

Abstract
In our efforts to develop novel small-molecule inhibitors for the treatment of influenza, we utilized molecular modeling and the X-ray crystal structure of the PB2 subunit of the influenza polymerase to optimize a series of acyclic β-amino acid inhibitors, highlighted by compound 4. Compound 4 showed good oral exposure in both rat and mouse. More importantly, it showed strong potency versus multiple influenza-A strains, including pandemic 2009 H1N1 and avian H5N1 strains and showed a strong efficacy profile in a mouse influenza model even when treatment was initiated 48 h after infection. Compound 4 offers good oral bioavailability with great potential for the treatment of both pandemic and seasonal influenza.
Keywords: PB2 inhibitor, azaindole, influenza
Seasonal and pandemic influenza outbreaks remain a significant challenge worldwide. Influenza is a common and potentially deadly infectious disease that has afflicted large human populations in terms of morbidity and mortality at a range of 3000 to 49000 deaths per year, in the US over the last 31 years.1 Recent statistics also suggest that each year in the United States, 5–20% of the population becomes infected with influenza resulting in more than 200,000 hospitalizations.2 Swine and avian hosts can also serve as a reservoir of influenza A that can occasionally transmit influenza variants from others species to humans, which can lead to a global epidemic such as the 2009 H1N1 swine flu pandemic.3,4 Finally, the emergence of H5N1 (“bird flu”), an extremely virulent strain to humans, could potentially become a global threat if it were to acquire the ability for human-to-human transmission. Since there are no efficient ways to accurately predict the exact viral strains prior to each influenza season or pandemic, targeting pathways to stop viral replication is an area of high interest for drug discovery. The most known and currently used standard of care (SOC) to treat influenza are the neuraminidase inhibitors (NA): oseltamivir 1 (Figure 1), zanamivir, and peramivir. While these small-molecule drugs can effectively treat a variety of type A and B influenza viruses, they suffer limitations from resistance and have to be administered within 24–48 h postinfection to be efficacious.5 Moreover, recent reports have shown that some H5N1 influenza strains are resistant to oseltamivir.6
Figure 1.

Inhibitors of influenza virus: NA inhibitor oseltamivir (1), azaindole (2), and JNJ-63623872 (3).
To circumvent some of these limitations and address unmet medical needs in this arena, we recently reported on the discovery of a novel class of compounds from a phenotypic cell protection (CPE) assay screen that have shown survival benefits in the mouse model when administered 48 h postinfection.7 Compound 2 was our initial hit from the CPE assay. Our early lead optimization efforts culminated in the discovery of a novel 7-azaindole based first-in-class inhibitor of influenza polymerase-B2 (PB2), JNJ-63623872 (3) (formerly known as VX-787) (Figure 1).7
PB2 is part of a heterotrimeric viral complex that is essential for viral RNA replication. This viral polymerase complex assembly, comprising PA, PB1, and PB2 subunits, is responsible for replication and transcription of the eight separate segments of the viral RNA genome in the nuclei of infected cells.8−10 The polymerase complex synthesizes viral mRNAs using short, capped primers derived from cellular transcripts by a unique “cap-snatching” mechanism, where the virus utilizes host pre-mRNA as a primer for transcription.11 The PB2 subunit binds the 5′ cap domain for 7-methyl GTP (m7GTP) of host pre-mRNAs and positions it for cleavage by the PA subunit, leaving a 10–13 nucleotide primer. Then, elongation of the chimeric viral mRNA in PB1 by polyadenylation of viral mRNA polymerase completes the replication cycle. Since compound 3 targets the viral transcription complex, it offers an alternative mechanism of action relative to oseltamivir and combination therapy with other antivirals.
Identity of PB2 as a target was further confirmed with the X-ray crystal structure of screening hit 2 bound to the PB2 cap-binding domain7 (Figure 2).
Figure 2.
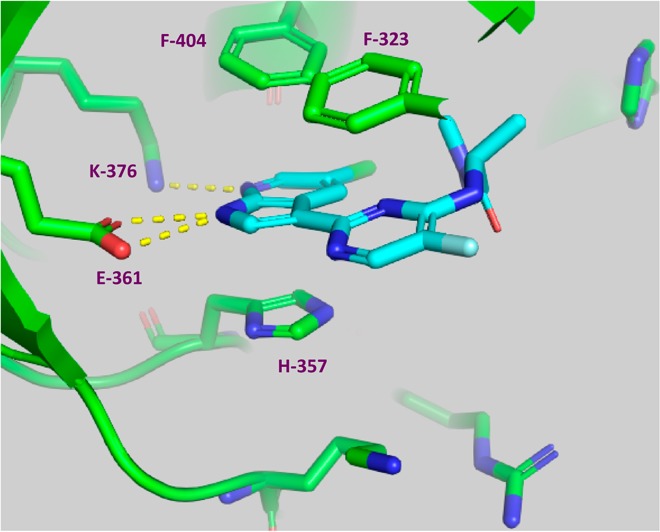
X-ray structure of cap-binding domain of PB2 containing PB2 inhibitor 2. PDB accession code is 4NCM. Reprinted from ref (7).
