

Abstract

Herein, we report the structure–activity relationships within a series of potent, selective, and orally bioavailable muscarinic acetylcholine receptor 4 (M4) positive allosteric modulators (PAMs). Compound 6c (VU0467485) possesses robust in vitro M4 PAM potency across species and in vivo efficacy in preclinical models of schizophrenia. Coupled with an attractive DMPK profile and suitable predicted human PK, 6c (VU0467485) was evaluated as a preclinical development candidate.
Keywords: Positive allosteric modulator (PAM), muscarinic acetylcholine receptor 4 (M4), VU0467485, schizophrenia
The rapid onset of clinical efficacy of the M1/M4 preferring agonist xanomeline in both schizophrenic and Alzheimer’s patients led to a revolution in muscarinic receptor drug discovery efforts targeting allosteric mechanisms to afford highly subtype selective M1 and M4 positive allosteric modulators (PAMs).1−6 Of these, M4 has emerged as the favored mAChR subtype responsible for antipsychotic-like efficacy as well as modest cognitive enhancement in multiple preclinical rodent models via a fundamentally new molecular mechanism.7−15 While M1 PAMs with diverse chemotypes (and without major species differences in pharmacology) have rapidly advanced to potentially address cognitive dysfunction and negative symptoms,5,6,16,17 M4 PAMs have progressed more slowly for many reasons, including species differences in M4 PAM potency (i.e., affinity and cooperativity), challenges with respect to M2 selectivity, and P-gp efflux as well as limited chemical diversity (Figure 1).9−14,18−20 Clearly, these are significant roadblocks en route to an M4 PAM preclinical candidate. In this Letter, we detail our navigation of these issues within the VU0467154 series of M4 PAMs, leading to the discovery of a potent, selective, and orally bioavailable M4 PAM 6c (VU0467485) with robust efficacy in behavioral models that was evaluated as a preclinical candidate. This is the first disclosure of the structure–activity relationships (SAR) and preclinical profile of 6c.
Figure 1.

Structures of reported M4 PAMs. LY2033298 (1) was the first reported M4 PAM but was human-preferring in potency. VU0152100 (2) was the first centrally active M4 PAM in rodents, and VU0467154 (3) has proven to be the best-in-class M4 PAM rodent in vivo tool compound.
Our discovery of 6c originated from an optimization campaign centered on 2, and from which 3 was identified, but discontinued due to a human and rat M4 PAM potency disconnect that precluded development.10,12−14 A major thrust of the initial lead optimization effort was to survey alternatives for the pyridine ring in 2, and while other pyridine regioisomers and pyrimidines afforded modest M4 potentiation, the pyridazine core, found in 3, proved optimal for imparting both potency, metabolic stability, and favorable physiochemical properties to the series.21 SAR was steep and divergent across rat and human M4, and CNS penetration was low in this series; however, exceptional DMPK properties could be achieved.21
