T. M. Combination of α-glucosidase inhibitor and ribavirin for the treatment of dengue virus infection in vitro and in vivo. Antiviral Res. 2011, 89, 26–34. 10.1016/j.antiviral.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia M.; Bao D.; Chang W.; Du J.; Nagarathnam D.; Rachakonda S.; Reddy P. G.; Ross B. S.; Wang P.; Zhang H.-R.; Bansal S.; Espiritu C.; Keilman M.; Lam A. M.; Steuer H. M. M.; Niu C.; Otto M. J.; Furman P. A. Discovery of a β-D-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem. 2010, 53, 7202–7218. 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- Li W.; Escarpe P. A.; Eisenberg E. J.; Cundy K. C.; Sweet C.; Jakeman K. J.; Merson J.; Lew W.; Williams M.; Zhang L.; Kim C. U.; Bischofberger N.; Chen M. S.; Mendel D. B. Identification of GS 4104 as an Orally Bioavailable Prodrug of the Influenza Virus Neuraminidase Inhibitor GS 4071. Antimicrob. Agents Chemother. 1998, 42, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furfine E. S.; Baker C. T.; Hale M. R.; Reynolds D. J.; Salisbury J. A.; Searle A. D.; Studenberg S. D.; Todd D.; Tung R. D.; Spaltenstein A. Preclinical pharmacology and pharmacokinetics of GW433908, a water-soluble prodrug of the human immunodeficiency virus protease inhibitor amprenavir. Antimicrob. Agents Chemother. 2004, 48, 791–798. 10.1128/AAC.48.3.791-798.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C. S.; Karabatsos P. J.; Schoenhard G. L.; Karim A. Species Dependent Esterase Activities for Hydrolysis of an Anti-HIV Prodrug Glycovir and Bioavailability of Active SC-48334. Pharm. Res. 1995, 8, 1158–1164. 10.1023/A:1016259826037. [DOI] [PubMed] [Google Scholar]
- Hosokawa M. Structure and catalytic properties of carbox-ylesterase isozymes involved in metabolic activation of prodrugs. Molecules 2008, 13, 412–431. 10.3390/molecules13020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakamata W.; Tamura S.; Hirano T.; Nishio T. Multicolor Imaging of Endoplasmic Reticulum-Located Esterase as a Prodrug Activation Enzyme. ACS Med. Chem. Lett. 2014, 5, 321–325. 10.1021/ml400398t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For data from bioanalysis, generally, compounds will be considered as stable when remaining percentages are between 80% and 120%, moderately stable when between 40% and 80%, and labile when the percentages are less than 40%.
- Karlssont G. B.; Butters T. D.; Dwek R. A.; Platt F. M. Effects of the Imino Sugar N-Butyldeoxynojirimycin on the N-Glycosylation of Recombinant gp120. J. Biol. Chem. 1993, 268, 570–576. [PubMed] [Google Scholar]
- The functional purity of the purified enzymes was evaluated using various 4-MU glycosides, and the result suggested that the two enzymes are specific for the hydrolysis of α-glucose, but not β-glucose, α-, β-mannose, or α-, β-galactose (data not shown).
- Shailubhai K.; Pratta M. A.; Vijay I. K. Purification and characterization of glucosidase I involved in N-linked glycoprotein processing in bovine mammary gland. Biochem. J. 1987, 247, 555–562. 10.1042/bj2470555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakamata W.; Muroi M.; Nishio T.; Oku T.; Takatsuki A. Recognition Properties of Processing α-Glucosidase I and α-Glucosidase II. J. Carbohydr. Chem. 2004, 23, 27–39. 10.1081/CAR-120030022. [DOI] [Google Scholar]
- Geisbert T. W.; Hensley L. E.; Larsen T.; Young H. A.; Reed D. S.; Geisbert J. B.; Scott D. P.; Kagan E.; Jahrling P. B.; Davis K. J. Pathogenesis of Ebola Hemorrhagic Fever in Cynomolgus Macaques. Am. J. Pathol. 2003, 163, 2347–2370. 10.1016/S0002-9440(10)63591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.