As shown in Figure 2, compound 2 forms hydrogen bonds to both Glu361 and Lys376 side chains, with the 7-azaindole ring sandwiched between His357 and Phe404. The pyrimidine ring pi-stacks with Phe323.
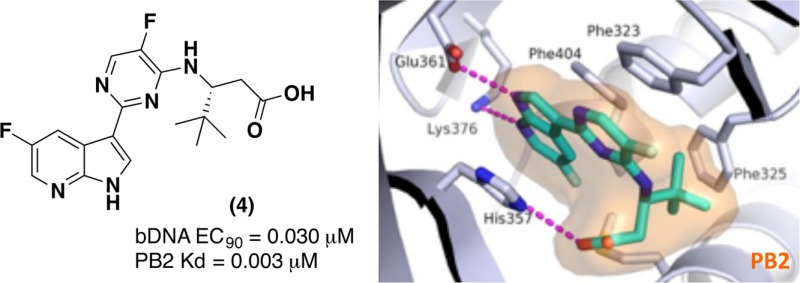
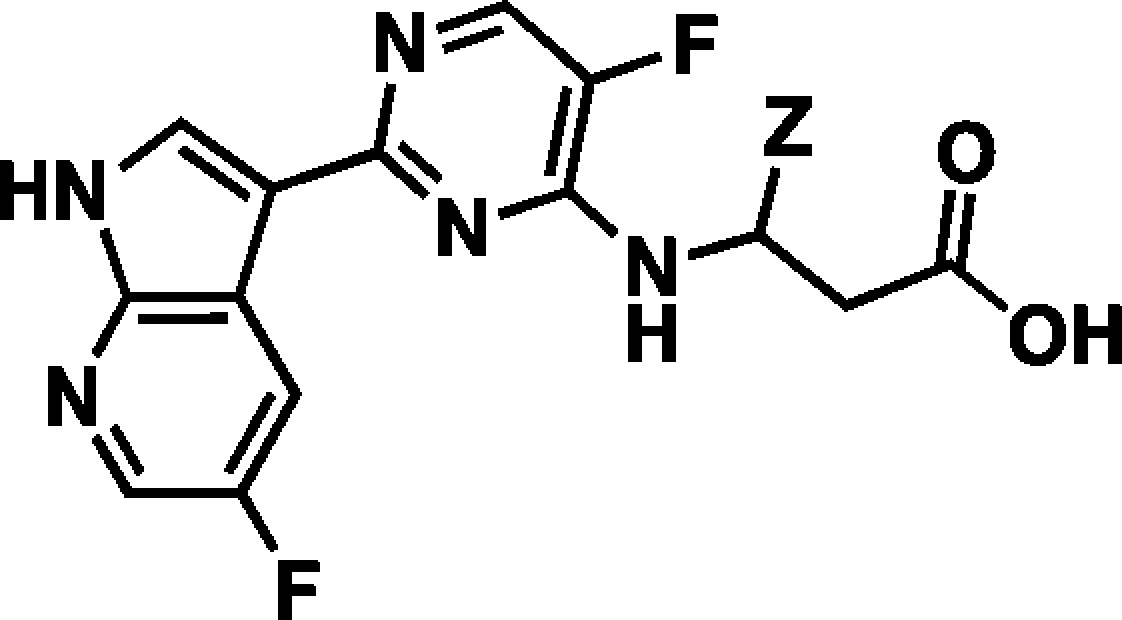
In our effort to generate novel inhibitors of PB2 we focused our attention on the alanine dimethylamide side chain. Our goal was to design simple acylic beta-amino acid analogues. We first envisioned that a (R)-t-butyl group could fill the hydrophobic space in the PB2 binding pocket, while retaining the key interactions of compound 2. To support this hypothesis, we docked our first proposed analogue, compound 4 (Table 1), into the active site of PB2 and then superimposed the docking model with the X-ray structure of PB2 in complex with 2 (Figure 3).