The synthesis of analogue 6 was straightforward and required only two steps from known materials (Scheme 1).12−14 Condensation of 3-chloro-5,6-dimethylpyridazin-4-carbonitrile 4 with methylthioglycolate under microwave irradiation delivered the core sodium 5-amino-3,4-dimethylthieno[2,3-c]pyridazine-6-carboxylate 5 in yields averaging 78%. Next, a HATU-mediated amide coupling reaction with various amines provides analogues 6 in yields ranging from 45 to 92% after HPLC purification.21 The direct congener of 2, VU0464090 (6a), demonstrated a ∼3-fold increase in M4 PAM potency (human M4 PAM EC50 = 130 nM, pEC50 = 6.89 ± 0.06, 83.7 ± 3.5 ACh Max and rat M4 PAM EC50 = 59.7 nM, pEC50 = 7.22 ± 0.06, 78.1 ± 1.7 ACh Max), increased fraction unbound in plasma (fuplasma(r,h) = 0.022, 0.035) and reduced hepatic microsomal intrinsic clearance (CLint(r,h) = 81 and 36 mL/min/kg; predicted CLhep(r,h) = 37 and 13 mL/min/kg) by virtue of the pyridazine ring system; however, metabolic instability of the PMB group precluded further advancement of 6a.14 As shown in Table 1, application of the fluorine walk strategy22−24 for allosteric modulator optimization proved fruitful, affording a number of potent M4 PAMs (6c–h). Heteroatom incorporation into the benzyl ring was met with limited success, with only 3-pyridyl congeners (6i and 6j) retaining PAM activity (other regioisomeric pyridines, pyrimidines, pyrazines, and pyridazines were inactive, M4 EC50s > 10 μM). Moreover, we suspected CYP450-mediated oxidative dealkylation of 6a led to 6b, an active putative metabolite (human M4 PAM EC50 = 96.7 nM, pEC50 = 7.01 ± 0.01, 69.8 ± 2.2 ACh Max), but with high clearance in vivo (rat) and undesired activity at hM2. As a battery of in vitro and in vivo DMPK assays (vide infra) quickly identified 6c as the most attractive PAM in the series, we prepared its putative dealkylated metabolite 6k, which also proved to be active (human M4 EC50 = 59 nM), but with no activity at hM2, suggesting the ortho-fluoro moiety enhances selectivity versus hM2. Incorporation of chiral methyl groups at the benzylic position of 6c led to (R)-6l (human M4 EC50 > 10 μM) and (S)-6m (human M4 EC50 = 508 nM), where enantiospecific activity was noted, but with unacceptable loss in human M4 PAM potency. Finally, based on previous beneficial disposition by virtue of the kinetic isotope effect, we evaluated a deuterated congener, but in this instance, there was no benefit to stability.
Scheme 1. Synthesis of Analogues 6.