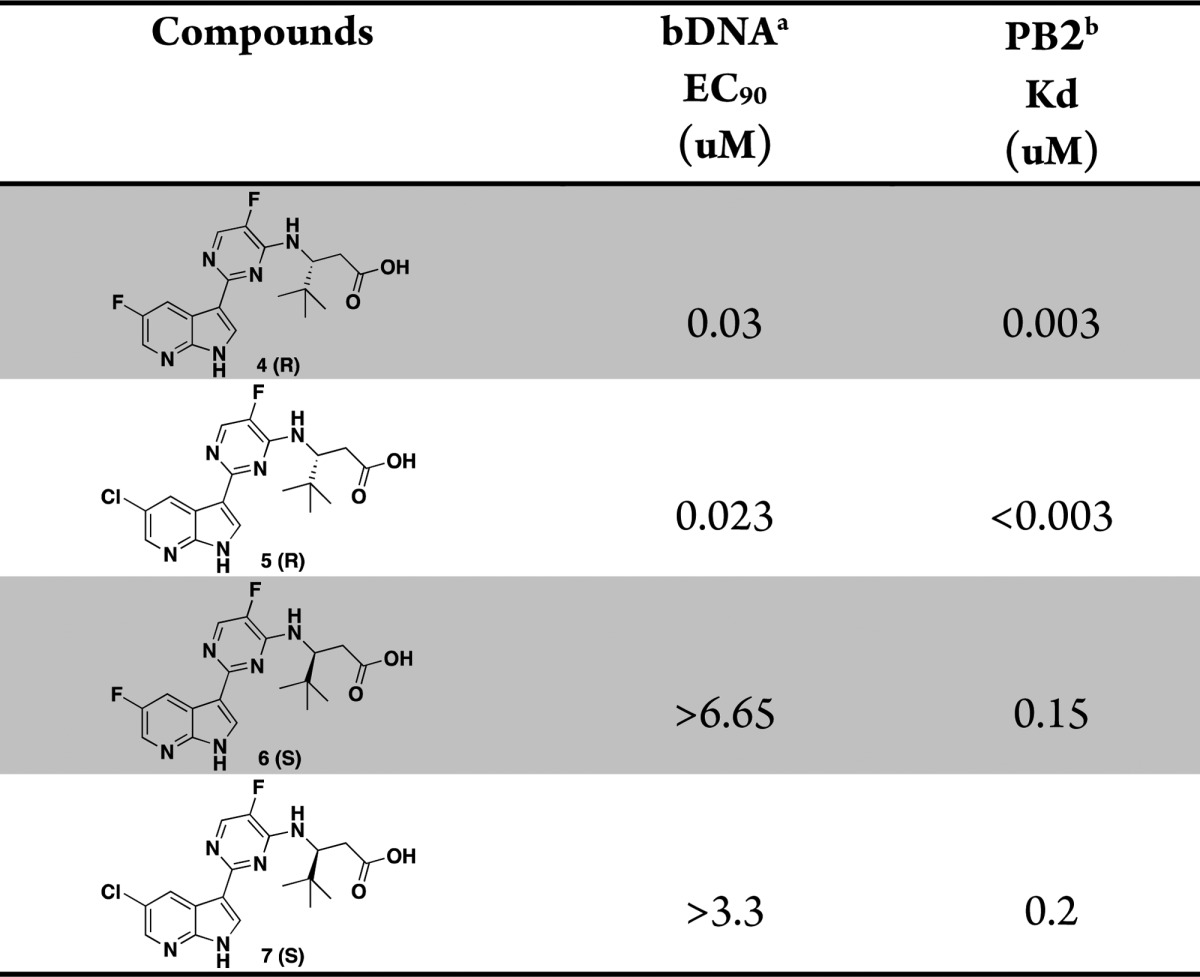
Table 1. In Vitro Potency of (R)-3-Amino-4,4-dimethylpentanoic Acids and Their Enantiomers.
The concentration of test compounds resulting in viral RNA levels equal to that of 10% of the control wells was reported as EC90.
Affinity for cap-binding domain of the PB2 subunit as measured in a fluorescence polarization competition binding assay.
Figure 3.
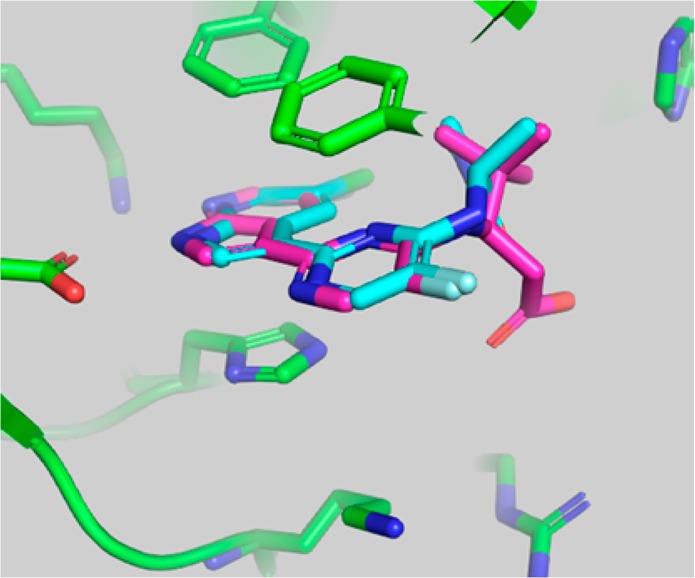
Superposition of the crystallographic pose of 2 and the docking model of compound 4 in the active site of PB2. Compound 4 was docked into the crystallographic conformation of PB2 from the complex with 2 using the Glide program (Schrodinger, Inc.).
The overlay of the two compounds in the active site of PB2 showed good superposition of the key pharmacophoric elements, within the azaindole moiety and carboxylic acid group. Additionally, the tert-butyl group of compound 4 occupied the same region occupied by the alpha-methyl dimethylamide functionality of compound 2. To test the validity of the model, two enantiomeric pairs (4 and 6, 5 and 7) were synthesized and evaluated for their binding affinity for PB2 (Kd) and their ability to inhibit viral RNA replication (EC90) in a branched DNA (bDNA) cell assay.11 Interestingly, and in agreement with the model (Figure 3), the expected (R)-enantiomers of both 5-fluoro (4) and 5-chloro (5) azaindole exhibited excellent binding activity and potency, whereas the (S)-enantiomers (6 and 7) showed reduced activity (Table 1).
The docked binding mode was subsequently confirmed by the X-ray structure of compound 4 bound to PB2 (Figure 4). Consistent with the model depicted in Figure 3, the compound 4 forms hydrogen bonds to both the Glu361 and Lys376 side chains, the azaindole ring is sandwiched between His357 and Phe404, while the pyrimidine ring forms a pi-stacking interaction with Phe323.
Figure 4.
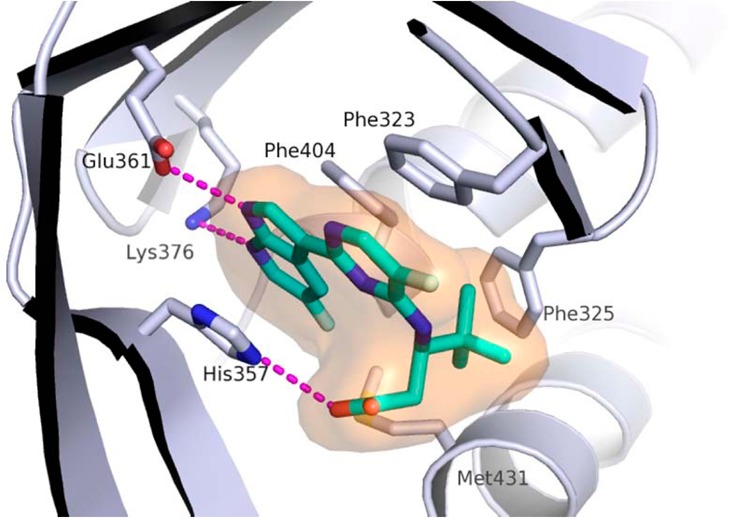
X-ray structure of cap-binding domain of PB2 containing PB2 inhibitor 4. PDB accession code is 5JUR.
Synthesis of the two enantiomeric pairs of compounds 4–7 from Table 1 was accomplished in a two-step protocol from the coupling of commercially available (R)- or (S)-3-amino-4,4-dimethyl pentanoic acid 18 or 19 via a facile displacement with the previously prepared sulfoxide 17(7) according to Scheme 1.
Scheme 1. Synthesis of Enantiomeric Pairs Compounds 4–7.

Reagents and conditions: (a) Na2CO3, THF, MeCN, MW, 135 °C, 30 min; (b) (i) MeONa, MeOH, 30 min, (ii) NH4Cl, 45–55% (2 steps).