Reagents and conditions: (a) methyl thioglycolate, MeOH, 1 M aq. NaOH, 150 °C, microwave, 30 min, 78%; (b) NH2CH2Ar(Het), HATU, DMF, DIEPA, 2 h, 45–92%.
Table 1. Structures and Activities of Analogues 6.
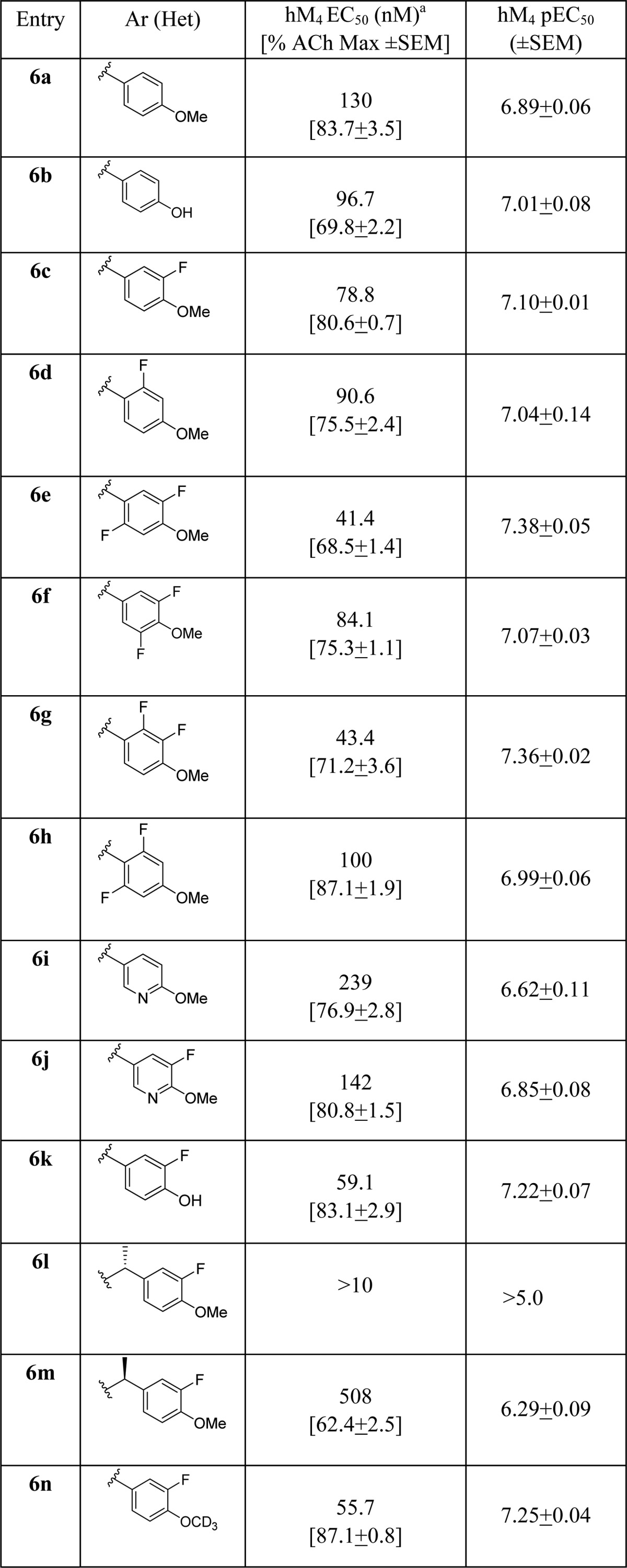
Calcium mobilization assays with hM4/Gqi5-CHO cells performed in the presence of an EC20 fixed concentration of acetylcholine; values represent means from three (n = 3) independent experiments performed in triplicate.
Thus, efforts focused on 6c as a potential M4 PAM preclinical candidate with minimal species differences in PAM potency.
The molecular pharmacology profile of M4 PAM 6c is shown in Figure 2. PAM 6c is inactive in the absence of acetylcholine (ACh), but in the presence of an EC20 concentration of ACh (Figure 2A), 6c potentiates M4 in a concentration-dependent manner, affording potent activity at both human and rat M4 (human M4 PAM EC50 = 78.8 nM, pEC50 = 7.10 ± 0.01, 80.6 ± 0.7 ACh Max and rat M4 PAM EC50 = 26.6 nM, pEC50 = 7.57 ± 0.05, 68.7 ± 3.4 ACh Max). For a preclinical candidate, comparable activity across species is essential for IND-enabling toxicology studies, and this was a major issue for previously reported M4 PAMs for which the species disconnect averaged 10–50-fold.9−14,18−20 Thus, we also evaluated 6c against dog and cynomolgous monkey M4 and the key antitarget, M2. Beyond rat and human, 6c displayed no major species differences in potency (dog M4 EC50 = 87 nM, 49% ACh Max, dog M2 > 30 μM and cyno M4 EC50 = 102 nM, 74% ACh Max, cyno M2 > 30 μM). In a progressive fold-shift assay with human M4 (Figure 2B), 6c afforded a maximal ∼45-fold leftward shift of the human M4 ACh concentration–response curve (CRC) at 10 μM (∼40-fold shift at rat M4). Moreover, PAM 6c was selective versus human and rat M1–3,5. The operational model of allosterism was applied to the fold-shift data to better understand the allosteric effects of this promising candidate.5,22 PAM 6c displayed robust potentiation of ACh and cooperativity (log αβ = 2.1, αβ = 134), significant intrinsic efficacy (log τB = 5.1), and an approximately 1 μM estimated affinity (pKB 6.025, KB = 944 nM) at human M4.
Figure 2.

Molecular pharmacology profile of M4 PAM 6c. (A) Enhanced intracellular calcium release induced by a subthreshold concentration of acetylcholine (EC20), a PAM CRC on both rat and human M4, with EC50s of 26.6 and 78.8 nM, respectively. (B) Compound 6c induces a ∼45-fold maximal leftward shift of the hM4 acetylcholine response curve at 10 μM. (C) Compound 6c is highly selective for hM4 over hM1–3,5. (D) Compound 6c is highly selective for rM4 over rM1–3,5. Data represent means from at least three independent determinations performed in triplicate using CHO cells stably transfected with the indicated mAChR.