This structure suggests that the hydrophobic pocket defined by the three phenylalanine residues (F323, F325, and F404) could potentially accommodate larger side chains. In order to test such a hypothesis, additional compounds with side chain variations to replace the t-butyl group were prepared to further explore this hydrophobic pocket. We focused our optimization on the preparation of analogues bearing a fluoro substitution at the 5-position of the azaindole ring system (Table 2).7 A slight increase in size with the addition of a methyl group as exemplified with the (R)-3-amino-4,4-dimethylhexanoic acid side chain based compound 8 led to comparable potency with compound 4. The removal of a methyl group as shown with the (R)-3-amino-4-methylpentanoic acid based compound 9 led to a 20-fold loss in cellular activity (EC90 = 0.59 μM) with respect to 4. Interestingly, the replacement of one methyl group on the side chain of 4 for the larger and more lipophilic trifluoromethyl in compound 10 was also less active. The iso-pentyl extended side chain version of analogue 9, as in compound 11, was slightly better tolerated (EC90 = 0.34 μM). Cyclizing both ethyl side chains of 11 as a cyclohexane ring 12 restored activity within the 100 nM range. With this result, more cycloalkyls were prepared to explore optimization. Cycloalkyl based side chains with the (R)-absolute configuration (compounds 13–16) all showed <1 μM potency (EC90 = 0.007 to 0.780 μM) in the bDNA assay. Spirocyclobutane analogue 16 was shown to have the highest anti-influenza activity (EC90 = 0.007 μM). It is worth noting that a single-point substitution from a methyl to hydrogen from compounds 16 to 15 led to over a 100-fold decrease in activity. The above results indicate that a balance of both size and shape of the side chains bearing a tertiary carbon appear to be important to fill this hydrophobic pocket. All the corresponding enantiomers showed less affinity for PB2 or weaker potency (data not shown), which correlated well with results initially observed with the 3-amino-4,4-dimethylpentanoic acids, based enantiomeric pairs (compound 4:6 and 5:7). Scheme 2 depicts the synthesis of 1-methyl-1-cyclobutane 16. Coupling of rac aminoester 21 (commercially available or prepared in 7 steps; see Supporting Information) with 2,4-dichloro-5-fluoropyrimidine 20 provided the desired intermediate 22.
Table 2. Optimization of in Vitro Potency of Various Substituted Butanoic Acid Side Chains.
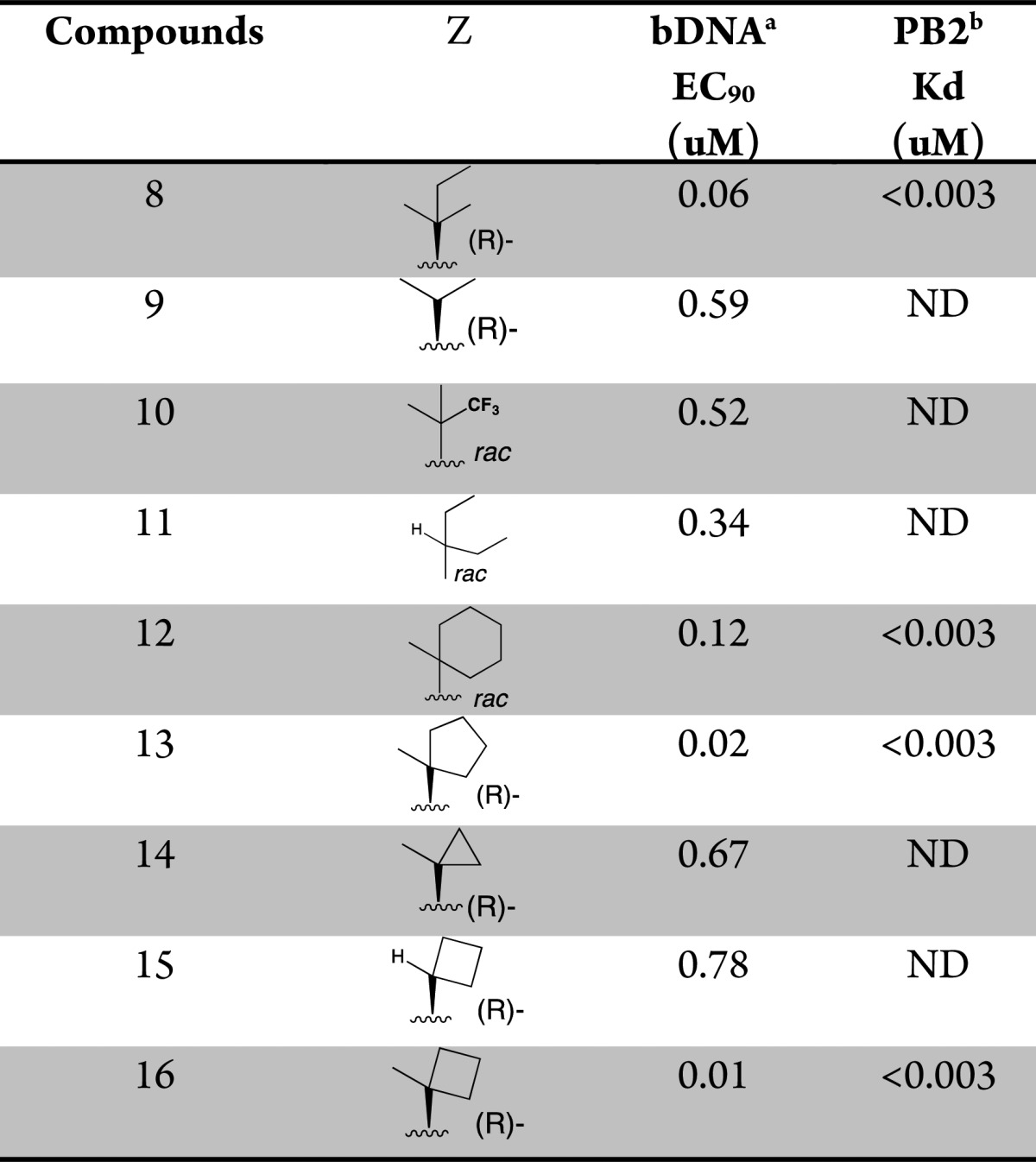
The concentration of test compounds resulting in viral RNA levels equal to that of 10% of the control wells was reported as EC90.