Compound 6c was then evaluated in a panel of in vitro DMPK assays23 where it displayed properties that supported continued progression. Not only did 6c possess an exceptionally clean CYP450 inhibition (3A4, 2D6, 2C9, 1A2 IC50s > 30 μM in human hepatic microsomes) and induction profile (3A4, 1A2, 2B6 EC50s > 50 μM, Emaxs ≤ 1.0 in cryopreserved human hepatocytes), but also moderate plasma protein binding was noted across species (fuplasma(r,h,c) = 0.031, 0.054, and 0.091) with moderate fraction unbound in rat brain homogenate (fubr = 0.037). Based on hepatic microsomal CLint data, moderate predicted hepatic clearance was also observed for 6c (predicted CLhep(r,h,c) = 73, 26, and 38 mL/min/kg). Moreover, 6c was not a P-gp substrate (ER = 1.4 in MDCK-MDR1) and showed good apparent permeability (Caco-2 Papp = 31 × 10–6 cm/s). In rat and dog brain distribution studies, 6c displayed moderate to high CNS penetration with Kps of 0.31 to 1.0 and Kp,uus of 0.37 to 0.84. With regard to toxicity evaluation in vitro, 6c was clean in GSH trapping experiments (human hepatic microsomes) and a mini-Ames test (data not shown). In vivo PK (Table 2) was assessed in three species (rat, dog, and cynomolgous monkey), which revealed moderate clearance, low to moderate volume of distribution at steady-state, and short to moderate elimination half-life with high oral bioavailability in rat but low bioavailability in dog (3 mg/kg suspension dose of a mono-HCl salt). In vitro metabolite identification experiments found good coverage of human metabolites in dog and cynomolgous monkey and no evidence for human-unique metabolites (Figure 3). Human CYP450 phenotyping experiments revealed that multiple CYPs contribute to 6c’s metabolism (Figure 4) with a generally low potential for drug–drug interactions (no metabolism-related DDI liabilities anticipated in Alzheimer’s disease clinical population as concomitant medications (acetylcholinesterase inhibitors possess a 3A4/2D6 phenotype) and, in schizophrenia populations, common antipsychotics (e.g., clozapineolanzapine) possess a 1A2/2D6/3A4 phenotype). Furthermore, in a functional hERG assay, 6c was inactive when tested at 11 μM, as well as against a larger cardiac ion channel panel (IC50s > 33 μM). Finally, ancillary pharmacology was assessed in an internal AZ/Cerep panel against 200 targets, and no significant off-target activities (IC50s or EC50s > 10 μM) were noted, including both binding and functional assays for cardiac ion channels, with the exception of a 1.2 μM IC50 (radioligand binding) at the rat GABAA receptor.21
Table 2. Pharmacokinetic Parameters of 6c.
| parameter | rata (SD) | doga (beagle or mongrel) | NHPa (cyno) |
|---|---|---|---|
| dose (mg/kg) i.v./p.o. | 1/3 | 1/3 | 0.2 (iv only) |
| CLp (mL/min/kg) | 29 | 21 | 25 |
| Vss (L/kg) | 1.5 | 1.5 | 0.88 |
| elimination t1/2 (h) | 4.2 | 1.3 | 0.48 |
| Cmax (μM) p.o. | 1.2 | 0.22 | |
| Tmax (h) p.o. | 1.5 | 0.75 | |
| AUCo-inf (μM·h) p.o. | 3.8 | 0.59 | |
| F (%) p.o. | 79 | 9.5 | |
| total brain/total plasma (Kp) | 0.31 | 1.0 | |
| unbound brain/unbound plasma (Kp,uu) | 0.37 | 0.84 |
Values represent means from two to three animals.
Figure 3.

Metabolite identification studies for 6c across species (rat, dog, cyno monkey, and human).
Figure 4.
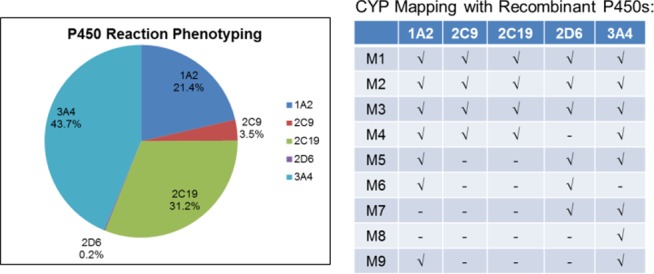
P450 phenotyping and CYP mapping for 6c, indicating that multiple CYP450s (3A4/2C19/1A2 and to a lesser extent, 2C9) catalyze biotransformation.
Evaluation in a rat amphetamine-induced hyperlocomotion (AHL) study, a traditional preclinical model of antipsychotic activity,10,12−14 revealed that 6c (Figure 5) showed robust activity with a minimum effective dose (MED) of 10 mg/kg p.o. (43.2% reversal) that correlates with terminal (time = 1.5 h) plasma and brain concentrations of 1.8 μM (0.06 μM unbound) and 0.56 μM (0.02 μM unbound), respectively, and showing greater efficacy than our previously reported rat tool M4 PAM, VU0467154.13,14 Beyond hyperdopaminergic states, we also assessed the ability of 6c to reverse hyperlocomotion induced by the N-Methyl-d-Aspartate (NMDA) receptor antagonist MK-801 (Figure 6) to pharmacologically model NMDA receptor hypofunction and the associated prefrontal cortex-mediate impairments.13,24,25 Here as well, 6c displayed a dose-responsive reversal of MK-801-induced hyperlocomotion, with an MED of 30 mg/kg p.o. and a 41.1% reversal.
Figure 5.

Compound 6c has antipsychotic-like activity in an AHL rat model. Compound 6c dose-dependently (1–10 mg/kg, po) reverses AHL (amphetamine, 0.75 mg/kg, s.c., *p < 0.05 vs vehicle + amphetamine, **p < 0.01 vs vehicle + amphetamine, ***p < 0.001 vs vehicle + amphetamine). N = 6–8 rats/group.
Figure 6.

Compound 6c has antipsychotic-like activity in an MK-801 hyperlocomotion rat model. Compound 6c dose-dependently (10−30 mg/kg, po) reverses MK-801-induced hyperlocomtion (MK-801, 0.18 mg/kg, s.c., *p < 0.05 vs vehicle + MK-801. N = 6−8 rats /group.
Based on the fact that 6c was the first M4 PAM discovered in our program with similar M4 PAM activity across all preclinical species and man, an attractive DMPK and ancillary pharmacology profile, as well as robust efficacy in a rodent model of antipsychotic activity, 6c was further profiled as a putative preclinical candidate, including evaluation in a rat modified Irwin test for effects on autonomic and somatomotor functions. In this study, following a single high dose (56.6 mg/kg, p.o., N = 6), no significant effects were observed over a 6 h observation period. However, based on these animal model acute concentration-effect data (rat AHL) and 6c’s predicted human PK profile (Table 3), efficacious human oral doses projected to provide 12-h daily coverage of the target/mechanism were undesirably high and frequent (e.g., >450 mg, TID).
Table 3. Predicted Human Pharmacokinetics of 6c.
| parameter | value |
|---|---|
| CL (mL/min/kg)a | 3.7–8.9 |
| Vss (L/kg)b | 1.5–2.1 |
| t1/2 (h) | 1.9–6.6 |
| F (%)c | 71 |
| ka (1/h)d | 0.55 |
Predicted by multispecies IVIVE.
Predicted by scaling of unbound Vss from rat and dog.
Predicted by assumption of an optimized form/formulation providing an fabs of 1.0 with an fgut of 1.0 and fhep of 0.71 (i.e., ERhep = 0.29 based on mean predicted human CL/Qhep).
Predicted by the MAT method using rat oral PK data from a 3 mg/kg dose of the mono-HCl salt formulated as a suspension in 0.1% tween80 and 0.5% methylcellulose in water.
Additionally, 6c displayed low aqueous solubility (2.4 μM at pH 7.4) and evidence for solubility-limited absorption in dog was observed, even at low doses and when administered as an HCl salt.