Affinity for cap-binding domain of the PB2 subunit as measured in a fluorescence polarization competition binding assay. ND = not determined.
Scheme 2. Synthesis of Varied β-Amino Acid Analogues.

Reagents and conditions: (a) Et3N, THF, t-BuOH, rt, 5 min.; (b) 55 °C, 62% (2 steps); (c) 23, K3PO4, X-Phos, 2-Me-THF, H2O, Pd2(dba)3, 75 °C, 3 h, 82%; (d) THF, MeOH, NaOH (2 M), 50 °C, 94%; (e) SFC chiral chromatography.
Suzuki cross coupling reaction with 5-fluoroazaindole boronate ester fragment 23(12) gave the fully assembled scaffold, which upon removal of the tosylate protecting group and hydrolysis of the ester followed by chromatographic chiral resolution delivered the desired product 16. The above general route was also used to prepare all other spirocyclic analogues (see Supporting Information).
The overall cellular potency and target-affinity of analogues 4 and 16 prompted us to further investigate their pharmacokinetic profiles and in vivo potency. The pharmacokinetic profile of compound 4 showed desirable iv and oral exposure in both rat (1 mg/kg iv, Cl = 8 mL min–1 kg–1 and t1/2 = 9.9 h; po (3 mg/kg) AUC = 3.3 μg·h/mL) and mouse (30 mg/kg dose, po AUC = 7.2 μg·h/mL and Cmax = 11.8 μg/mL). Compound 16 also showed good oral exposure in mouse (30 mg/kg dose, po AUC = 6.7 μg·h/mL and Cmax = 7.4 μg/mL). Additionally, evaluation in S9 fractions shows similar stability in rat (100% remaining at 1 μM) vs human (94% remaining).
Compound 4 demonstrates potent, antiviral activity in vitro (cell protection assay; CPE) against a broad range of influenza type A strains,13,14 including oseltamivir carboxylate resistant isolates and current pandemic H1N1 and H5N1 strains (Table 3; see also Table 4 in Supporting Information for comparative zanamivir and oseltamivir carboxylate activities in neuraminidase enzyme assay with these influenza strains/types). Compound 4 shows limited activity against influenza B (data not shown).
Table 3. Compound 4 Activity Against Oseltamivir Sensitive and Resistant Influenza A Viruses.
| virus name | type | oseltamivir R/S | 4 CP EC50 (μM)a |
|---|---|---|---|
| A/Georgia/17/2006 | A(H1N1)b | S | 0.009 |
| A/Georgia/20/2006 | A(H1N1)b | R | 0.002 |
| A/Puerto Rico/8/34 | A(H1N1)b | S | 0.014 |
| A/Henan/Jinshui/147/2007 | A(H3N2)b | S | 0.019 |
| A/Texas/12/2007 | A(H3N2)b | R | 0.030 |
| A/California/07/2009 | A(H1N1)c | S | 0.004 |
| A/Texas/48/2009 | A(H1N1)c | R | 0.002 |
| A/Vietnam/1203/2004 | A(H5N1)d | S | 0.004 |
Mean MDCK cell 3-day CP (cell protection) assay (MDCK cells incubated with test compounds and influenza A virus for 72 h and the concentration of test compound resulting in 50% cell protection was reported as EC50). N = 3.