Still, 6c represents a major advance in the field, as the first potent M4 PAM to overcome major species differences in potency while maintaining high selectivity versus M2 (rat, dog, cyno, and human EC50s > 30 μM), CNS penetration, and in vivo efficacy. However, given the projected human efficacious dosing and solubility issues, together with an anticipated challenge in achieving sufficiently high oral exposure in IND-enabling safety studies to establish acceptable margins, further advancement of 6c was halted (pending pharmaceutical sciences work), and optimization efforts shifted toward improvement of aqueous solubility and longer predicted human t1/2 while retaining all the desirable properties of 6c. Results from this ongoing work will be reported in due course.
Acknowledgments
The authors would also like to thank NIH (U01MH087965, Vanderbilt NCDDG). We also thank William K. Warren, Jr. and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.).
Glossary
ABBREVIATIONS
- DCM
dichloromethane
- AHL
amphetamine-induced hyperlocomotion
- MED
minimum effective dose
- mAChR
muscarinic acetylcholine receptor
- PAM
positive allosteric modulator
- HATU
O-(7-azabenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DIEPA
N,N-diisopropylamine
- MED
minimum effective dose
- AHL
amphetamine-induced hyperlocomotion
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00461.
General methods for the synthesis and characterization of all compounds, and methods for the in vitro and in vivo DMPK protocols and supplemental tables(PDF)
Author Contributions
⊥ M.R.W. and M.J.N contributed equally to this work. C.W.L., T.M.B., M.R.W., P.J.C., C.M.N., N.J.B., M.E.D., and C.J.K. drafted/corrected the manuscript. B.J.M., K.D.N., M.S.P., M.A.H., and D.W.E. performed the chemical synthesis. C.W.L., M.W.R., T.M.B., P.J.C., C.M.N., C.J.K., M.J.N., N.J.B., M.W.W., M.E.D., and A.L.R. oversaw the target selection and interpreted the biological data. M.J.N., A.L.R., A.L., V.B.L., and R.L.W. performed the in vitro molecular pharmacology studies. T.M.B., A.L.B., and S.C. performed the in vitro and in vivo DMPK studies. C.K.J. and M.B. performed the in vivo experiments. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): The authors are developing M4 PAMs for the treatment of schizophrenia and hold patents on the same.
Supplementary Material
References
- Bodick N. C.; Offen W. W.; Levey A. I.; Cutler N. R.; Gauthier S. G.; Satlin A.; Shannon H. E.; Tollefson G. D.; Rasmussen K.; Bymaster F. P.; Hurley D. J.; Potter W. Z.; Paul S. M. Effects of Xanomeline, a Selective Muscarinic Receptor Agonist, on Cognitive Function and Behavioral Symptoms in Alzheimer Disease. Arch. Neurol. 1997, 54, 465–473. 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- Shekhar A.; Potter W. Z.; Lightfoot J.; Lienemann J.; Dube S.; Mallinckrodt C.; Bymaster F. P.; McKinzie D. L.; Felder C. C. Selective muscarinic receptor agonist Xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry 2008, 165, 1033–1039. 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Raedler T. J.; Bymaster F. P.; Tandon R.; Copolov D.; Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry 2007, 12, 232–246. 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- Bridges T. M.; LeBois E. P.; Hopkins C. R.; Wood M. R.; Jones J. K.; Conn P. J.; Lindsley C. W. Antipsychotic potential of muscarinic allosteric modulation. Drug News Perspect. 2010, 23, 229–240. 10.1358/dnp.2010.23.4.1416977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J.; Lindsley C. W.; Meiler J.; Niswender C. M. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for the treatment of CNS disorders. Nat. Rev. Drug Discovery 2014, 13, 692–708. 10.1038/nrd4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menniti F. S.; Lindsley C. W.; Conn P. J.; Pandit J.; Zagouras P.; Volkmann R. A. Allosteric modulation for the treatment of schizophrenia: Targeting glutamatergic networks. Curr. Top. Med. Chem. 2013, 13, 26–54. 10.2174/1568026611313010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubser M.; Byun N.; Wood M. R.; Jones C. K. Muscarinic receptor pharmacology and circuitry for the modulation of cognition. Handb. Exp. Pharmacol. 2012, 208, 121–166. 10.1007/978-3-642-23274-9_7. [DOI] [PubMed] [Google Scholar]
- Jones C. K.; Byun N.; Bubser M. Muscarinic and nicotinic acetylcholine receptor agonists and allosteric modulators for the treatment of schizophrenia. Neuropsychopharmacology 2012, 37, 16–42. 10.1038/npp.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan W. Y.; McKinzie D. L.; Bose S.; Mitchell S. N.; Witkin J. M.; Thompson R. C.; Christopoulos A.; Lazareno S.; Birdsall N. J.; Bymaster F. P.; Felder C. C. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10978–10983. 10.1073/pnas.0800567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady A. E.; Jones C. K.; Bridges T. M.; Kennedy J. P.; Thompson A. D.; Heiman J. U.; Breininger M. L.; Gentry P. R.; Yin H.; Jadhav S. B.; Shirey J. K.; Conn P. J.; Lindsley C. W. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J. Pharmacol. Exp. Ther. 2008, 327, 941–953. 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K.; Loiacono R. E.; Felder C. C.; McKinzie D. L.; Mogg A.; Shaw D. B.; Sexton P. M.; Christopoulos A. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology 2010, 35, 855–869. 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun N. E.; Grannan M.; Bubser M.; Barry R. L.; Thompson A.; Rosanelli J.; Gowrishankar R.; Kelm N. D.; Damon S.; Bridges T. M.; Melancon B. J.; Tarr J. C.; Brogan J. T.; Avison M. J.; Deutch A. Y.; Wess J.; Wood M. R.; Lindsley C. W.; Gore J. C.; Conn P. J.; Jones C. K. Antipsychotic drug-like effects of the selective M4 muscarinic acetylcholine receptor positive allosteric modulator VU0152100. Neuropsychopharmacology 2010, 39, 1578–1593. 10.1038/npp.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubser M.; Bridges T. M.; Thorbeck D. D.; Gould R. W.; Grannan M.; Noetzel M. J.; Niswender C. M.; Daniels J. S.; Melancon B. J.; Tarr J. C.; Wess J.; Duggan M. E.; Brandon N. J.; Dunlop J.; Wood M. W.; Wood M. R.; Lindsley C. W.; Conn P. J.; Jones C. K. Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem. Neurosci. 2014, 5, 920–942. 10.1021/cn500128b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M. R.; Noetzel M. J.; Melancon B. J.; Nance K. D.; Poslunsey M. S.; Hurtado M. A.; Luscombe V. B.; Weiner R. L.; Rodriguez A. L.; Lamsal A.; Chang S.; Engers D. W.; Niswender C. M.; Jones C. K.; Brandon N. J.; Wood M. W.; Duggan M. E.; Conn P. J.; Bridges T. M.; Lindsley C. W. Challenges in the development of an M4 PAM in vivo tool compound: The discovery of VU0467154 and unexpected DMPK profiles of close analogs. Bioorg. Med. Chem. Lett. 2016, 10.1016/j.bmcl.2016.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster D. J.; Wilson J. M.; Wess J.; Jones C. K.; Xiang Z.; Lindsley C. W.; Rook J. M.; Conn P. J. M4 activation reduces striatal dopamine release and has antipsychotic-like effects via a CB2 cannabinoid receptor-dependent mechanism. Neuron 2016, 91, 1244–1252. 10.1016/j.neuron.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melancon B. J.; Tarr J. C.; Panarese J. D.; Wood M. R.; Lindsley C. W. Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discovery Today 2013, 18, 1185–1199. 