Seasonal subtype.
Pandemic subtype.
Highly pathogenic avian influenza (H5N1) strain Oseltamivir R, resistant (determined via externally validated phenotypic or genetic analysis); S, sensitive.
Compound 4 was advanced into a mouse influenza model (Figure 5). It was dosed orally at 10, 30, and 60 mg/kg twice a day for 10 days starting 48 h after infection, and it showed complete survival benefit at all three doses. Additionally, compound 4 provided a dose-dependent decrease in body weight loss as compared to untreated controls.14 It is worth noting that oseltamivir (dosed 10 mg/kg BID) is known to be devoid of efficacy in this +48 h delay to treatment model.7,8,15
Figure 5.

In vivo activity of 4 and oseltamivir in mouse influenza A model when administered 48 h postinfection. Survival and body weight curves of male BALB/C mice (8 mice/group) inoculated with mouse-adapted influenza viruses A/PR/8/34 (5e3 TCID50/mouse) by intranasal instillation.
Similarly compound 16 showed complete survival benefit in the mouse influenza model when administered at 3, 10, and 30 mpk BID 48 h postinfection (see Figure 6; Supporting Information).
In summary, structure-guided optimization led to a set of novel tertiary beta-amino acid analogues. The most potent compounds 4 and 16 were found to be orally bioavailable and efficacious inhibitors in vivo. Further testing showed that compound 4 is very potent against all influenza A strains tested, including pandemic H1N1 and avian H5N1 flu strains. These data clearly indicate that both 4 and 16 are promising inhibitors of PB2 mediated viral influenza replication with respect to overall potency, efficacy, and extended treatment window. Both compounds possess therapeutic potential.
Acknowledgments
The authors thank Barry Davis for analytical chemistry support and Greg May for SFC chiral separation.
Glossary
ABBREVIATIONS
- DMF
dimethylformamide
- THF
tetrahydrofuran
- TFA
trifluoroacetic acid
- TEA
triethylamine
- LDA
lithium diisopropylamide
- X-Phos
2-dicyclohexylphosphino-2′4′6′-triisopropylbiphenyl
- DME
dimethoxyethane
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00486.
Protocols for PB2 binding assay; bDNA cellular assay; experimentals for key compounds; Figure 6 (PDF)
Author Present Address
† Paraza Pharma., Inc., 7171 Frederick-Banting, Montreal, Quebec H4S 1Z9, Canada.
Author Present Address
‡ Contrafect Corporation, 28 Wells Avenue, 3rd Floor, Yonkers, New York 10701, United States.
Author Present Address
§ Goldfinch Bio., 215 First Street, 4th Floor, Cambridge, Massachusetts 02142, United States.
Author Present Address
∥ DE Synthetics, Inc., 30 Dineen Drive, Fredericton, New Brunswick E3B 5A3, Canada.
Author Present Address
⊥ Arbutus Biopharma, Inc., 3805 Old Easton Road, Doylestown, Pennsylvania 18902, United States.