10.1016/j.drudis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grannan M. D.; Mielnik C. A.; Moran S. P.; Gould R. W.; Ball J.; Bubser M.; Ramsey A. J.; Abe M.; Cho H. P.; Nance K. D.; Blobaum A. L.; Niswender C. M.; Conn P. J.; Lindsley C. W.; Jones C. K. Prefrontal cortex-mediated impairments in a genetic model of NMDA receptor hypofunction are reversed by the novel M1 PAM VU6004256′. ACS Chem. Neurosci. 2016, 10.1021/acschemneuro.6b00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le U.; Melancon B. J.; Bridges T. M.; Utley T. J.; Lamsal A.; Vinson P. N.; Sheffler D. J.; Jones C. K.; Morrison R.; Wood M. R.; Daniels J. S.; Conn P. J.; Niswender C. M.; Lindsley C. W.; Hopkins C. R. Discovery of a selective M4 positive allosteric modulator based on the 3-amino-thieno[2,3-b]pyridine-2-carboxamide scaffold: development of ML253, a potent and brain penetrant compound that avoids species bias. Bioorg. Med. Chem. Lett. 2013, 23, 346–350. 10.1016/j.bmcl.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salovich J. M.; Sheffler D. J.; Vinson P. N.; Lamsal A.; Utley T. J.; Blobaum A. L.; Bridges T. M.; Le U.; Jones C. K.; Wood M. R.; Daniels J. S.; Conn P. J.; Niswender C. M.; Lindsley C. W.; Hopkins C. R. ‘Discovery of N-(4-methoxy-7-methylbenzo[d]thiazol-2-yl)isonicatinamide, ML293, as a novel, selective and brain penetrant positive allosteric modulator of the muscarinic 4 (M4) receptor. Bioorg. Med. Chem. Lett. 2012, 22, 5084–5088. 10.1016/j.bmcl.2012.05.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M. R.; Noetzel M. J.; Engers J. L.; Bollinger K. A.; Melancon B. J.; Tarr J. C.; Han C.; West M.; Gregro A. R.; Lamsal A.; Chang S.; Ajmera S.; Smith E.; Chase P.; Hodder P. S.; Bubser M.; Jones C. K.; Hopkins C. R.; Emmitte K. A.; Niswender C. M.; Wood M. W.; Duggan M. E.; Conn P. J.; Bridges T. M.; Lindsley C. W. Discovery and optimization of a novel series of highly CNS penetrant M4 PAMs based on a 5,6-dimethyl-4-(piperdin-1-yl)thieno[2,3-d]pyrimidine core. Bioorg. Med. Chem. Lett. 2016, 26, 3029–3033. 10.1016/j.bmcl.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information for full details.
- Lindsley C. W.; Emmitte K. A.; Hopkins C. R.; Bridges T. M.; Gregory K. A.; Niswender C. M.; Conn P. J. Practical strategies and concepts in GPCR allosteric modulator discovery: Recent advances with metabotropic glutamate receptors. Chem. Rev. 2016, 116, 6707–6741. 10.1021/acs.chemrev.5b00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde-Ceide S.; Martin-Martin M. L.; Alcazar J.; Manka T.; Tong H. M.; Garcia-Barrantes P. M.; Lavreysen H.; Mackie C.; Vinson P. N.; Daniels S. J.; Menges A.; Niswender C. M.; Jones C. K.; Macdonald G. J.; Steckler T.; Conn P. J.; Stauffer S. R.; Bartolome-Nebreda J. M.; Lindsley C. W. Discovery of VU0409551/JNJ-46778212: An mGlu5 Positive Allosteric Modulator Clinical Candidate Targeting Schizophrenia. ACS Med. Chem. Lett. 2015, 6, 716–720. 10.1021/acsmedchemlett.5b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagmann W. K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- Ghoshal A.; Rook J.; Dickerson J.; Roop G.; Morrison R.; Jalan-Sakrikar N.; Lamsal A.; Noetzel M.; Poslunsey M. S.; Wood M. R.; Melancon B. J.; Stauffer S. R.; Xiang Z.; Daniels J. S.; Niswender C. M.; Jones C. K.; Lindsley C. W.; Conn P. J. Selective potentiation of M1 muscarinic receptors reverses deficits in plasticity and negative and cognitive symptoms in repeated phencyclidine treated mouse model of schizophrenia. Neuropsychopharmacology 2016, 41, 598–610. 10.1038/npp.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.