Author Present Address
# Moderna Therapeutics, Inc., 200 Technology Square, Cambridge, Massachusetts 02139, United States.
Author Present Address
∇ Vertex Pharmaceuticals (Canada) Inc., 275 Armand-Frappier, Laval, Quebec H7 V 4A7, Canada.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Centers for Disease Control and Prevention. Estimates of Deaths Associated with Seasonal Influenza: United States, 1976–2007; 2010, Vol. 59 ( (33), ), pp 1057–1062. [PubMed] [Google Scholar]
- Thompson W. W.; Shay D. K.; Weintraub E.; Brammer L.; Bridges C. B.; Cox N. J.; Fukuda K. Influenza-associated hospitalizations in the United States. JAMA 2004, 292, 1333–1340. 10.1001/jama.292.11.1333. [DOI] [PubMed] [Google Scholar]
- Cohen J. Surprising Twist in Debate Over Lab-Made H5N1. Science 2012, 335, 1155–1156. 10.1126/science.335.6073.1155. [DOI] [PubMed] [Google Scholar]
- Herfst S.; Schrauwen J. A.; Linster M.; Chutinimitkul S.; deWit E.; Munster V. J.; Sorrell E. M.; Bestebroer T. M.; Burke D. F.; Smith D. J.; Rimmelzwaan G. F.; Osterhaus A. D. M. E.; Fouchier R. A. M. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 2012, 336, 1534–1541. 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A. Neuraminidase inhibitors for influenza. N. Engl. J. Med. 2005, 353, 1363–1373. 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- Nitsch-Osuch A.; Byrdak L. B. Influenza viruses resistant to neuraminidase inhibitors. Acta Biochim Pol 2014, 61, 505–508. [PubMed] [Google Scholar]
- Clark M. P.; Ledeboer M. W.; Davies I.; Byrn R. A.; Jones S. M.; Perola E.; Tsai A.; Jacobs M.; Nti-Addae K.; Bandarage U. K.; Boyd M. J.; Bethiel R. S.; Court J. J.; Deng H.; Duffy J. P.; Dorsch W. A.; Farmer L. J.; Gao H.; Gu W.; Jackson K.; Jacobs D. H.; Kennedy J. M.; Ledford B.; Liang J.; Maltais F.; Murcko M.; Wang T.; Wannamaker M. W.; Leeman J. R.; McNeil C.; Taylor W. P.; Memmott C.; Rijnbrand R.; Bral C.; Germann U.; Nezami A.; Zhang Y.; Salituro F. G.; Bennani Y. L.; Charifson P. S. Discovery of a novel, first-in-class, orally bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2. J. Med. Chem. 2014, 57 (15), 6668–6678. 10.1021/jm5007275. [DOI] [PubMed] [Google Scholar]
- Guilligay D.; Tarendeau F.; Resa-Infante P.; Coloma R.; Crepin T.; Sehr P.; Lewis J.; Ruigrok R. W.; Ortin J.; Hart D. J.; Cusack S. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. 10.1038/nsmb.1421. [DOI] [PubMed] [Google Scholar]
- Pautus S.; Sehr P.; Lewis J.; Fortune A.; Wolkerstorfer A.; Szolar O.; Guilligay D.; Lundardi T.; Decout J.-L.; Cusack S. New 7- methylguanine derivatives targeting the influenza polymerase PB2 cap- binding domain. J. Med. Chem. 2013, 56, 8915–8930. 10.1021/jm401369y. [DOI] [PubMed] [Google Scholar]
- Fechter P.; Mingay L.; Sharps J.; Chambers A.; Fodor E.; Brownlee G. G. Two aromatic residues in the PB2 subunit of influenza A RNA polymerase are crucial for cap binding. J. Biol. Chem. 2003, 278, 20381–20388. 10.1074/jbc.M300130200. [DOI] [PubMed] [Google Scholar]
- Palese P.; Shaw M. L.. Orthomyxoviridae: The viruses and their Replication. In Fields Virology; Knipe D. M., Howley P. M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, 2007; pp 1647–1689. [Google Scholar]
- Charifson P. S.; Clark M. P.; Bandarage U. K.; Bethiel R. S.; Boyd M. J.; Davies I.; Deng H.; Duffy J. P.; Farmer L. J.; Gao H.; Gu W.; Kennedy J. M.; Ledford B.; Ledeboer M. W.; Maltais F.; Perola E.; Wang T.. Inhibitors of influenza viruses replication. WO 2013/019828, February 2, 2013.
- Wagaman P. C.; Leong M. A.; Simmen K. A. Development of a novel influenza A antiviral assay. J. Virol. Methods 2002, 105, 105–114. 10.1016/S0166-0934(02)00088-5. [DOI] [PubMed] [Google Scholar]
- Byrn R. A.; Jones S.; Bennett H. B.; Bral C.; Clark M. P.; Jacobs M. D.; Kwong A. D.; Ledeboer M. W.; Leeman J. R.; McNeil C. F.; Murcko M. A.; Nezami A.; Perola E.; Rijnbrand R.; Saxena K.; Tsai A. W.; Zhou Y.; Charifson P. S. Preclinical activity of VX-787, a first-in-class, orally bioavailable inhibitor of the influenza viral polymerase PB2 subunit. Antimicrob. Agents Chemother. 2015, 59 (3), 1569–1582. 10.1128/AAC.04623-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A. W.; McNeil C. F.; Leeman J. R.; Bennett H. B.; Nti-Addae K.; Huang C.; Germann U. A.; Byrn R. A.; Berlioz-Seux F.; Rijnbrand R.; Clark M. P.; Charifson P. S.; Jones S. M. Novel ranking system for identifying efficacious anti-influenza virus PB2 inhibitors. Antimicrob. Agents Chemother. 2015, 59 (10), 6007–6016. 10.1128/AAC.00781-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.